

Abstract
Benzoxaboroles are a class of boron-containing compounds with a broad range of biological activities. A subset of benzoxaboroles have antimicrobial activity due primarily to their ability to inhibit leucyl-tRNA synthetase (LeuRS) via the oxaborole tRNA trapping mechanism, which involves formation of a stable tRNALeu–benzoxaborole adduct in which the boron atom interacts with the 2′- and 3′-oxygen atoms of the 3′-terminal tRNA adenosine. We sought to identify other antibacterial targets for this promising class of compounds by means of mode of action studies, and we selected a nitrophenyl sulfonamide-based oxaborole (PT638) as a probe molecule because it had potent antibacterial activity (MIC of 0.4 μg/mL against methicillin-resistant Staphylococcus aureus) but did not inhibit LeuRS (IC50 > 100 μM). Analogues of PT638 were synthesized to explore the importance of the sulfonamide linker and the impact of altering the functionalization of the phenyl ring. These structure–activity relationship studies revealed that the nitro substituent was essential for activity. To identify the target for PT638, we raised resistant strains of S. aureus and whole genome sequencing revealed mutations in leuRS, suggesting that the target for this compound was indeed LeuRS, despite the lack of enzyme inhibition. Subsequent analysis of PT638 metabolism demonstrated that bacterial nitroreductases readily converted this compound into the amino analogue, which inhibited LeuRS with an IC50 of 3.0 ± 1.2 μM demonstrating that PT638 is thus a prodrug.
Keywords: LeuRS, oxaborole, nitroreductase, nitro prodrug, S. aureus, resistance
Graphical Abstract
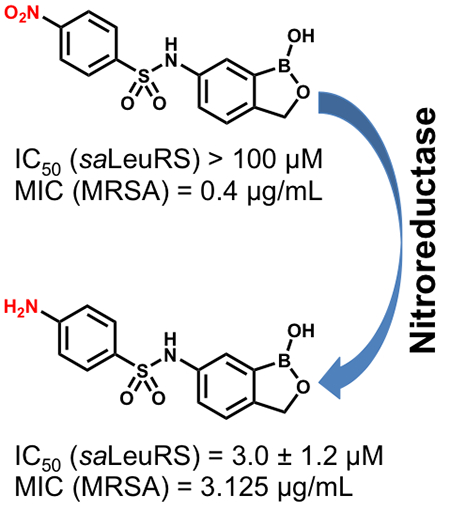
Multidrug-resistant bacteria are a major threat to human health because they cause infections that are hard to treat and often life-threatening.1,2 Drug resistance arises by a variety of mechanisms, including conversion of a drug into inactive metabolites, modification of drug binding sites, changes in cell permeability or drug efflux, and the formation of bacterial populations, such as biofilms, that are less susceptible to antibiotics.3,4 One particularly problematic pathogen is methicillin-resistant Staphylococcus aureus (MRSA), which is resistant to numerous antibiotics, including most β-lactams,5 macrolides, fluoroquinolones, and aminoglycosides.6 The threat posed by multidrug-resistant pathogens such as MRSA underscores the need to develop antibiotics with novel mechanisms of action.
The benzoxaboroles are a versatile class of small molecules with potential utility as antibiotics because their selectivity and specificity can be tuned by minor structural modifications. Targets for these compounds include β-lactamases,7 PDE4 nucleotide phosphodiesterase,8 ROCK kinase,9 carbonic anhydrase,10 and leucyl-tRNA synthetase (LeuRS).11 The oxaborole scaffold can reversibly form covalent tetrahedral complexes with nucleophiles such as hydroxyl groups owing to the presence of the heterocyclic boron atom, which acts as a Lewis acid because it has an empty p orbital.12,13 Formation of such complexes is involved in LeuRS inhibition, which occurs via the oxaborole tRNA trapping (OBORT) mechanism (Figure 1), whereby the boron atom forms a tetrahedral complex with both hydroxyl groups of the ribose diol of the terminal 3′ tRNA adenosine. Enzyme inhibition via the formation of an enzyme–substrate adduct is also observed in other drug classes, such as the bacterial enoyl-ACP reductases, which are inhibited by isoniazid and diazaborines.14,15 Anacor Pharmaceuticals has developed a number of oxaborole-based inhibitors of LeuRS from bacteria, fungi, protozoa, and other pathogens (Figure 1). AN2690,11 which has broad-spectrum antifungal activity, is one of the most effective US Food and Drug Administration–approved treatments for onychomycosis,16 while AN6426 is an inhibitor of the Mycobacterium tuberculosis LeuRS (minimum inhibitory concentration, MIC 0.13 μM, M. tuberculosis LeuRS IC50 0.09 μM),17 which also has antimalarial activity,18 and inhibits the growth of Cryptosporidium and Toxoplasma.19,20 Finally, AN3365, an aminomethylbenzoxaborole, binds to the editing site of Escherichia coli LeuRS with an IC50 value of 0.31 μM and has broad-spectrum activity against Gram-negative pathogens (MIC 0.5–4 μg/mL).21,22
Figure 1.
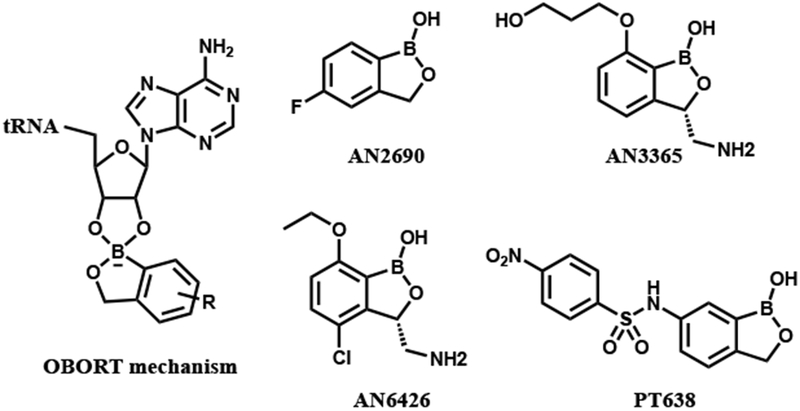
OBORT mechanism and oxaborole-based enzyme inhibitors.
