

Abstract
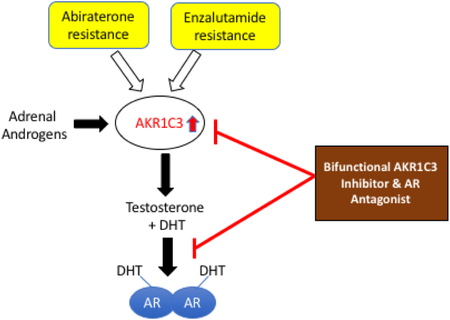
Drugs used for the treatment of castration resistant prostate cancer (CRPC) include Abiraterone acetate (Zytiga®) and Enzalutamide (XTANDI®). However, these drugs provide clinical benefit in metastatic disease for only a brief period before drug resistance emerges. One mechanism of drug resistance involves the overexpression of type 5 17-β-hydroxysteroid dehydrogenase (aldo-keto reductase 1C3 or AKR1C3), a major enzyme responsible for the formation of intratumoral androgens that activate the androgen receptor (AR). 3-((4-Nitronaphthalen-1-yl)amino)benzoic acid 1 is a “first-in-class” AKR1C3 competitive inhibitor and AR antagonist. Compound 1 was compared in a battery of in vitro studies with structurally related N-naphthyl-aminobenzoates, and AKR1C3 targeted therapeutics e.g. GTx-560 and ASP9521, as well as with R-bicalutamide, enzalutamide and abiraterone acetate. Compound 1 was the only naphthyl derivative that was a selective AKR1C3 inhibitor and AR antagonist in direct competitive binding assays and in AR driven reporter gene assays. GTx-560 displayed weak activity as a direct AR antagonist but had high potency in the AR reporter gene assay consistent with its ability to inhibit the co-activator function of AKR1C3. By contrast ASP9521 did not act as either an AR antagonist or block AR reporter gene activity. Compound 1 was the only compound that showed comparable potency to inhibit AKR1C3 and act as a direct AR antagonist. Compound 1 blocked the formation of testosterone in LNCaP-AKR1C3 cells, and the expression of PSA driven by the AKR1C3 substrate (4-androstene-3,17-dione) and by an AR agonist, 5α-dihydrotestosterone consistent with its bifunctional role. Compound 1 blocked the nuclear translocation of the AR at similar concentrations to enzalutamide and caused disappearance of the AR from cell lysates. R-biaclutamide and enzalutamide inhibited AKR1C3 at concentrations 200x greater than compound 1, suggesting that its bifunctionality can be explained by a shared pharmacophore that can be optimized.
Keywords: Aldo-keto reductase (AKR), non-steroidal anti-inflammatory drugs (NSAIDs), N-Phenylanthranilic acids, adaptive androgen biosynthesis, androgen receptor antagonist, competitive inhibition, enzalutamide, abiraterone
GraphicalAbstract
1. Introduction
Androgen deprivation therapy (ADT) has been a mainstay for advanced prostate cancer [1-3]. Following a period of remission, the cancer invariably returns despite castrate levels of circulating androgens and is accompanied by an increase in prostatic specific antigen (PSA). This form of the disease is known as castration resistant prostate cancer (CRPC). CRPC is invariably fatal and accounts for 30,000 deaths annually in the US alone [4]. CRPC arises due to adaptive responses in the tumor which includes among others, increased intratumoral androgen biosynthesis and amplification of the androgen receptor (AR) [5, 6]. Both these mechanisms are targeted by new agents. Abiraterone acetate targets CYP17A1 predominately in the adrenal depriving the tumor of adrenal androgens e.g. dehydroepiandrosterone (DHEA)-SO4 and DHEA, which can be converted to the potent androgens testosterone (T) and dihydrotestosterone (DHT) [7-9]. By contrast AR is targeted by a super-antagonist, enzalutamide (ENZ) which also prevents AR from translocating to the nucleus and binding to chromatin [10, 11]. After an initial response, drug resistance occurs with both agents within 3-4 months [12, 13]. While many mechanisms of drug resistance occur, one involves the overexpression of type 5 17β-hydroxysteroid dehydrogenase or AKR1C3 [14, 15].
