

Abstract
Background:
Mechanisms of resistance to immune-modulating cancer treatments are poorly understood. Using a novel cohort of patients with head and neck squamous cell carcinoma (HNSCC), we investigated mechanisms of immune escape from epidermal growth factor receptor-specific monoclonal antibody (mAb) therapy.
Methods:
HNSCC tumors (n = 20) from a prospective trial of neoadjuvant cetuximab monotherapy underwent whole-exome sequencing. Expression of killer-cell immunoglobulin-like receptor (KIR) and human leukocyte antigen-C (HLA-C) and the effect of KIR blockade were assessed in HNSCC cell lines.
Results:
Nonresponders to cetuximab had an increased rate of mutations in HLA-C compared to responders and HNSCC tumors (n = 528) in The Cancer Genome Atlas (P < 0.00001). In vitro, cetuximab-activated natural killer (NK) cells induced upregulation of HLA-C on HNSCC cells (P < 0.01) via interferon gamma. Treatment of NK cells with the anti-KIR mAb lirilumab increased killing of HNSCC cells (P < 0.001).
Conclusions:
Alterations in HLA-C may provide a mechanism of immune evasion through disruption of NK activation.
Keywords: cetuximab, head and neck cancer, HLA, immunogenomics, NK cells
1. | INTRODUCTION
The majority of patients with head and neck squamous cell carcinoma (HNSCC) were seen with advanced locoregional disease.1–4 Despite significant advances in our understanding of the biology of HNSCC, little progress has been made in extending the survival of patients with advanced disease. Immunotherapeutic approaches to treating HNSCC, as well as other cancers, are of great interest to the scientific and oncologic communities. Although excitement surrounding the concept of harnessing the host’s own immune response to eradicate a tumor has been present for decades, only recently have immunotherapy approaches been shown to improve survival in patients with advanced stage cancer.5 Currently, interest is focused on immunomodulation with tumor antigen (TA)-specific monoclonal anti-bodies (mAb). Many of these approaches have concentrated on reversing the suppressive phenotype of cytotoxic T lymphocytes (CTL) through immune checkpoint receptor (ICR) inhibition with mAbs, such as anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab). Although initial results with ICRs have been promising, only a small percentage of patients attain a durable response.5 Interestingly, immune modulation in HNSCC has been achieved by targeted mAb therapy as well. Cetuximab, an immunoglobulin G-1 (IgG1) mAb specific for the epidermal growth factor receptor (EGFR), effectively inhibits ligand binding. Although the EGFR is upregulated in approximately 90% of HNSCC,6 less than 20% of patients achieve a lasting response to cetuximab.7,8 Initially, this was thought to be as a result of alterations in the EGFR or downstream molecules; however, such alterations are infrequent in HNSCC9,10
The hypothesis that the antitumor effects of cetuximab are driven by immune mechanisms is supported by a number of lines of evidence, including that lymphocytes are necessary for cetuximab to induce tumor killing in vitro11,12 and that cetuximab’s effects are influenced by Fcγ receptors present on natural killer (NK) cells.13 Work done by our lab11,13–21 and others12,22–27 supports the hypothesis that NK cells mediate cetuximab’s antitumor effects via antibody-dependent cellular cytotoxicity (ADCC) and through upregulation of EGFR-specific CTL via NK: dendritic cell (DC) crosstalk. NK-dependent ADCC is mediated by binding of FcγRDIa (CD16) to the Fc portion of cetuximab-coated EGFR+ tumor cells. Additionally, NK cell activation involves the interaction of killer-cell immunoglobulin-like receptors (KIRs) on NK cells with human leukocyte antigen-C (HLA-C) expressed on tumor cells. Antigen presentation through HLA class I molecules activates CTL as part of the adaptive immune response. However, HLA molecules also interact with KIRs on NK cells modulating the balance between inhibitory and activating signals and thus tolerance versus killing. Although incompletely understood, previous work has shown that KIRs on NK cells can be activating or inhibitory, depending on both the HLA-C allele and KIR molecule.28,29 Interestingly, HNSCC appears to have a particularly robust NK cell response compared to other tumor types.30
Mechanisms of resistance to cetuximab in HNSCC are poorly understood. Failure of NK cell activation or immune evasion of NK cell killing has been hypothesized to be mechanisms of resistance. However, direct evidence in HNSCC is currently lacking. To investigate the mechanisms mediating response or resistance to cetuximab therapy, we recently conducted a prospective clinical trial of neoadjuvant cetuximab monotherapy (UPCI #08–013, NCT #01218048) in previously untreated patients with stage III/IV HNSCC. Response to treatment was assessed by change in tumor size on CT from precetuximab treatment to postcetuximab treatment and before definitive surgery (%ΔCT). This format has been used successfully in previous trials designed to evaluate brief exposure to a study drug, in which categorical Response evaluation criteria in solid tumors (RECIST) would not be informative. Response status was defined using well-characterized RECIST measurement techniques to establish the sum of index lesions at baseline and after treatment and then used the proportional change in tumor size, which resulted in a 35% response rate (see Supporting Information for trial protocol). This novel cohort is optimized to characterize the immunogenomic correlates of cetuximab treatment response. Herein, we report results of this genomic analysis as well as in vitro experimental correlates.
2. | METHODS
Clinical trial UPCI #08–013 sample collection and processing:
Informed consent:
All patients underwent informed consent as part of UPCI #08–013. The clinical trial, and this study, was approved by the UPMC Institutional Review Board.
Sequencing library preparation and whole-exome sequencing:
Tumor DNA was extracted using the DNeasy kit (Qiagen). Peripheral blood DNA was extracted using the PAXgene DNA extraction kit (Qiagen). Sequencing libraries were prepared using SureSelect Human All Exon V5 and SureSelect Reagent Kit (Agilent). Sequencing was performed on an Illumina HiSeq 4000 or 2500 as 100-bp-paired end reads.