Given the good drug-like properties of the oxaborole scaffold and given that both laboratory and clinical isolates show resistance to LeuRS-based inhibitors (arising mainly from mutations in the LeuRS editing domain),23,24 we sought to identify new antibacterial targets for this promising class of compounds. Building on the extensive medicinal chemistry efforts conducted by Anacor, we identified the nitrophenylsulfonyl-substituted 6-aminobenzoxaborole PT638 as a probe molecule (Figure 1). This compound was previously reported to have a MIC value of < 0.2 μg/mL against S. aureus but to not inhibit LeuRS (IC50 > 200 μM).25 We conducted structure–activity relationship (SAR) studies to explore the importance of the nitro group, sulfonamide linker region, and oxaborole ring for biological activity. These studies revealed that the nitro group was essential for activity. However, whole genome sequencing of resistant bacterial strains suggested that this compound did in fact target LeuRS, despite the lack of enzyme inhibition. Investigation of the mode of action of PT638 revealed that this compound is reduced to the active species by nitroreductases in MRSA cells.
Results and Discussion
SAR for inhibition of bacterial growth
We began by determining the antibacterial activity of PT638 toward MRSA (ATCC BAA-1762) and found that the MIC was 0.4 μg/mL (Table 1). Similarly, we assessed the cytotoxicity of PT638 to Vero cells using an MTT assay and determined the IC50 to be ≥ 100 μg/mL (Table 1). We subsequently performed SAR studies by synthesizing three series of PT638 analogues; specifically, we introduced modifications to the substituent on the phenyl ring (SAR1), to the sulfonamide linker (SAR2), and to the oxaborole ring (SAR3) and determined the antibacterial activity of the analogues, as well as their ability to inhibit S. aureus LeuRS (saLeuRS) (Figure 2, Table 1).
Table 1.
Biochemical activities of oxaboroles with MIC values <10 μg/mL
| Compound | Structure | IC50 (μM)a | MICb MRSA (μg/mL) | Cytotoxicityc Vero cells (IC50 μg/mL) |
|---|---|---|---|---|
| PT649 | 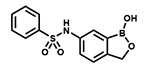 |
94 ± 2 | 6.25 | N.D. |
| PT638 | 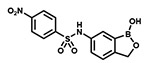 |
>100 | 0.4 | ≥100 |
| PT650 | 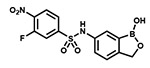 |
>100 | 1.56 | N.D. |
| PT659 | 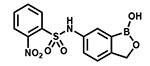 |
>100 | 6.25 | N.D. |
| PT662 | 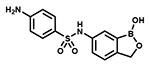 |
3.0 ± 1.2 | 3.125 | ≥100 |
| PT660 | 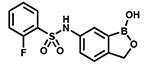 |
28 ± 1 | 6.25 | N.D. |
IC50 values were measured by means of a radiolabeled LeuRS aminoacylation assay.
MIC against MRSA ATCC BAA-1762 was determined using a broth microdilution method.
Cytotoxicity was determined with Vero cells using an MTT cell viability assay. All measurements were performed in triplicate and standard errors are reported.
N.D., not determined.
Figure 2. Structure–activity relationships (SARs) for inhibition of bacterial growth.
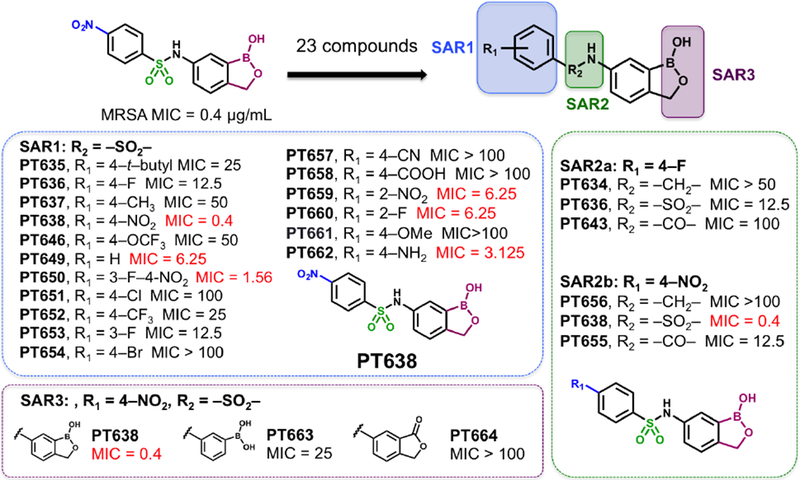
Three series of analogues were synthesized to explore SAR associated with the substituent on the phenyl ring (SAR1), the sulfonamide linker (SAR2), and the oxaborole ring (SAR3). MIC values (μg/mL) against MRSA (ATCC BAA-1762) were determined by broth microdilution; values of < 10 μg/mL are indicated in red.
With the SAR1 analogues, we explored the importance of the 4-nitro group for antibacterial activity (Figure 2). We expected that a strongly electron-withdrawing group like the nitro group would be required for activity, and, in fact, removing the nitro group (PT649) or replacing it with an electron-donating methyl group (PT637) or methoxy group (PT661) did lead to 10-, 100-, and >250-fold increases in MIC, respectively. However, neither replacement of the 4-nitro group with another electron-withdrawing group such as a cyano moiety (PT657, MIC >100 μg/mL) nor relocation of the nitro group to the 2-position (PT659, MIC 6.25 μg/mL) restored the activity to that of the parent compound. When we replaced the 4-nitro group with various 4-halogen atoms, we found that increasing the size of the halogen atom decreased the antibacterial activity: specifically, the 4-fluoro (PT636), 4-chloro (PT651), and 4-bromo (PT654) analogues had MICs of 12.5, 100, and >100 μg/mL, respectively. However, even the most active analogue (4-fluoro) had a MIC value that was 30-fold higher than that of PT638. In addition, activity did not depend on the position of the fluorine atom: the 2-fluoro (PT660) and 3-fluoro (PT653) analogues had MICs similar to that of PT636. Other substituents were also explored, but none of the resulting analogues, including 3-fluoro-4-nitro analogue PT650 and 4-amino analogue PT662, were as active as PT638. Collectively, these data attest to the critical importance of the 4-nitro group for antibacterial activity.