AKR1C3 plays a role in all biosynthetic pathways to DHT within the prostate. It converts 4-androstene-3,17-dione to testosterone by the canonical pathway [16, 17]; it converts 5α-androstane-3,17-dione to DHT [17, 18] by a pathway that bypasses T altogether [19]; and it converts androsterone to 5α-androstane-3α,17β-diol, [17, 18]in the penultimate step in the backdoor pathway [20]. AKR1C3 also catalyzes the conversion of DHEA to Δ5-androstene-3β, 17β-diol an immediate precursor of testosterone. AKR1C3 inhibitors would act downstream of Abiraterone acetate and other CYP17A1 inhibitors and block terminal steps in androgen biosynthesis. They would also not require co-administration with prednisone since they would not cause adrenal insufficiency. With this mind, we and others have embarked on developing AKR1C3 inhibitors for eventual clinical use in CRPC[21-26].
3-(4-Nitronaphthlen-1-yl) amino-benzoate (compound 1) is one compound that emerged as a promising lead in that it displayed nanomolar affinity for inhibition of AKR1C3 and also functioned as a AR antagonist based on its ability to block AR driven reporter gene activity [27]. We now report extended structure-activity relationships on this lead and compare its potency and efficacy with other targeted therapies for AKR1C3, including GTx-560 and ASP9521 that have been reported [24, 26]. We find that compound 1 is the only N-naphthylamino-benzoate that has the desired bifunctional activity to inhibit AKR1C3 and antagonize the AR. We also find that R-biaclutamide and enzalutamide are weak inhibitors of AKR1C3 supporting the presence of a shared pharmacophore between AKR1C3 inhibitors and AR antagonists that could be exploited in further optimization.
2. Materials and Methods:
2. 1. Reagents.
Reagents were of ACS grade or higher and were purchased from Fisher Scientific (Pittsburgh, PA, USA) and used without further purification. [1β-3H(N)]-Δ4-androstene-3,17-dione (4-AD) (Specific Radioactivity 96.2 Ci/mole) and [3H-R1881 (85.1 Ci/mmole) were obtained from Perkin-Elmer. 4-Androstene-3,17-dione and dihydrotestosterone were purchased from Steraloids and Sigma, respectively. Unlabeled R1881 was obtained from Perkin-Elmer. Nicotinamide adenine dinucleotide, Grade I reduced form (NADH) and nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) were obtained from Roche Diagnostics (Indianapolis, IN, USA). R-biaclutamide was purchased from Sigma; Enzalutamide was obtained from Selleckchem; and abiraterone was purchased Euroasia Chemicals (Mumbai - 400 013, India ). Charcoal dextran stripped fetal bovine serum (CD-FBS) was from Atlanta Biologicals (Lawrenceville, GA, USA).
2. 2. Synthesis of Compounds.
GTx560 6-hydroxy-4-(3,4,5-trifluorophenyl)- 1(2H)-Isoquinolinone, and ASP9521 1-(-{1-[(5-methoxy-1H-indol-2-yl)carbonyl] piperidin-4-yl}-2-methylpropan-2-ol) were synthesized according to published methods [28, 29]. The following compounds: 3-((4-nitronaphthalen-1-yl)amino)benzoic acid (1); 3-(naphthalen-1-ylamino)benzoic acid (2); 3-((4,5-dinitronaphthalen-1-yl)amino)benzoic acid (3); 3-((4-acetylnaphthalen-1-yl)amino)benzoic acid (4); 5-((4-acetylnaphthalen-1-yl)amino)isophthalic acid (5); 3-((4-cyanonaphthalen-1-yl)amino)benzoic acid (6); 3-((4-(trifluoromethyl)naphthalen-1-yl)amino)benzoic acid (7); 3-((4-fluoronaphthalen-1-yl)amino)benzoic acid (8); 3-(quinolin-4-ylamino)benzoic acid (9); 4-(naphthalen-1-ylamino)benzoic acid (10); 4-((4-acetylnaphthalen-1-yl)amino)benzoic acid (11); 2-(naphthalen-1-ylamino)benzoic acid (12); 4-methoxy-2-(naphthalen-1-ylamino)benzoic acid (13); 5-acetyl-2-(naphthalen-1-ylamino)benzoic acid (14); 5-acetyl-2-((4-nitronaphthalen-1-yl)amino)benzoic acid (15); dimethyl-3-(4-nitronaphthalen-1-yl)imidazolidine-2,4-dione (16); and 5,5-dimethyl-3-(4-nitronaphthalen-1-yl)-2-thioxoimidazolidin-4-one (17) were synthesized as described in the e-component containing supporting information.
2. 3. Cell Lines and Antibodies.