Data analysis:
FASTQ files were mapped to human reference genome (hg19) using the BWA mem v0.7.12 algorithm at default parameters. Removal of duplicate reads, local realignment, and base recalibration was done using GATK (v3.4) and Picard (v1.138) tools. Somatic variant calling was done using MuTect (v1.1.7) and VarScan2 (v2.3.7), and a union of these variants was used for all further analysis. Maftools was used to annotate and visualize these variants. MutSigCV v1.4 was used to identify significantly overly mutated genes. Canonical pathway analysis was performed using ingenuity pathway analysis (IPA; Qiagen). The top 50 genes identified by MutSig were uploaded into IPA, and an over-representation analysis was performed with standard settings. HLA-C mutation status in HNSCC from The Cancer Genome Atlas (TCGA) datasets was obtained from https://portal.gdc.cancer.gov/.
Fresh Frozen Paraffin-Embedded (FFPE) DNA extraction for confirmatory sequencing, pyrosequencing, and targeted next generation sequencing:
Peripheral blood mononuclear cell (PBMC) and NK isolation from peripheral blood:
After approval by our Institutional Review Board (UPCI protocol 99–069), informed consent was obtained from each subject before blood withdrawal. Blood from healthy donors or patients with HNSCC was drawn and lymphocytes were purified by Ficoll-Paque PLUS centrifugation following standard protocol (Amersham Biosciences). NK cells were purified using NK negative selection magnetic EasySep kits (Stem cell technologies). Purity of the selection was more than 95% FcγRIIIa+, CD56+, and CD3−.
Tumor cell lines:
JHU029 was a kind gift from Dr James Rocco (Harvard Medical School) in January of 2006. 93-VU-147 T (called 93VU in this report) was a kind gift from Dr Henning Bier (Technische Universitat Munchen) in October of 2013.
Flow cytometry analysis:
Surface flow cytometry was performed as previously described.31
Antibodies and treatments:
Mouse anti-human HLA-C-PE mAb (clone DT9) was purchased from Millipore. The CD3-Alexa Fluor 700, CD56-FITC, and KIR-PE (CD158b, clone DX27) were purchased from BD Pharmingen. Cetuximab and lirilumab were kindly provided by Bristol-Meyers Squibb and used at 10 μg/mL. IgG1 isotype control was purchased from Thermo Fisher and used at 10 μg/mL.
Enzyme-linked immunosorbent assay (ELISA):
Interferon gamma (IFNγ) production was determined by IFNγ ELISA kit (R&D systems) according to the manufacturer’s instructions.
Flow cytometry-based cellular cytotoxicity assay:
Purified NK cells (CD3–CD56+) were cocultured either alone or with tumor target cells (JHU029 or 93VU) in the presence or absence of lirilumab (10 μg/mL) for 24 hours in a 1:1 ratio. Tumor cells were harvested and stained using the 7-AAD and Annexin V cell death kit purchased from (BD biosciences). Cell death was determined by flow cytometry within 30 minutes of staining. Data were collected and analyzed using FloJo v10 software.
Cellular cytoxicity assay:
51Cr-labeled tumor target cells (JHU029 or 93VU) were added to NK cells in the presence or absence of lirilumab (10 μg/mL) at a 20:1 effector: target ratio. Following a 4 hour incubation, supernatants were harvested, and chromium release and relative percent lysis determined as previously described.32
3. | RESULTS
3.1. | Whole-exome sequencing of responders and nonresponders to cetuximab
Twenty post-treatment HNSCC samples (9 nonresponders and 11 responders) underwent whole-exome sequencing (WES) at an average depth of ×52. Patient demographics and variant data can be found in Table 1. The median overall mutational and missense mutational burden were not statistically different between nonresponders (1583, 224) and responders (1428, 163), respectively (P < 0.29, t test). A summary of mutation type, distribution, and most commonly mutated genes can be found in Supporting Information Figure S1. MutSig was used to identify statistically overly mutated genes, revealing five genes (U2AF2, TP53, INPP5E, RBMXL3, and KIR3DL2; P < 0.05, q < 0.05; Supporting Information Figure S2). Other than TP53, none of these genes are in the MutSig top 100 genes in 528 HNSCC from TCGA (http://firebrowse.org/?cohort=HNSC#). U2AF2 (U2 small nuclear RNA auxiliary factor 2), INPP5E (inositol polyphosphate-5-phosphatase E), and RBMXL3 (RNA binding motif protein, X-linked-like 3) have no previously reported role in HNSCC or cetuximab escape. KIR3DL2 is a transmembrane glycoprotein expressed by NK cells and subsets of T cells.
TABLE 1.