We subsequently explored the contributions of the sulfonamide linker and the oxaborole ring with the analogues in the SAR2 and SAR3 series, respectively (Figure 2). We found that regardless of whether the phenyl ring carried a 4-nitro or 4-fluoro substituent, replacement of the sulfonamide linker with an amine or amide linker substantially reduced antibacterial activity. All three linker analogues are able to adopt similar, although not identical conformations, while replacement of the sulfonyl moiety with a carbonyl (amide) or methylene (amine) group reduces the number of hydrogen bond acceptors at this position to 1 and 0, respectively (Figure S1). Thus, either the change in geometry and/or alteration in hydrogen bonding propensity may perturb binding to the target. In addition, the oxaborole ring was also critical for activity, as expected for molecules that might exert their activity via the OBORT mechanism; replacement of the oxaborole ring with a lactone (PT664) or a boronic acid group (PT663) dramatically reduced the activity.
Time-kill experiments and post-antibiotic effect of PT638
To further assess the antibacterial activity of PT638, we conducted time-kill experiments and determined the duration of the post-antibiotic effect following removal of the test compound from the medium. Treatment of MRSA cells with PT638 at concentrations 1, 4, and 16 times the MIC led to 1.8 log10, 2.2 log10, and 2.7 log10 cfu/mL reductions in cell numbers over 8 h, respectively (Figure 3A). That is, PT638 showed bacteriostatic activity over the course of 8 h, as indicated by the fact that the cfu reduction was <3 log10.26–28 In addition, the duration of the post-antibiotic effect due to the treatment of PT638 increased from 0.2 h at 0.25 × MIC to 1.5 h at 4 × MIC (Figure 3B). However, no further increase in the duration of post-antibiotic effect was observed above 4 × MIC, perhaps because this concentration was sufficient to saturate the target.
Figure 3. Time-kill experiments and post-antibiotic effect of PT638.
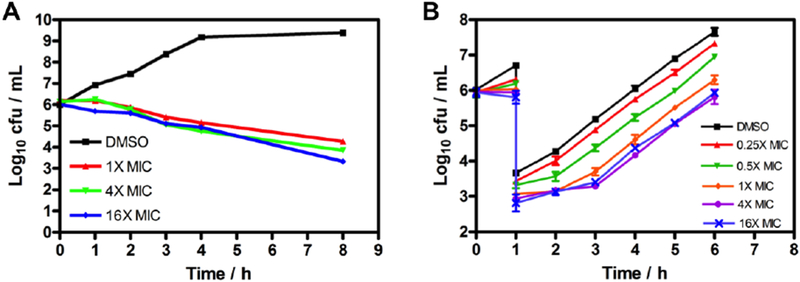
(A) Cell viability of MRSA (ATCC BAA-1762) treated with PT638 at 1, 4, and 16 times the MIC or with vehicle (DMSO) over the course of 8 h. (B) Post-antibiotic effect of PT638. Recovery of bacterial growth was measured after 1 h of exposure to PT638 at 0.25, 0.5, 1, 4, and 16 times the MIC and subsequent washout. Values are means of three independent replicates, and error bars indicate the standard deviations.
Inhibition of saLeuRS and target identification
Next, we used an aminoacylation assay to determine the ability of compounds with MICs of <10 μg/mL to inhibit saLeuRS. In agreement with previous reports, our data clearly show that PT638 did not inhibit saLeuRS (IC50 > 100 μM, Table 1). In addition, other nitro-substituted analogues, such as PT659 and PT650, also did not inhibit saLeuRS; and PT649, which lacks any substituents, showed only weak activity (IC50 = 94 ± 2 μM). In contrast, the 2-fluoro analogue (PT660) and the 4-amino analogue (PT662) had better activity than PT638, with IC50 values of 28 ± 1 and 3.0 ± 1.2 μM, respectively (Table 1, Figure S2).
The potent cellular activity of PT638 combined with its inability to inhibit saLeuRS suggested that it might have some other target in MRSA. To assess this possibility, we generated resistant mutants by plating MRSA (ATCC BAA-1762) on Mueller–Hinton agar plates containing PT638 at 10 times the MIC. Colonies were obtained with a single-step selection of resistant strains. The frequency of resistance was determined to be 1.5 × 10−8. Three colonies were picked and were shown to have MICs of ≥ 3.125 μg/mL, which is at least 8 times the MIC of the parent strain. Genomic DNA was extracted, purified, and subjected to whole genome sequencing. However, in each case, the only mutations found were within the leuRS gene; the mutations were D343Y, G303S, and F233I, all of which are within the editing domain of saLeuRS (Table 2). Sequence alignment revealed that these three residues are conserved in various pathogens (Figure S3) and that the counterparts of D343 in E. coli, Streptococcus pneumonia, and M. tuberculosis are directly involved in binding the oxaborole inhibitors.17,22,29,30 The other two residues, F233 and G303, are not located in the binding pocket, and it is less clear how mutation of these residues leads to PT638 resistance. Nevertheless, our results clearly pointed to saLeuRS as the target for PT638, which led us to hypothesize that a metabolite of PT638 might be the cellular inhibitor of saLeuRS.
Table 2.
PT63-resistant mutant stains
| Strain | MIC shifta | Mutation | Protein |
|---|---|---|---|
| 1 | 16-fold | D343Y | saLeuRS editing domain |
| 2 | 8-fold | G303S | |
| 3 | 8-fold | F233I |
The MIC shift was determined from the MIC of the mutant strain compared to that of the wild-type MRSA strain (ATCC BAA-1762).