LNCaP-AKR1C3 cells were obtained and grown as described [16] and HeLa-AR3a-PSA-(ARE)4-Luc 13 cells were a kind gift from Dr. Elizabeth Wilson. Anti-human-PSA antibody (SC-7638) and murine anti-human AR (sc-441) were obtained from Santa Cruz Biotechnology; monoclonal anti-human β-tubulin (clone AA2 was from EMD Millipore); murine monoclonal anti-human β-actin (was from Sigma A5441) and anti-human GAPDH (was from Millipore MAB374).
2. 4. Molecular Simulation
Ligand binding poses generated using a LigAlign v1.0 plugin (University of Toronto) installed onto Pymol v1.7.6 (Schrödinger). The crystal structure of AKR1C3 in complex with NADP+ and 3’-[(4-nitronaphthalen-1-yl)amino]benzoic acid (PBD codes: 4DBS) was used as a template to determine interactions of compounds in the AKR1C3 binding pockets. The crystal structures of the AR-LBD in complex with S-1 (PDB code: 2AXA) and AR-LBD W741L mutant complex with R-bicalutamide (PBD code: 1Z95) were used as templates to superimpose AKR1C3 inhibitors in AR-LBD and compare these poses to those observed with known AR antagonists. Water molecules and original ligands were removed from the structures and hydrogens were manually added using AutoDockTools-1.5.6 (Scripps Research Institute). Ligands (Compounds 2, 3, 4) were generated as PDB files using Chem 3D Pro 14.0 (Cambridgesoft). All ligand bonds were identified as flexible. Parameters for the gridbox were determined from the original ligand binding sites with the size of 40 for all the coordinates (see Table 1 in Supporting Information). Docking experiments were performed using AutoDock Vina (Scripps Research Institute) [30].
Table 1.
Inhibitory Properties of Compounds on Other Human AKR1C Enzymes
| Compound | IC50 values (μM) | |||
|---|---|---|---|---|
| AKR1C3 | AKR1C1 | AKR1C2 | AKR1C4 | |
| 1 | 0.086 ± 0.011 | 41.3 ± 1.64 | 24.7 ± 1.07 | 30.6 ± 3.87 |
| 3 | 0.053 ± 0.002 | 15.4 ± 1.9 | 15.6 ± 1.1 | 5.34 ± 0.29 |
| 4 | 0.082 ± 0.006 | 50.0 ± 3.89 | 19.5 ± 0.47 | 17.7 ± 2.08 |
All values are mean + SE (n= 3)
2. 5. Enzyme Purification
Homogenous recombinant AKR1C1-4 were expressed and purified as previously described [31]. Enzymes were stored at −80 °C in 20 mM potassium buffer pH 8.0 containing 30% glycerol, 1 mM EDTA and 1 mM β-mercaptoethanol. Enzymes gave the predicted specific activities observed for the purified enzymes.
2. 6. Enzyme Assays
Inhibition of S-Tetralol oxidation: The half maximal inhibitory concentration (IC50) for each analog was estimated by measuring the oxidation of S-tetralol to tetralone catalyzed by the AKR1C isoform in the presence of saturating concentrations of NADP+ in the absence and presence of different inhibitor concentrations. Assay conditions and fluorescence measurements were conducted using our published procedure [21-23]. Patterns of enzyme inhibition were determined at five different fixed concentrations of S-tetralol while the inhibitor concentration was varied. Data were fit to the equations for COMP, NONCOMP, and UNCOMP using the Grafit program to obtain the best fit.
Inhibition of Δ4-AD Reduction: The ability of compounds to inhibit the AKR1C3 catalyzed reduction of [3H] Δ4-AD to [3H] testosterone was monitored in the presence of saturating concentrations of NADPH in the presence and absence of different inhibitor concentrations according to the published procedure [21]. The assay system contained 5 nM [3H] Δ4-AD, fixed concentrations of non-radioactive Δ4-AD to bring the final concentration of substrate to 1.25-20 μM, varied concentrations 0.3-10 μM inhibitor, 0.9 mM NADPH, and 1.65 μM recombinant AKR1C3 in 100 mM phosphate buffer pH 7.0,
2. 7. Cell-Based Assays
2 .7. 1. Inhibition of androgen metabolism in LNCaP-AKR1C3 cells.