Patient demographics and mutational load
| ID | Sex | Race | Age | Smoking history |
Alcohol history |
Prior treatment |
Cancer site | HPV status | Final pTNM stage |
Responder status |
SNV | Missense mutations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | Men | White | 51 | Yes | Yes | None | Oral cavity | Negative | T4AN0MX | Yes | 1285 | 118 |
| 11 | Women | White | 58 | No | Yes | None | Oropharynx | Not tested | T2N2BMX | Yes | 1363 | 148 |
| 16 | Women | White | 51 | Yes | Yes | None | Oropharynx | Negative | T2N)MX | Yes | 1486 | 194 |
| 22 | Men | White | 62 | Yes | Yes | None | Oral cavity | Not tested | T2N0MX | Yes | 1248 | 107 |
| 23 | Men | White | 54 | Yes | Yes | None | Oral cavity | Not tested | T2N2CMX | Yes | 1388 | 149 |
| 24 | Men | White | 62 | Yes | Yes | None | Oral cavity | Not tested | T2N1MX | Yes | 1375 | 177 |
| 27 | Women | White | 47 | Yes | Yes | None | Oral cavity | Not tested | T4AN1MX | Yes | 1340 | 136 |
| 29 | Men | White | 64 | Yes | Yes | None | Oropharynx | Positive | T1N1MX | Yes | 2851 | 366 |
| 36 | Men | White | 69 | Yes | Yes | None | Oral cavity | Negative | T4AN0MX | Yes | 1566 | 148 |
| 37 | Men | White | 64 | Yes | Yes | None | Larynx | Not tested | T4AN1MX | Yes | 2297 | 340 |
| 39 | Men | White | 65 | Yes | Yes | None | Oral cavity | Not tested | T2N0MX | Yes | 1467 | 185 |
| 6 | Men | White | 69 | Yes | Yes | None | Oropharynx | Positive | T3N2BMX | No | 1589 | 206 |
| 10 | Women | White | 40 | No | No | None | Oropharynx | Negative | T2N2BMX | No | 1384 | 174 |
| 13 | Men | White | 60 | Yes | No | None | Oropharynx | Positive | T2N1MX | No | 1568 | 229 |
| 14 | Men | African American | 55 | Yes | Yes | None | Larynx | Not tested | T3N1MX | No | 1814 | 238 |
| 15 | Men | White | 64 | Yes | Yes | None | Oral cavity | Not tested | T1N1MX | No | 1313 | 134 |
| 18 | Men | White | 52 | Yes | Yes | None | Oropharynx | Negative | T2N)MX | No | 1550 | 181 |
| 19 | Men | White | 59 | No | Yes | None | Oropharynx | Positive | T2N2AMX | No | 5489 | 1635 |
| 20 | Men | White | 64 | Yes | Yes | None | Oropharynx | Positive | T2N2BMX | No | 2360 | 476 |
| 21 | Men | White | 51 | No | Yes | None | Oropharynx | Positive | T1N2BMX | No | 1583 | 224 |
Abbreviations: SNV, Single Nucleotide Variant; HPV, Human Papilloma Virus.
3.2. | Somatic HLA-C alterations are over-represented in nonresponders to cetuximab
We next compared gene mutation rates between responders and nonresponders, to identify genes that were statistically over-represented in either group. HLA-C was the most disproportionately mutated gene with 10 mutations in six nonresponders as compared with 1 mutation in responders (Figure 1A,B). The mutation rate in HLA-C in nonresponders compared to responders, and 528 untreated HNSCC specimens from TCGA, was statistically increased (67%, 9%, and 2%, respectively, P < 0.00001 by x2 test with P < 0.01 considered significant; Figure 1C). All HLA-C mutations in nonresponders were missense mutations and four out of the six nonresponder samples with HLA-C mutations possessed deleterious mutations on protein function, as determined by SIFT and/or PolyPhen predictions (Figure 1D; Supporting Information Figure S3). As the HLA locus can produce higher sequencing error rates because of high polymorphism and guanine cytosine (GC) content, we first manually curated HLA-C reads in IGV (Supporting Information Figure S4) and, after confirmation, attempted to validate these mutations using pyrosequencing of the remaining tumor/normal DNA. Of the 11 mutations identified in HLA-C, 10 failed pyrosequencing as a result of either nonspecific amplification or insufficient tumor DNA quantity (Supporting Information Methods section). One sample (TS-19) was able to be amplified and sequenced with validation of the mutation. We next attempted targeted next generation sequencing (tNGS) with HLA-C specific primers (NGSgo, GenDx). Only three samples (TS-6, −13, and −19) had adequate tumor DNA remaining. Of these, TS-6 had poor sequencing quality, TS-13’s mutation was not identified, and TS-19’s mutation was validated. Interestingly, in both TS-6 and −13, we found loss of heterozygosity (LOH) at HLA-C (Supporting Information Figure S5). As all DNA had been used at this point, additional DNA was extracted from FFPE for all samples for which we had FFPE blocks (five samples). DNA TapeStation (Agilent) analysis showed degradation of all samples, and no further analysis was possible.
FIGURE 1.