Activation of prodrug PT638 by bacterial nitroreductases
Previous studies have shown that bacteria contain nitroreductases, such as NfsA and NfsB, that can reduce a variety of nitroheterocyclic compounds.31 For example, the antibacterial agent nitrofurantoin undergoes a reduction that is required for its activity against E. coli.32 Enzymatic reduction of nitroaromatic compounds proceeds through a one- or two-electron mechanism33 in which the nitro group is reduced to an amine via nitroso and hydroxylamine intermediates.33,34 On the basis of this previous research, we hypothesized that PT638 might in fact be a prodrug that needs to be activated by one or more nitroreductases in MRSA (Figure 4A). To test this hypothesis, we evaluated the metabolism of PT638 following incubation with MRSA cell lysate for 48 h. Analysis by HPLC and LC-UV/MS revealed the formation of only one major metabolite, which had a shorter retention time than PT638 and which co-eluted with amino analogue PT662 (Figure 4B). LC-MS showed that this metabolite had a mass of 305.0771, which corresponds to the molecular ion of PT662 (Figure 4C). These results indicate that PT638 was metabolized to PT662 by MRSA cell lysate.
Figure 4. Formation of PT662 in MRSA cell lysate treated with PT638.
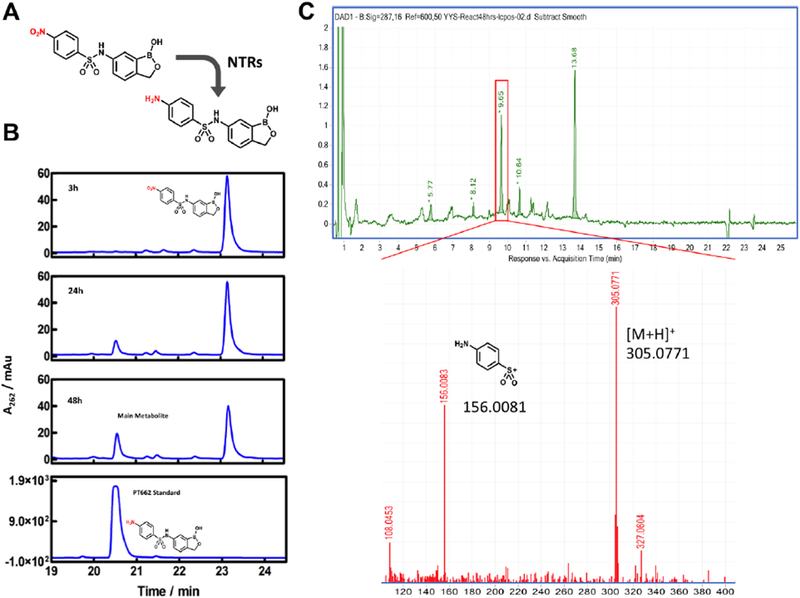
(A) Reduction of PT638 to aniline PT662 by nitroreductases in MRSA cells. (B) HPLC analysis of PT638 metabolites extracted from MRSA cell lysate at 3, 24, and 48 h. PT638 had a retention time of 23.2 min, and the major metabolite had a retention time of 20.7 min, which was identical to that of synthetic PT662 standard. (C) High-resolution LC-UV/MS spectra of sample obtained after incubation for 48 h. The major metabolite had a retention time of 9.65 min with [M + H]+ = 305.0771 and m/z = 156.0081. NTRs, nitroreductases.
Because NfsA and NfsB are the two major nitroreductases in bacteria,35 these enzymes were cloned and purified from MRSA, and their catalytic activities toward PT638 were assayed using NADH or NADPH as the co-substrate (Figure S4). PT638 was found to be a substrate for both nitroreductases but was reduced much more effectively by NfsB than by NfsA, with kcat/Km values of 1.0 × 106 and 5.2 × 104 M−1s−1, respectively (Table 3). Finally, although PT638 did not inhibit saLeuRS, amino analogue PT662 had an IC50 value of 3.0 μM (Table 1). Thus, we propose that PT638 is converted into an active saLeuRS inhibitor in MRSA cells primarily by the action of NfsB.
Table 3.
Reduction of PT638 by NfsA and NfsBa
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (M−1s−1) |
|---|---|---|---|
| NfsA | 43.8±7.0 | 840±260 | 5.2×104 |
| NfsB | 45.3±0.9 | 45±3 | 1×106 |
Initial velocities were monitored at 340 nm at a fixed concentration of NAD(P)H (60 μM NADPH for NfsA, 60 μM NADH for NfsB) and varied concentrations of PT638 (6.25–1500 μM). Data were fit to the Michaelis–Menten equation.
Other antibacterial prodrugs have been discovered such as nitrofurantoin,32 mentioned above, and the anti-tuberculosis nitroimidazole PA-824.36 In these cases, antibacterial activity is due to reactive intermediates, such as NO in the case of PA-824, formed during reduction of the nitro group that likely have multiple targets in the cell, and selection for resistance results in mutations in the enzymes responsible for prodrug activation including nfsA and nfsB for nitrofurantoin and a deazaflavin-dependent nitroreducase Ddn for PA-824.37 In contrast, no mutations were observed in nfsA and nfsB when selecting for resistance to PT638 but in the LeuRS target, supporting a specific mode of action in which reactive intermediates are not formed and in which mutations in the primary target are sufficient for resistance. In this regard, we note that the MIC of the ortho-nitro analog PT659 is ~ 10-fold higher than that of PT638 (MIC 0.4 and 6.25 μg/mL, respectively, Table 1) and the IC50 value for inhibition of saLeuRS is reported to be ~10-fold greater for the corresponding amines in Xia et al.25 (IC50 1.9 and 15.4 μM, respectively). The 8-fold increase in antibacterial of PT638 compared to the aniline PT662 (MIC 0.4 and 3.1 μg/mL, respectively) is thus likely because the former is better at penetrating the bacterial cells and accumulates following activation. However, although PT662 is the only stable metabolite formed during activation of PT638, we cannot rule out the possibility that the activity of PT638 might partially be due to one or more reactive intermediates formed during bioactivation of PT638.