The ability of analogs to inhibit the reduction of [3H] Δ4-AD to testosterone in LNCaP-AKR1C3 cells was monitored according to our previously reported procedure [21]. LNCaP-AKR1C3 cells were plated in 6 well flat bottom tissue culture plates with a low evaporation Lid (Corning Inc., #353046) at a density of 1.5 × 106 cells in phenol red free RPMI-1640 media supplemented with 5% CD-FBS, 1% Pen/Strep and 2 mM L-Glutamine (CSS Media). The cells were allowed to attach for 24 hr, after which time CSS media was replaced by fresh media. The cells were pre-treated with varying concentrations of inhibitor dissolved in 100% DMSO for 30 min, and then treated with 100 nM Δ4-AD radioactive mixture, consisting of 7.5 nM (1.26 μCi) of [3H] Δ4-AD and 92.5 nM of non-radioactive Δ4-AD in a final concentration of 0.75% DMSO. Treated cells were incubated with the reaction mixture for 48 hr, after which time the cell supernatant was collected for analysis by radiochromatography as previously described [16, 21].
2. 7. 2. Displacement of [3H]-R1881 Binding to AR.
HeLa-13 cells that are stably transfected with the AR and a luciferase reporter gene (HeLa-AR3A-PSA-(ARE)4-Luc13 cell, Hela-13) were used for this assay [32]. HeLa-13 cells were plated in 12 well flat bottom plates with lid (Corning Inc., #3513) containing 1 mL phenol red free MEM media supplemented with 5% CD-FBS, 1% Pen/Strep and 2 mM L-Glutamine at a density of 4 × 105 cells per well. Cells were allowed to attach for 24 hr, after which time the media was aspirated and replaced by fresh MEM containing 0.1 nM [3H]-R1881. 100% DMSO inhibitor solution at varying concentration was added to each well to achieve a final concentration of 0.5% DMSO. Cells were incubated in the absence and presence of inhibitors for 2 hr. The extraction of the bound [3H]- R1881 from each well was initiated by incubating the cells with 200 μL of Lysis Buffer A (2% SDS, 10% glycerol, 10 mM Tris-HCl, pH 6.8) at 37 °C. Lysed cells were shaken vigorously before they are neutralized by 20-min incubation with 300 μL of Buffer B (10 mM Tris-HCl, pH 8.). Bound ligand was quantified using scintillation counting. A portion of each well was added to Ultima Gold scintillation fluid (Perkin Elmer Life Sciences) and analyzed on a TriCarb 2100 (Packard Instruments, Perkin Elmer Life Sciences) and radioactivity reported as corrected cpm. Samples were normalized to total protein measured using Pierce BCA Protein Assay Kit (Thermo Scientific, #23225). Binding that remained after a 10 nM DHT treatment was defined as non-specific binding and was subtracted.
2. 7. 3. AR-Luciferase Reporter Gene Assays:
HeLa-13 cells that are stably transfected with the AR and a luciferase reporter gene (HeLa-AR3A-PSA-(ARE)4-Luc13 cell, Hela-13) were used for this assay. The cells (1.5 × 104) were platted in a 96-well plate in phenol-red-free-MEM supplemented with 5% charcoal stripped FBS media. After attachment (6-8 h) the media was aspirated and replaced with fresh media containing 0.1 nM DHT in the presence and absence of increasing concentrations of drug and luciferase activity measured using the BightGlo luciferase kit (PRO E2610 [32].
3. Results
Compound 1, was previously identified as a lead bifunctional agent that inhibited AKR1C3 and antagonized the AR [27]. Here we describe our medicinal chemistry efforts (Figure 1) to optimize this bifunctionality. Class 1 compounds have the closest resemblance to compound 1, in that the carboxylic acid was retained at the metci- position on the A-ring and substituents on the B and B’ rings were varied (compounds 1-9), where compound 9 was a quinoline derivative. In class 2 compounds, the carboxylic acid was moved to the para- position on the A-ring (compounds 10-11).
Figure 1.

Classes of N-naphthylaminobenzoate analogs derived from compound 1.
In class 3 compounds, the carboxylic acid was moved to the ortho-position and substituents varied on both the A and B rings (compounds 12-15). Class 4 compounds were bioisosteres of compound 1 and included 5,5-dimethyl-1-(4-nitronaphthalen-1-yl) imidazolidine-2,4-dione (16) and 5,5-dimethyl-1-(4-nitronaphthalen-1-yl)-2-thioxoimidazolidin-4-one (17).
3. 1. Class 1: 3-(Naphthen-1-yl)aminobenzoate Analogues with B and/or B’-Ring Substitution.
Compounds were screened for their ability to inhibit the NADP+ dependent oxidation of S-tetralol catalyzed by AKR1C3 and AKR1C2. AKR1C2 was used in counter screens due to its high sequence identity with AKR1C3 (> 86%) and because its inhibition is not desired in prostate cancer since it eliminates DHT [33]. We found that the presence of an electron-withdrawing group (EWG) significantly increases potency and selectivity for AKR1C3 within this class (Figure 2).