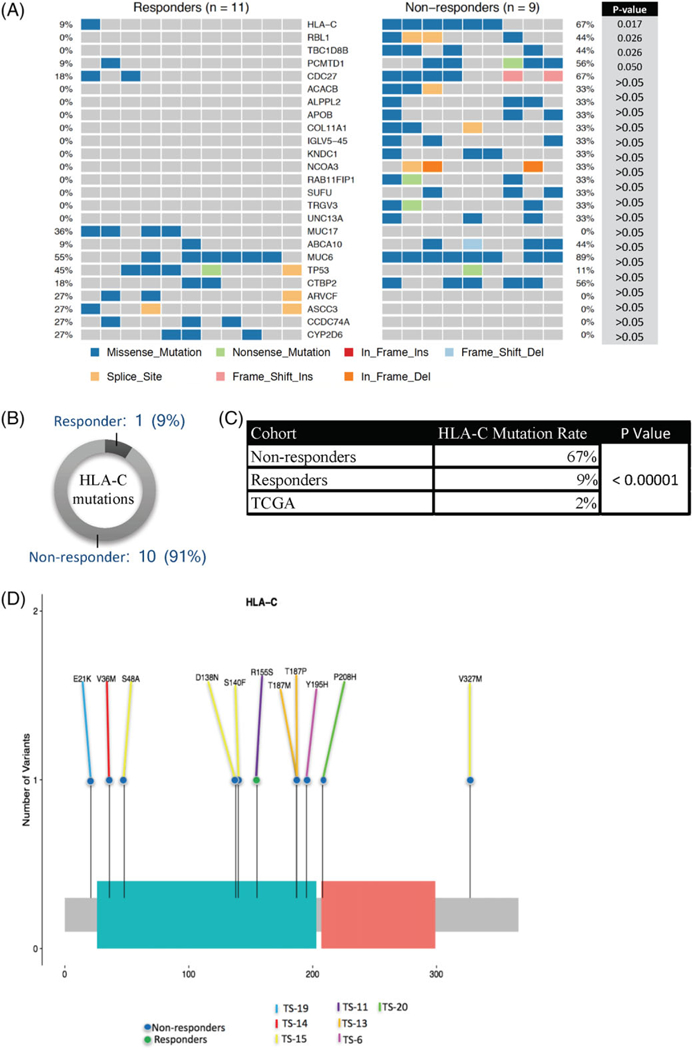
HLA-C mutations are over-represented in nonresponders to cetuximab. A, Most differentially mutated genes between responders and nonresponders by Fishers exact test. Percentages represent the percent of tumors within the cohort that possess a potentially deleterious mutation within a given gene. B, Graphical representation of total HLA-C mutations within each cohort. C, Comparison of HLA-C mutation rates among responders, nonresponders, and TCGA by x2 test. D, Lollipop plot demonstrating distribution of mutations within HLA-C. Colored lines correspond to sample, and colored dots correspond to responder/nonresponder category. Abbreviations: HLA-C, human leukocyte antigen-C; TCGA, The Cancer Genome Atlas [Color figure can be viewed at wileyonlinelibrary.com]
3.3. | Mutations in NK signaling pathways are over-represented in tumors treated with cetuximab
In order to investigate if commonly mutated genes occurred in similar biologic pathways in patients treated with cetuximab, we conducted a somatic mutation canonical pathway analysis (IPA) using the top 50 genes from MutSig analysis. Interestingly, the pathway most statistically over-represented was crosstalk between DCs and NK cells (Supporting Information Figure S7). The genes within this pathway, which were mutated in our cohort, were HLA-A, KIR3DL2, and MICA (MHC class I polypeptide-related sequence A). The most likely upstream regulator of the disrupted pathways was predicted to be the IFN group, including IFNγ.
3.4. | KIRs are expressed on NK cells from patients with HNSCC
As noted previously, HLA-C is the ligand for KIR on NK cells. In light of the findings that HLA-C alterations were statistically increased in nonresponders to cetuximab and that pathway analysis of frequently mutated genes identified NK cell signaling pathways as statistically overly mutated, we hypothesized that NK cells may play a critical role in killing HLA-C expressing tumors. First, we characterized the expression of KIRs (see section 2) on NK cells isolated from peripheral blood of patients with HNSCC by flow cytometry. We found that KIR expression was observed only on CD56dim NK cells (Figure 2A), which is known to be the main cytotoxic NK cell subset.33,34 Furthermore, we found that KIR expression is variable among patients ranging from 2% to 63% of CD56dim NK cells (Figure 2B; n = 20, mean = 33.5 ± SEM = 2.7). When we compared the frequency of KIR expressing NK cells of patients with HNSCC with that of healthy individuals, there was no significant difference (Figure 2C; two variable Student’s t test; ns P > 0.05). These findings suggest that expression of KIR in patient with HNSCC- derived NK cells is preserved, when compared to that of healthy donors, and thus, that NK cell modulation through HLA-C: KIR interaction could potentially induce tumor cell killing.
FIGURE 2.
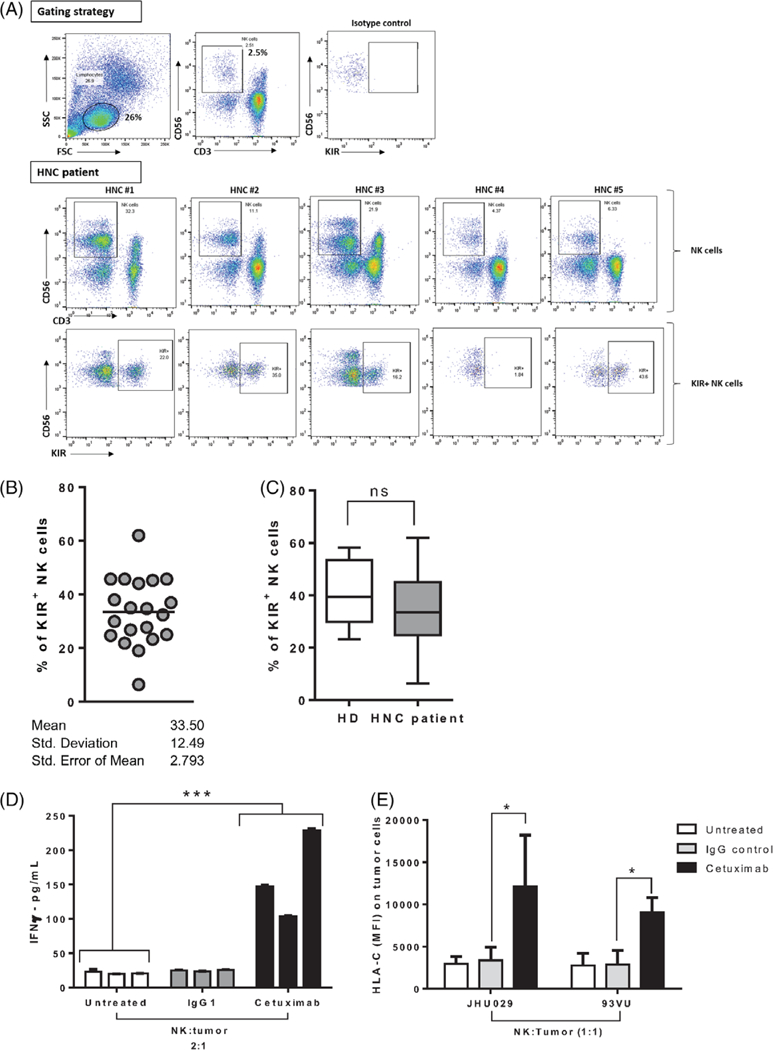