Conclusion
LeuRS is a common target for the oxaborole class of compounds, which inhibit this enzyme by forming an adduct with the terminal adenosine of the bound tRNA substrate. In an attempt to identify additional antibacterial targets for the oxaboroles, we selected the nitrophenylsulfonamide-substituted benzoxaborole PT638 as a probe for mode of action studies on the basis of reports that it has potent antibacterial activity toward S. aureus but did not inhibit LeuRS. We confirmed the antibacterial activity of PT638 toward MRSA (MIC = 0.4 μg/mL) and then synthesized a series of 6-substituted benzoxaborole analogues for SAR studies. These studies showed that the nitro group, the sulfonamide linker, and oxaborole ring were all important for antibacterial activity and that any modification of the nitro group led to at least a 4-fold increase in the MIC relative to that of the parent compound. Although a LeuRS aminoacylation assay confirmed that PT638 did not inhibit saLeuRS, selection for resistance to PT638 and subsequent whole genome sequencing revealed mutations in the editing domain of saLeuRS. Bacterial cell lysate was found to convert PT638 into amino analogue PT662, which inhibited saLeuRS with an IC50 of 3 μM. In addition, PT638 was shown to be a substrate for the bacterial nitroreductase enzymes NfsA and NfsB, confirming that the nitro analogue is a prodrug that is reduced into the active pharmacophore in bacterial cells.
Materials and Methods
Antibacterial activity
Minimum inhibitor concentrations (MICs) were determined using MRSA strain ATCC BAA-1762. Bacteria were cultured to mid-log phase (OD600 = 0.6, 108 cfu/mL) in cation-adjusted Mueller–Hinton (CAMH) medium at 37 °C in an orbital shaker. An inoculum of 106 cells per well was placed in transparent 96-well plates and treated with inhibitor at final concentrations ranging from 0.2 to 100 μg/mL. The MIC was defined as the minimum concentration at which a well showed no obvious growth by visual inspection.
Time-kill experiments
MRSA (ATCC BAA-1762) was grown in CAMH medium to mid-log phase (OD600 = 0.6, 108 cfu/mL) at 37 °C in a shaker and then diluted 100-fold into fresh medium. PT638 or an equal volume of vehicle (DMSO) was then added to give concentrations 1, 4, or 16 times the MIC. Cultures were then shaken at 37 °C for 8 h. Kill curves were obtained by sampling the cell cultures at various times and plating serial dilutions on Mueller–Hinton agar. Colony forming units (cfus) were counted after incubation of the plates overnight at 37 °C. Each experiment was repeated at least twice.
Post-antibiotic effect
MRSA (ATCC BAA-1762) was grown in CAMH medium to mid-log phase (OD600 = 0.6, 108 cfu/mL) at 37 °C in a shaker and diluted 100-fold into fresh medium containing either PT638 or an equal volume of DMSO. After the culture was shaken for 1 h at 37 °C, PT638 was washed out by diluting the culture 1000-fold into fresh CAMH medium. The diluted cells were incubated in a shaker at 37 °C for an additional 5 h. The regrowth of MRSA cells was monitored by taking 100 μL of cell culture per hour and plating serial dilutions on Mueller–Hinton agar plates. The plates were incubated at 37 °C overnight, after which cfus were counted. The post-antibiotic effect was calculated as the time difference required for the number of compound-treated cells (cfus) to increase 1 log compared to the number of untreated cells. Each experiment was repeated in triplicate.
Generation of PT638-resistant strains and whole genome sequencing
MRSA (ATCC BAA-1762) was grown in CAMH medium to a cell density of 109 cfu/mL and then 100 μL of the cell culture was plated on Mueller–Hinton agar containing PT638 (4 μg/mL, 10 × MIC). After incubation at 37 °C for 48 h, three colonies were picked, and the genomic DNA was purified using a GenElute Bacterial Genomic DNA Kit (Sigma). Whole genome sequencing was performed by Admera Health.
Cloning, expression, and purification of saLeuRS
The leuRS gene from MRSA cells (ATCC BAA-1762) was amplified with primers 5′CGCGGATCCGTGTTGAATTACAACCACAATC3′ and 5′CCGCTCGAGTTATTTAGCTACAATATTGAC3′. The amplified leuRS gene was cloned into a pET28a vector so that a His-tag was encoded at the N-terminus of the protein. After sequencing, the pET28a-leuRS plasmid was transformed into E. coli BL21(DE3) cells, which were then grown overnight in 10 mL of Luria-Bertani (LB) medium containing 50 μg/mL kanamycin. Then the overnight culture was inoculated into 1 L of LB medium containing kanamycin at the same concentration, and the cells were grown until the OD600 reached 0.6. One mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to induce protein expression, and the culture was shaken overnight at 20 °C. Cells were harvested by centrifugation at 5000 rpm for 10 min at 4 °C. The cell pellet was resuspended in 30 mL of His-binding buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM Imidazole, pH 7.9), and the bacteria were disrupted by sonication. Cell debris was removed by centrifugation at 40,000 rpm for 60 min at 4 °C, and the clear supernatant was loaded onto a His-bind column (1.5 cm × 15 cm) containing 4 mL of His-bind resin (Novagen) that had been charged with 10 mL of charge buffer (Ni2+). The column was washed with wash buffer containing 60 mM imidazole, and the protein was eluted from the column with elution buffer containing 500 mM imidazole. Fractions containing protein were loaded onto a size-exclusion column (Superdex 75, GE Healthcare) and eluted with 60 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 8.0) containing 30 mM NaCl and 30 mM MgCl2 to remove imidazole.
LeuRS aminoacylation activity assay
The aminoacylation reaction was performed in a 50 μL reaction volume with 7 nM saLeuRS, 15 μM E. coli total tRNA (Roche), 20 μM 3H-leucine (174.6 mCi/mmol), and 4 mM ATP in 50 mM HEPES-KOH buffer (pH 8.0) containing 30 mM MgCl2, 30 mM KCl, 0.02% (w/v) bovine serum albumin, and 1 mM dithiothreitol. Unless stated otherwise, the test compound, saLeuRS, and E. coli total tRNA were pre-incubated for 20 min before the reaction was initiated with 4 mM ATP. At specific times, tRNA was precipitated by the addition of 10% (w/v) trichloroacetic acid. The precipitate was collected with a 0.45 μm microcentrifugal filter (Thomas Scientific F2517-2) and washed twice with 100 μL of 5% trichloroacetic acid, and then the filter was counted with a liquid scintillation analyzer (Tri-Carb 2900TR, PerkinElmer).
IC50 values were determined at various concentrations of compound (Figure S2) and data were fit to a 4 parameter IC50 equation in GraphPad Prism 4 after constraining the Hill slope to a value of 1. Each experiment was performed in triplicate.
Cytotoxicity
The in vitro cytotoxicity of PT638 and PT662 was determined using Vero cells (ATCC CCL-81). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum and then aliquoted into a 96-well tissue culture plate to give 2 × 104 cells/well. After incubation for 24 h at 37 °C in 5% CO2, the media was aspirated and replaced with serum-free DMEM. After an additional 1 h of incubation, PT638 or PT662 was added to give a final concentration ranging from 0.2 to 100 μg/mL. Following incubation for 24 h at 37 °C in 5% CO2, cell viability was assessed by means of an MTT assay (Vybrant MTT Cell Proliferation Assay Kit). The absorbance of each well was measured at 570 nm and the data were used to determine compound cytotoxicity (IC50).
Cloning, expression, and purification of NfsA and NfsB
The nfsA gene from MRSA (ATCC BAA-1762) was amplified with primers 5′CTAGCTAGCGTGTCAGATCATGTATATAATC3′ and 5′CGCGGATCCCTATCGCTGTATTAAGCCTG3′. The nfsB gene from the same strain was amplified with 5′CTAGCTAGCATGAGCAATATGAATCAAACAATTATG3′ and 5′CGCGGATCCTTATTCTTTTGGTCCAACCC3′. The amplified nfsA (nfsB) gene was cloned into a pET28a vector so that a His-tag was encoded at the N-terminus of the protein. After sequencing, the pET28a-nfsA (pET28a-nfsB) plasmid was transformed into E. coli BL21(DE3) cells, which were then grown overnight in 10 mL of LB medium containing 50 μg/mL kanamycin. Then the overnight culture was inoculated into 1 L of LB medium containing kanamycin at the same concentration, and the cells were grown until the OD600 reached 0.6. IPTG (1 mM) was added to induce protein expression, and the culture was shaken overnight at 20 °C. Cells were harvested by centrifugation at 5000 rpm for 10 min at 4 °C. The cell pellet was resuspended in 30 mL of His-binding buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM imidazole, pH 7.4), and the bacteria were disrupted by sonication. Cell debris was removed by centrifugation at 40,000 rpm for 60 min at 4 °C, and the clear supernatant was loaded onto a His-bind column (1.5 cm × 15 cm) containing 4 mL of His-bind resin (Novagen) that had been charged with 10 mL of charge buffer (Ni2+). The column was washed with washing buffer containing 30 mM imidazole, and the protein was eluted from the column with elution buffer containing 500 mM imidazole. Fractions containing protein were loaded onto a size-exclusion column (Superdex 75, GE Healthcare), which was eluted with 50 mM Tris-HCl buffer (pH 7.4) containing 150 mM NaCl to remove the imidazole. The concentrations of NfsA and NfsB were determined from the flavin absorbance at 450 nm.
Nitroreductase activity assay
The nitroreductase assay was performed in 50 mM Tris-HCl buffer (pH 7.4), containing 5 mM EDTA at RT in a total reaction volume of 500 μL. Reactions were initiated by the addition of NfsA or NfsB to a final concentration of 45 nM and the consumption of NAD(P)H was continuously monitored at 340 nm. The kcat and Km values were determined at a fixed concentration of NAD(P)H (60 μM NADPH for NfsA, 60 μM NADH for NfsB) and by varying the concentration of PT638 (6.25–1500 μM). Initial velocities as a function of substrate concentration were fit to the Michaelis–Menten equation using GraphPad Prism 4. Each experiment was performed for triplicate.
PT638 metabolite analysis
MRSA (ATCC BAA-1762) was grown in 20 mL of CAMH medium to log phase (OD600 = 0.6). Cells were harvested by centrifugation and then resuspended in 5 mL of lysis buffer (50 mM Tris-HCl, 100 mM NaCl, pH 8.0). Cells were lysed by sonication, and cell debris was removed by centrifugation. The supernatant was then incubated with 4 μg/mL PT638 at 37 °C. After 3, 24, and 48 h, 500 μL of cell lysate was removed and extracted twice with ethyl acetate. The organic layers were combined, and the solvent was removed by rotary evaporation. The residue after rotovapping was then dissolved in 300 μL of 50% MeCN/H2O and analyzed by HPLC with a Phenomenex C18 column (250 × 4.6 mm). A linear gradient at a flow rate of 0.8 mL/min was used, with absorbance detection at 254 nm. Solvent A was water, and solvent B was acetonitrile. The percentages of solvent B at times t were as follow: t = 0–5 min, B = 5%; t = 5–35 min, B = 5–100%; t = 35–40 min, B = 100%; t = 40–50 min, B = 100–5%; t = 50–55 min, B = 5%.
The sample taken at 48 h was also analyzed by LC-MS using an Agilent LC-UV-TOF system consisting of a 1260 uPLC, a UV–vis diode-array detector, and a TOF mass analyzer. The LC eluent consisted of a gradient of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile). The percentages of solvent B at times t were as follows: t = 0–1 min, B = 5%; t = 1–31 min, B = 5–95%; t = 31–33 min, B = 95–99%. Compounds were detected by monitoring absorbance at 287 nm. Mass data were collected with an in-line mass spectrometer (G6224A oaTOF) in positive-ion mode.
Supplementary Material
Acknowledgements
This work was supported by NIH grants GM102864 and AI119316 to PJT and GM092714 to JI (T32 Fellowship). We thank Dr. Bela Ruzsicska for his help with LC-UV/MS and high-resolution MS.
Footnotes
Supporting Information
Supporting information contains IC50 plots, sequence alignment of the LeuRS proteins from various bacteria, and the synthetic procedures and characterization data for the 6-substituted benzoxaboroles.