KIR is expressed uniquely in CD56dim NK cells from patients with HNSCC and healthy individuals and cetuximab-activated NK cells-induced HLA-C upregulation on tumor cells in vitro. A, Frequency of NK cells (CD3–CD56+ cells) from peripheral blood lymphocytes (PBLs) in patients with HNSCC. Top row shows gating strategy, second row shows CD56dim and CD56bnght NK cell populations, and bottom row shows KIR (CD158b) expression on CD56dim NK cells. Representative gating dot plots shown for five patients with HNSCC. B, Percent of circulating KIR+ (CD158b/KIR2DL2/KIR2DL3) NK cells (CD3-CD56+ cells) in PBL from patients with HNSCC, n = 20. C, Percent of circulating KIR+ NK cells (CD3-CD56+ cells) in PBL from patients with head and neck cancer (HNSCC; n = 20) versus healthy individuals (n = 5) (Student’s t test ns = P > 0.05). D, NK cells were isolated from PBMC and cocultured with JHU029 tumor cells at 2 to 1 ratio for 24 hours in the absence of mAb or with IgG1 isotype control (10 μg/mL) or cetuximab (10 μg/mL). IFNγ concentration in culture supernatants was determined by ELISA and normalized to number of NK cells in each condition (data from triplicate experiments from three different donors, ANOVA, ***P < 0.001). E, NK cells from five healthy donors were cocultured with JHU029 or 93VU tumor targets for 24 hours in the absence of mAb, IgG1 control (10 ĝ/mL), or cetuximab (10 μg/mL), harvested, and HLA-C expression on tumor cells was determined by flow cytometry (ANOVA, *P < 0.05). Abbreviations: ANOVA, analysis of variance; ELISA, enzyme-linked immunosorbent assay; HLA-C, human leukocyte antigen-C; HNSCC, head and neck squamous cell carcinoma; IFNγ, Interferon gamma; IgG1, immunoglobulin G-1; KIR, killer-cell immunoglobulin-like receptor; mAb, monoclonal antibodies; NK, natural killer [Color figure can be viewed at wileyonlinelibrary.com]
3.5. | Cetuximab-activated NK cells upregulate HLA-C expression on tumor cells via IFNγ
We next hypothesized that cetuximab-activated NK cells could upregulate HLA-C expression on tumor cells via IFNγ release. Indeed, in vitro coculture of NK cells and tumor cells in the presence of cetuximab induced a significant increase in IFNγ secretion to the culture supernatants (Figure 2D). Moreover, cetuximab-activated NK cells induced a significant upregulation of HLA-C on tumor cells while maintaining KIR expression (data not shown), when compared to untreated or IgG1 control treated NK cells (Figure 2E; *P < 0.01). Overall, these results suggest that cetuximab-activated NK cells not only eliminate tumor targets as previously shown but also upregulate HLA-C expression on tumor cells, which could further enhance NK cell activation.
3.6. | Lirilumab-activated NK cells increase killing of HNSCC cells
Lirilumab is an anti-KIR mAb. We cocultured tumor cells (JHU029 and 93VU) and freshly isolated NK cells either alone or in the presence of lirilumab for 24 hours and determined cell death by flow cytometry and chromium release (see section 2). As shown in Figure 3, NK-cell-mediated lysis of tumor targets was significantly higher when cocultured in the presence of lirilumab (analysis of variance [ANOVA]; ***P < 0.001). Interestingly, JHU029 cells, that express higher levels of HLA-C than 93VU cells at baseline (data not shown), and when cocultured with cetuximab-activated NK cells (Figure 2E), showed higher lysis when cocultured with lirilumab compared to 93VU cells (Figure 3B; ANOVA, **P < 0.01). The lytic activity of NK cells in the presence of lirilumab was enhanced compared to spontaneous lysis when cultured with JHU029, but not 93VU, as determined by a 4 hours chromium release assay (P < 0.001; Figure 3C). Taken together, these in vitro findings support our view that the HLA-C-KIR axis is important in NK-cell-mediated cytotoxicity of HNSCC cells.
FIGURE 3.
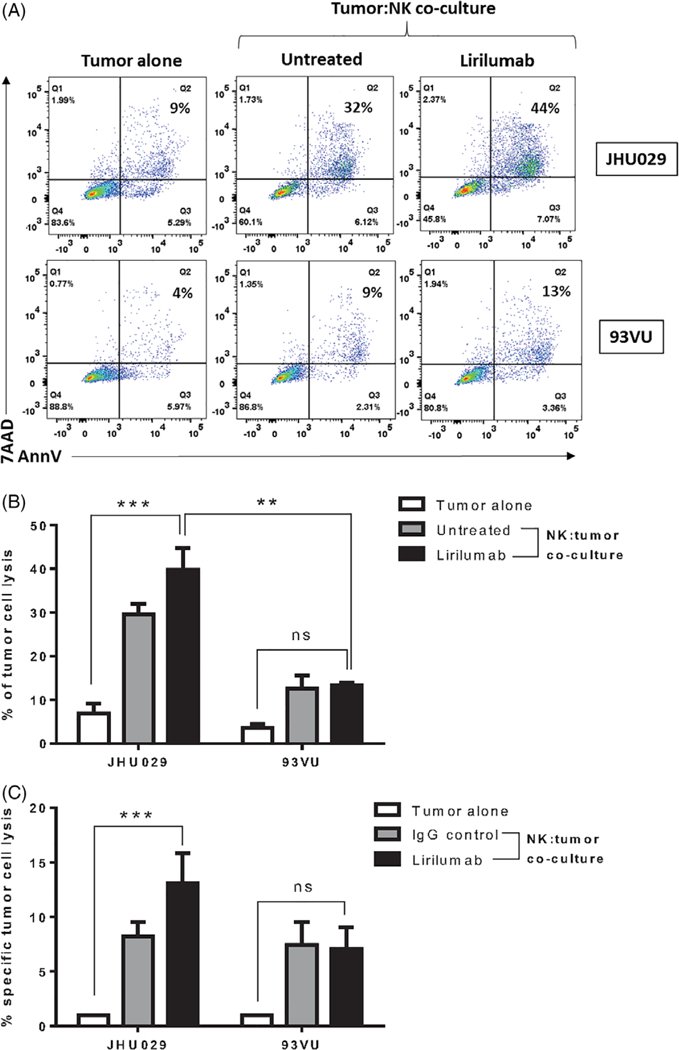
Lirilumab increased cell killing of HNSCC target cells. Tumor cells (JHU029 and 93VU) and freshly isolated NK cells cocultured either alone or in the presence of lirilumab (10 μg/mL) for 24 hours and with cell death determined by flow cytometry. A, Representative dot plot of the gating strategy. B, Graphical summary of cell death data determined by flow cytometry (n = 2) (ANOVA, ***P < 0.001). JHU029 cells showed higher lysis when cocultured with lirilumab than 93VU cells (ANOVA, **P < 0.01). C, The lytic activity of NK cells was assessed in a standard 4 hours chromium release assay with JHU029 or 93VU cell lines as targets at a 20:1 E:T ratio in the presence or absence of lirilumab. Abbreviations: ANOVA, analysis of variance; HNSCC, head and neck squamous cell carcinoma; NK, natural killer [Color figure can be viewed at wileyonlinelibrary.com]
4. | DISCUSSION
Immune evasion is a sine qua non for cancer development. Mechanisms of resistance to immune modulating therapeutics in cancer are poorly understood. For example, less than 20% of patients with HNSCC have a lasting response to cetuximab, despite initial effectiveness.7,8 Existing work supports that the mechanism of action of cetuximab in HNSCC may be mediated by NK cells.11–27 Similarly, failure of NK cell activation or immune evasion from NK-cell-mediated tumor lysis have been suggested as mechanisms of resistance. We recently completed a novel prospective trial of neoadjuvant cetuximab monotherapy with post-treatment tumor biopsies and well-annotated response data. Using this unique cohort of patients, we examined the relationship between response to cetuximab therapy and somatic alterations as a mechanism of immune escape. Of primary interest was the finding that HLA-C was the most disproportionately mutated gene between cohorts of nonresponders and responders (P < 0.017; Fishers exact) with 6 of 9 nonresponders possessing at least one potentially deleterious HLA-C mutation, compared to only 1 of 11 in responders (Figure 1; Supporting Information Figure S3). When compared to 528 HNSCC from TCGA, the HLA-C nonresponder mutation rate was significantly elevated (67% vs 2%; P < 0.00001; x2 test).
HLA-C mutations are of particular interest as HLA-C is the ligand for KIR on NK cells, modulating NK cell activity.29 Although the specifics of this interaction remain poorly defined, there are nine recognized inhibitory KIRs and six excitatory KIRs.35 Further, dimorphism at position 80 in HLA-C defines two epitopes, C1 and C2, which interact variably with different KIRs.35,36
HLA mutations have been demonstrated in numerous cancer types but most commonly occur in HLA-A and -B.37–39 Loss of HLA expression has been hypothesized to be a means of immune escape employed by tumors by downregulating TA presentation.38,40 This process is proposed to be as a result of a combination of genetic instability and immunoselective pressure.37 HLA-A and -B mutations are both relatively common in HNSCC, whereas HLA-C mutations are infrequent, as reported in the TCGA (HLA-A: 6%, HLA-B: 5%, and HLA-C: 1%, respectively; https://portal.gdc.cancer.gov/). HLA-A and -B predominantly bind antigen peptides and modulate TA-specific CTL activation and killing.38 HLA-C has garnered less attention because of its low mutation rate in many cancers and its perceived minor role in antigen-induced T cell responses. The finding that HLA-C mutations were identified in 67% of non-responders to cetuximab is intriguing, due to the existing hypotheses that NK cells are vital to successful HNSCC cetuximab therapy and that failure of mAb therapy may be mediated by immune escape from NK-cell-mediated killing. Recently, Leidner et al41 demonstrated a significant improvement in response when a first in class anti-KIR mAb (lirilumab) was added to checkpoint blockade, suggesting that augmentation of NK cell killing may be a mechanism to overcome immunoresistance.