References
- (1).Karras G; Giannakaki V; Kotsis V; Miyakis S (2012) Novel antimicrobial agents against multi-drug-resistant gram-negative bacteria: an overview Recent Pat Antiinfect Drug Discov, 7, 175–81. DOI: 10.2174/157489112803521922 [DOI] [PubMed] [Google Scholar]
- (2).Giannakaki V; Miyakis S (2012) Novel antimicrobial agents against multi-drug-resistant gram-positive bacteria: an overview Recent Pat Antiinfect Drug Discov, 7, 182–8. DOI: 10.2174/157489112803521959 [DOI] [PubMed] [Google Scholar]
- (3).Santajit S; Indrawattana N (2016) Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens Biomed Res Int, 2016, 2475067 DOI: 10.1155/2016/2475067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mathur P; Singh S (2013) Multidrug resistance in bacteria: a serious patient safety challenge for India J Lab Physicians, 5, 5–10. DOI: 10.4103/0974-2727.115898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fisher JF; Meroueh SO; Mobashery S (2005) Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity Chem Rev, 105, 395–424. DOI: 10.1021/cr030102i [DOI] [PubMed] [Google Scholar]
- (6).Theuretzbacher U (2013) Global antibacterial resistance: The never-ending story J Glob Antimicrob Resist, 1, 63–69. DOI: 10.1016/j.jgar.2013.03.010 [DOI] [PubMed] [Google Scholar]
- (7).Xia Y; Cao K; Zhou Y; Alley MR; Rock F; Mohan M; Meewan M; Baker SJ; Lux S; Ding CZ; Jia G; Kully M; Plattner JJ (2011) Synthesis and SAR of novel benzoxaboroles as a new class of beta-lactamase inhibitors Bioorg Med Chem Lett, 21, 2533–6. DOI: 10.1016/j.bmcl.2011.02.024 [DOI] [PubMed] [Google Scholar]
- (8).Dong C; Virtucio C; Zemska O; Baltazar G; Zhou Y; Baia D; Jones-Iatauro S; Sexton H; Martin S; Dee J; Mak Y; Meewan M; Rock F; Akama T; Jarnagin K (2016) Treatment of Skin Inflammation with Benzoxaborole Phosphodiesterase Inhibitors: Selectivity, Cellular Activity, and Effect on Cytokines Associated with Skin Inflammation and Skin Architecture Changes J Pharmacol Exp Ther, 358, 413–22. DOI: 10.1124/jpet.116.232819 [DOI] [PubMed] [Google Scholar]
- (9).Akama T; Dong C; Virtucio C; Sullivan D; Zhou Y; Zhang YK; Rock F; Freund Y; Liu L; Bu W; Wu A; Fan XQ; Jarnagin K (2013) Linking phenotype to kinase: identification of a novel benzoxaborole hinge-binding motif for kinase inhibition and development of high-potency rho kinase inhibitors J Pharmacol Exp Ther, 347, 615–25. DOI: 10.1124/jpet.113.207662 [DOI] [PubMed] [Google Scholar]
- (10).Adamczyk-Wozniak A; Borys KM; Sporzynski A (2015) Recent developments in the chemistry and biological applications of benzoxaboroles Chem Rev, 115, 5224–47. DOI: 10.1021/cr500642d [DOI] [PubMed] [Google Scholar]
- (11).Rock FL; Mao W; Yaremchuk A; Tukalo M; Crepin T; Zhou H; Zhang YK; Hernandez V; Akama T; Baker SJ; Plattner JJ; Shapiro L; Martinis SA; Benkovic SJ; Cusack S; Alley MR (2007) An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site Science, 316, 1759–61. DOI: 10.1126/science.1142189 [DOI] [PubMed] [Google Scholar]
- (12).Dowlut M; Hall DG (2006) An improved class of sugar-binding boronic acids, soluble and capable of complexing glycosides in neutral water J Am Chem Soc, 128, 4226–7. DOI: 10.1021/ja057798c [DOI] [PubMed] [Google Scholar]
- (13).Berube M; Dowlut M; Hall DG (2008) Benzoboroxoles as efficient glycopyranoside-binding agents in physiological conditions: Structure and selectivity of complex formation J Org Chem, 73, 6471–6479. DOI: 10.1021/jo800788s [DOI] [PubMed] [Google Scholar]
- (14).Rawat R; Whitty A; Tonge PJ (2003) The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance Proc Natl Acad Sci U S A, 100, 13881–13886. DOI: 10.1073/pnas.2235848100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baldock C; Rafferty JB; Sedelnikova SE; Baker PJ; Stuitje AR; Slabas AR; Hawkes TR; Rice DW (1996) A mechanism of drug action revealed by structural studies of enoyl reductase Science, 274, 2107–2110. DOI: 10.1126/science.274.5295.2107 [DOI] [PubMed] [Google Scholar]
- (16).Sarkar J; Mao W; Lincecum TL Jr.; Alley MR; Martinis SA (2011) Characterization of benzoxaborole-based antifungal resistance mutations demonstrates that editing depends on electrostatic stabilization of the leucyl-tRNA synthetase editing cap FEBS Lett, 585, 2986–91. DOI: 10.1016/j.febslet.2011.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Palencia A; Li X; Bu W; Choi W; Ding CZ; Easom EE; Feng L; Hernandez V; Houston P; Liu L; Meewan M; Mohan M; Rock FL; Sexton H; Zhang S; Zhou Y; Wan B; Wang Y; Franzblau SG; Woolhiser L; Gruppo V; Lenaerts AJ; O’Malley T; Parish T; Cooper CB; Waters MG; Ma Z; Ioerger TR; Sacchettini JC; Rullas J; Angulo-Barturen I; Perez-Herran E; Mendoza A; Barros D; Cusack S; Plattner JJ; Alley MR (2016) Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase Antimicrob Agents Chemother, 60, 6271–80. DOI: 10.1128/AAC.01339-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sonoiki E; Palencia A; Guo D; Ahyong V; Dong C; Li X; Hernandez VS; Zhang YK; Choi W; Gut J; Legac J; Cooper R; Alley MR; Freund YR; DeRisi J; Cusack S; Rosenthal PJ (2016) Antimalarial Benzoxaboroles Target Plasmodium falciparum Leucyl-tRNA Synthetase Antimicrob Agents Chemother, 60, 4886–95. DOI: 10.1128/AAC.00820-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Palencia A; Liu RJ; Lukarska M; Gut J; Bougdour A; Touquet B; Wang ED; Li X; Alley MR; Freund YR; Rosenthal PJ; Hakimi MA; Cusack S (2016) Cryptosporidium and Toxoplasma Parasites