In the light of this novel finding, we attempted to validate and further characterize the identified HLA-C mutations. Genotyping HLA-C presents numerous, well known, obstacles. High polymorphism of HLA-C prevents accurate identification of mutations due to suboptimal alignments. HLA genes are GC rich and typically have lower sequencing coverage as a result of lower efficiency in capture and amplification, leading to sequencing errors. Additionally, although the ability to leverage tissue samples from a tightly curated, patient cohort is advantageous for optimizing the chances of identifying meaningful findings, the use of clinical trial specimens has unique challenges because of the limited tissue availability and an inability to obtain additional specimens because of the temporal importance of when the biopsy was taken in relation to treatment. Although validation of some of these mutations was hampered by these obstacles, of significant interest was the finding that two of the three samples that underwent HLA-C-specific tNGS harbored LOH of an HLA-C allele (Supporting Information Figure S5). LOH, in addition to the mutations identified in HLA-C, could serve as a means of immune escape. McGranahan et al recently described allele-specific HLA class I loss in nonsmall cell lung cancer as a mechanism of immune escape.42 Similarly, Tran et al demonstrated that LOH of HLA-C in a patient undergoing adoptive T cell therapy provided a mechanism of immune evasion for a single nonresponsive lesion.43
Further supporting our finding that alterations in HLA-C may contribute to immune evasion in cetuximab therapy was the identification of NK signaling pathways as the most statistically over-represented biologic pathway across the 20 patient cohort, on canonical pathway analysis (Supporting Information Figure S7). This finding is despite the fact that HLA-C was not included in the analysis, as it fell outside the MutSig gene list. This finding suggests that, in addition to HLA-C, alterations in HLA class I signaling pathway genes (HLA-A, KIR3DL2, and MICA) occur at an unexpected rate in patients who have received cetuximab therapy. Consistent with these findings, our lab has previously done work supporting the importance of NK-DC crosstalk in cetuximab therapy.16–18
Acquisition of HLA-C alterations may be through one of two potential mechanisms: (1) alterations are acquired de novo after cetuximab treatment as a mechanism of immune escape or (2) HLA-C mutations exist at low allelic frequencies and treatment with cetuximab causes expansion of these HLA-C possessing clones through selective pressure. Although both mechanisms are possible37,38, the recent report by McGranahan et al demonstrating HLA LOH in subclones of lung cancers, with higher rates of HLA loss in metastatic lesions compared to primary lesions, suggests that selective pressure may induce expansion of pre-exiting mutations later in the tumors’ natural history.
To explore the hypothesis that HLA-C disruption could be a mechanism of immune escape from cetuximab therapy in HNSCC, we first characterized the expression of KIRs on NK cells (Figure 2A,B). This confirmed our view that KIR expressing NK cells are present in the circulation of patients with HNSCC to the same extent as in healthy individuals (Figure 2C) and may play an important role in tumor rejection. Previously, our laboratory has shown that cetuximab-activated NK cells increase HLA class I expression on tumor cells in an in vitro coculture system,13,16 however, whether cetuximab-activated NK cells could specifically upregulate HLA-C was not known. Herein, we report that cetuximab-activated NK cells induce a significant upregulation of HLA-C expression on tumor cells in an IFNγ-dependent fashion (Figure 2D,E). Of note, canonical pathway analysis of our WES data identified IFN signaling as the most likely upstream signaling pathway over-represented. These findings suggest that cetuximab-mediated NK cell activation would not only enhance ADCC of tumor targets through FcγRIIla stimulation but also potentiate KIR-mediated cytotoxicity of HLA-C-upregulated tumor targets. In this context, the identification of HLA-C alterations in tumors from cetuximab nonresponders may suggest an escape mechanism of tumor cells by which HLA-C alteration prevents NK cell activation. We further tested this hypothesis by coculturing tumor cells and freshly isolated NK cells in the presence of lirilumab, finding that lirilumab-treated NK cells showed a higher specific tumor cell lysis than the tumor alone condition or to the coculture control condition, although the latter in a nonstatistically significant fashion. Interestingly, tumor targets that expressed higher levels of HLA-C such as JHU029 cells showed significantly increased lysis (Figure 3B,C). Taken together, these results strongly suggest that HLA-C-KIR interaction is indeed important for NK cell activation. Examining HLA-C-KIR alterations at a single-cell level, for example, through single-cell RNA-Seq, could help further elucidate these immune escape mechanisms. In summary, the identification of HLA-C alterations in nonresponders to cetuximab and in vitro correlative data supports a role for HLA-C-NK cell-mediated immune evasion in cetuximab treatment failure in HNSCC.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UPMC Hillman Center shared resource facility (Cancer Genomics Facility) supported in part by award P30CA047904. Pyrosequencing was conducted at the Genetic Resources Core Facility, Johns Hopkins Institute of Genetic Medicine.
Funding information
Mosites Initiative for Personalized H&N Cancer Therapy (RLF); National Institutes of Health, Grant/Award Numbers: P50 CA097190–13, R01 CA206517–02 (RLF); AHNS Pilot (DLF)
This study was presented at the AACR-AHNS meeting in San Diego, CA, April 2017.