Are Inhibited by a Benzoxaborole Targeting Leucyl-tRNA Synthetase Antimicrob Agents Chemother, 60, 5817–27. DOI: 10.1128/AAC.00873-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Monteferrante CG; Jirgensons A; Varik V; Hauryliuk V; Goessens WH; Hays JP (2016) Evaluation of the characteristics of leucyl-tRNA synthetase (LeuRS) inhibitor AN3365 in combination with different antibiotic classes Eur J Clin Microbiol Infect Dis, 35, 1857–1864. DOI: 10.1007/s10096-016-2738-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mendes RE; Alley MR; Sader HS; Biedenbach DJ; Jones RN (2013) Potency and spectrum of activity of AN3365, a novel boron-containing protein synthesis inhibitor, tested against clinical isolates of Enterobacteriaceae and nonfermentative Gram-negative bacilli Antimicrob Agents Chemother, 57, 2849–57. DOI: 10.1128/AAC.00160-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hernandez V; Crepin T; Palencia A; Cusack S; Akama T; Baker SJ; Bu W; Feng L; Freund YR; Liu L; Meewan M; Mohan M; Mao W; Rock FL; Sexton H; Sheoran A; Zhang Y; Zhang YK; Zhou Y; Nieman JA; Anugula MR; Keramane el M; Savariraj K; Reddy DS; Sharma R; Subedi R; Singh R; O’Leary A; Simon NL; De Marsh PL; Mushtaq S; Warner M; Livermore DM; Alley MR; Plattner JJ (2013) Discovery of a Novel Class of Boron-Based Antibacterials with Activity against Gram-Negative Bacteria Antimicrob Agents Chemother, 57, 1394–403. DOI: 10.1128/AAC.02058-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).O’Dwyer K; Spivak AT; Ingraham K; Min S; Holmes DJ; Jakielaszek C; Rittenhouse S; Kwan AL; Livi GP; Sathe G; Thomas E; Van Horn S; Miller LA; Twynholm M; Tomayko J; Dalessandro M; Caltabiano M; Scangarella-Oman NE; Brown JR (2015) Bacterial Resistance to Leucyl-tRNA Synthetase Inhibitor GSK2251052 Develops during Treatment of Complicated Urinary Tract Infections Antimicrob Agents Chemother, 59, 289–98. DOI: 10.1128/AAC.03774-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhao H; Palencia A; Seiradake E; Ghaemi Z; Cusack S; Luthey-Schulten Z; Martinis S (2015) Analysis of the Resistance Mechanism of a Benzoxaborole Inhibitor Reveals Insight into the Leucyl-tRNA Synthetase Editing Mechanism ACS Chem Biol, 10, 2277–85. DOI: 10.1021/acschembio.5b00291 [DOI] [PubMed] [Google Scholar]
- (25).Xia Y; Alley MKR; Zhou Y; Hernandez VS; Plattner JJ; Ding CZ; Cao K; Zhang YK; Benowitz A; Akama T; Sligar J; Jia G; Ou L; Saraswat N; Ramachandran S; Diaper C; Zhang Y; Banda GR; Nieman JA; Keramane M; Mohammed R; Subedi R; Liang H; Singh R (2010) Boron-containing small molecules, US 2010.0256092A1
- (26).Petersen PJ; Jones CH; Bradford PA (2007) In vitro antibacterial activities of tigecycline and comparative agents by time-kill kinetic studies in fresh Mueller-Hinton broth Diagn Microbiol Infect Dis, 59, 347–9. DOI: 10.1016/j.diagmicrobio.2007.05.013 [DOI] [PubMed] [Google Scholar]
- (27).Silva F; Lourenco O; Queiroz JA; Domingues FC (2011) Bacteriostatic versus bactericidal activity of ciprofloxacin in Escherichia coli assessed by flow cytometry using a novel far-red dye J Antibiot (Tokyo), 64, 321–5. DOI: 10.1038/ja.2011.5 [DOI] [PubMed] [Google Scholar]
- (28).Pankey GA; Sabath LD (2004) Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections Clin Infect Dis, 38, 864–70. DOI: 10.1086/381972 [DOI] [PubMed] [Google Scholar]
- (29).Hu QH; Liu RJ; Fang ZP; Zhang J; Ding YY; Tan M; Wang M; Pan W; Zhou HC; Wang ED (2013) Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase Sci Rep, 3, 2475 DOI: 10.1038/srep02475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Palencia A; Crepin T; Vu MT; Lincecum TL Jr.; Martinis SA; Cusack S (2012) Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase Nat Struct Mol Biol, 19, 677–84. DOI: 10.1038/nsmb.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Whiteway J; Koziarz P; Veall J; Sandhu N; Kumar P; Hoecher B; Lambert IB (1998) Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli J Bacteriol, 180, 5529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sandegren L; Lindqvist A; Kahlmeter G; Andersson DI (2008) Nitrofurantoin resistance mechanism and fitness cost in Escherichia coli J Antimicrob Chemother, 62, 495–503. DOI: 10.1093/jac/dkn222 [DOI] [PubMed] [Google Scholar]
- (33).Patterson S; Wyllie S (2014) Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects Trends Parasitol, 30, 289–98. DOI: 10.1016/j.pt.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Miller AF; Park JT; Ferguson KL; Pitsawong W; Bommarius AS (2018) Informing Efforts to Develop Nitroreductase for Amine Production Molecules, 23 DOI: 10.3390/molecules23020211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tavares AF; Nobre LS; Melo AM; Saraiva LM (2009) A novel nitroreductase of Staphylococcus aureus with S-nitrosoglutathione reductase activity J Bacteriol, 191, 3403–6. DOI: 10.1128/JB.00022-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Manjunatha U; Boshoff HI; Barry CE (2009) The mechanism of action of PA-824: Novel insights from transcriptional profiling Commun Integr Biol, 2, 215–8. DOI: 10.4161/cib.2.3.7926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Manjunatha UH; Boshoff H; Dowd CS; Zhang L; Albert TJ; Norton JE; Daniels L; Dick T; Pang SS; Barry CE 3rd. (2006) Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis Proc Natl Acad Sci U S A, 103, 431–6. DOI: 10.1073/pnas.0508392103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.