Robert L. Ferris: Amgen—Advisory Board; Astra-Zeneca/ MedImmune—Advisory Board, Clinical Trial, Research Funding; Bain Capital Life Sciences—Consulting; Bristol-Myers Squibb—Advisory Board, Clinical Trial, Research Funding; EMD Serono—Advisory Board; Iovance Biotherapeutics, Inc—Consulting; Lilly—Advisory Board; Merck— Advisory Board, Clinical Trial; Oncorus, Inc—Advisory Board; Ono Pharmaceutical Co Ltd—Consulting; Pfizer— Advisory Board; PPD—Advisory Board (Benitec, Immuni- cum); Regeneron Pharmaceuticals, Inc—Advisory Board; Tesaro—Advisory Board, Research Funding; TTMS—Consulting; VentiRx Pharmaceuticals—Research Funding.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
CONFLICT OF INTERESTS
REFERENCES
- 1.Bernier J, Domenge C, Ozsahin M, et al. Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med. 2004;350:1945–1952. [DOI] [PubMed] [Google Scholar]
- 2.Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med. 2004;350:1937–1944. [DOI] [PubMed] [Google Scholar]
- 3.Pignon JP, le Maître A, Maillard E, Bourhis J, Group M-NC. Metaanalysis of chemotherapy in head and neck cancer (MACH- NC): an update on 93 randomised trials and 17,346 patients. Radiother Oncol. 2009;92:4–14. [DOI] [PubMed] [Google Scholar]
- 4.Saloura V, Cohen EE, Licitra L, et al. An open-label single-arm, phase II trial of zalutumumab, a human monoclonal anti-EGFR antibody, in patients with platinum-refractory squamous cell carcinoma of the head and neck. Cancer Chemother Pharmacol. 2014; 73:1227–1239. [DOI] [PubMed] [Google Scholar]
- 5.Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53:3579–3584. [PubMed] [Google Scholar]
- 7.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. [DOI] [PubMed] [Google Scholar]
- 8.Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–1127. [DOI] [PubMed] [Google Scholar]
- 9.McBride SM, Rothenberg SM, Faquin WC, et al. Mutation frequency in 15 common cancer genes in high-risk head and neck squamous cell carcinoma. Head Neck. 2014;36:1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science (New York, NY). 2011;333:1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopez-Albaitero A, Ferris RL. Immune activation by epidermal growth factor receptor specific monoclonal antibody therapy for head and neck cancer. Arch Otolaryngol Head Neck Surg. 2007; 133:1277–1281. [DOI] [PubMed] [Google Scholar]
- 12.Roda JM, Joshi T, Butchar JP, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res. 2007;13:6419–6428. [DOI] [PubMed] [Google Scholar]
- 13.López-Albaitero A, Lee SC, Morgan S, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Concha-Benavente F, Srivastava R, Ferrone S, Ferris RL. Immunological and clinical significance of HLA class I antigen processing machinery component defects in malignant cells. Oral Oncol. 2016;58:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jie HB, Schuler PJ, Lee SC, et al. CTLA-4(+) regulatory T cells increased in Cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75:2200–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SC, Srivastava RM, Lopez-Albaitero A, Ferrone S, Ferris RL. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivastava RM, Lee SC, Andrade Filho PA, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19:1858–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava RM, Trivedi S, Concha-Benavente F, et al. CD137 stimulation enhances Cetuximab-induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin. Cancer Res. 2017;23:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava RM, Trivedi S, Concha-Benavente F, et al. STAT1-induced HLA class I upregulation enhances immunogenicity and clinical response to anti-EGFR mAb Cetuximab therapy in HNC patients. Cancer Immunol Res. 2015;3:936–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trivedi S, Concha-Benavente F, Srivastava RM, et al. Immune biomarkers of anti-EGFR monoclonal antibody therapy. Ann Oncol. 2015;26:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trivedi S, Srivastava RM, Concha-Benavente F, et al. Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients. Clin Cancer Res. 2016;22:5229–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertino EM, McMichael EL, Mo X a. A phase I trial to evaluate antibody-dependent cellular cytotoxicity of cetuximab and lenali-domide in advanced colorectal and head and neck cancer. Mol Cancer Ther. 2016;15:2244–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luedke E, Jaime-Ramirez AC, Bhave N, et al. Cetuximab therapy in head and neck cancer: immune modulation with interleukin-12 and other natural killer cell-activating cytokines. Surgery. 2012; 152:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMichael EL, Jaime-Ramirez AC, Guenterberg KD, et al. IL-21 enhances natural killer cell response to cetuximab-coated pancreatic tumor cells. Clin Cancer Res. 2017;23:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohrt HE, Colevas AD, Houot R, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124:2668–2682. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Taylor RJ, Chan SL, Wood A, et al. FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2009;58:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veluchamy JP, Spanholtz J, Tordoir M, et al. Combination of NK cells and cetuximab to enhance anti-tumor responses in RAS mutant metastatic colorectal cancer. PloS One 2016; 11:e0157830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89:216–224. [DOI] [PubMed] [Google Scholar]
- 29.Blais ME, Dong T, Rowland-Jones S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology. 2011;133:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mandal R, Senbabaoglu Y, Desrichard A, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Concha-Benavente F, Srivastava RM, Trivedi S, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Res. 2016;76:1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMichael EL, Jaime-Ramirez AC, Guenterberg KD, et al. IL-21 enhances natural killer cell response to cetuximab-coated pancreatic tumor cells. Clin Cancer Res. 2017;23:489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. [DOI] [PubMed] [Google Scholar]
- 34.De Maria A, Bozzano F, Cantoni C, Moretta L. Revisiting human natural killer cell subset function revealed cytolytic CD56(dim) CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci USA. 2011;108:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao X, Jiao Y, Wang L, et al. Inhibitory KIR and specific HLA-C gene combinations confer susceptibility to or protection against chronic hepatitis B. Clin Immunol (Orlando, Fla). 2010;137:139–146. [DOI] [PubMed] [Google Scholar]
- 36.Hilton HG, Vago L, Older Aguilar AM, et al. Mutation at positively selected positions in the binding site for HLA-C shows that KIR2DL1 is a more refined but less adaptable NK cell receptor than KIR2DL3. J Immunol (Baltimore, MD: 1950). 2012;189:1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda H, Lethe B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6: 199–208. [DOI] [PubMed] [Google Scholar]
- 38.Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Margulies L, Hicklin DJ, Ferrone S. Molecular and functional phenotypes of melanoma cells with abnormalities in HLA class I antigen expression. Tissue Antigens. 1996;47: 382–390. [DOI] [PubMed] [Google Scholar]
- 40.Koopman LA, van Der Slik AR, Giphart MJ, Fleuren GJ. Human leukocyte antigen class I gene mutations in cervical cancer. J Natl Cancer Inst. 1999;91:1669–1677. [DOI] [PubMed] [Google Scholar]
- 41.Leidner R Preliminary efficacy from a phase ½ study of the natural killer cell-targeted antibody, lirilumab in combination with nivolumab in squamous cell carcinoma of the head and neck. In: Society of Cancer Immunotherapy Annual Meeting; 2016; National Harbor, MD Abstract 456. [Google Scholar]
- 42.McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171: 1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tran E, Robbins PF, Lu YC, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.