Figure 2.

IC50 values of 3-(naphthalen-1-ylamino)benzoic acid analogs for the inhibition of S-tetralol oxidation catalyzed by AKR1C3 and AKR1C2.
The presence of the p-nitro group in the B ring of compound 1 displays a significant improvement in the desired properties over compound 2 which lacks the p-nitro group. Improvements in potency increase according to the strength of the EWG in the B and B’ rings. Analogs with strong EWGs (compounds 1, 4, and 7) displayed greater inhibitory potency and selectivity for AKR1C3 than compounds that did not (compounds 8) and when the naphthalene ring was replaced with a quinoline ring (compound 9), where the ring nitrogen would be electron donating, the potency and selectivity was reversed in favor of AKR1C2. The presence of two nitro groups in compound 3 increased the potency when compared to the lead compound 1. Furthermore, introduction of an additional carboxylic group into the A ring decreased the inhibition potency of the compound against AKR1C3, thus the dicarboxylate compound 5 had an IC50 value 10 times higher than the corresponding monocarboxylate, compound 4.
3. 2. Classes 2 and 3: 4- and 2-(Nitronaphthen-1-yl)aminobenzoate Analogues (NAB) with A and/or B Ring Substitution.
The inhibitory profile of class 2 and 3 compounds are shown in Figure 3. Notably, p-and o-carboxylic acid NAB analogs inhibit AKR1C3 in the low micromolar range but display a decrease in the AKR1C3 potency and selectivity versus compound 1, see compounds 10, 11. Introduction of substituents in the A ring of N-phenylamino benzoate-based analogs compounds 12 versus 13 did not significantly alter these properties. However, the presence of an EWG in the B ring of compound 15 showed a slight improvement in both AKR1C3 inhibitory potency and selectivity over compound 14.
Figure 3.

IC50 values of 2- and 4-(napththalen-1-ylamino)benzoic acid analogs for the inhibition of S-tetralol oxidation catalyzed by AKR1C3 and AKR1C2.
3. 3. Class 4: Bioisostere Compounds.
Replacement of the A ring with its bioisosteres such as imidazolidine-2,3-dione and thiooimidazolidine-4-one eliminates AKR1C3 inhibition (Figure 4). However, compounds 16 and 17 displayed an improvement in AKR1C2 inhibition potency compared to the lead compound 2.
Figure 4.

IC50 values of 5,5-dimethyl-1-(4-nitronaphthalen-1-yl) imidazolidine-2,4-dione 16 and 5,5-dimethyl-1-(4-nitronaphthalen-1-yl)-2-thioxoimidazolidin-4-one 17 for the inhibition of S-tetralol oxidation catalyzed by AKR1C3 and AKR1C2.
The most promising potent AKR1C3 inhibitors developed from lead compound 1 were compounds 3 and 4. These analogs displayed low nanomolar AKR1C3 inhibition activity and displayed selectivity for the inhibition of AKR1C3 over other human AKR1C isoforms by at least 2 orders of magnitude (Table 1).
3. 4. Mode of AKR1C3 inhibition by NAB analogs.
The patterns of AKR1C3 inhibition by lead naphthylaminobenzoates 1, 3, 4 were evaluated in two assays, inhibition of the NADP+ dependent oxidation of S-tetralol and inhibition of NADPH dependent reduction of Δ4-AD catalyzed by recombinant AKR1C3. Compounds 1, 3, and 4 displayed competitive inhibition against AKR1C3 with Ki values for the oxidation reaction ranging from 41 to 76 nM and displayed competitive inhibition against AKR1C3 with Ki values for the reduction of Δ4-AD ranging from 0.87 to 2.4 μM (Figure 5 and Table 2). The difference in the binding constants in the two assays (20-40 fold) suggests that the inhibitors exhibit preference for the E•NADP complex in the oxidation direction over the E•NADPH complex in the reduction direction similar to that previously reported for other AKR1C3 inhibitors [21].
Figure 5.

Lead N-naphthylaminobenzoates display competitive enzyme inhibition for the oxidation of S-tetralol and reduction of 4-Androstene-3,17-dione catalyzed by AKR1C3. Panels (A-C) competitive inhibition of S-tetralol oxidation by compounds 1, 3 and 4, respectively. Panels (D-F) competitive inhibition of 4-androstene-3,17-dione reduction by compounds 1, 3 and 4, respectively.
Table 2.
Inhibitory Properties of Drug Candidates
| Drug | IC50 value S-Tetralol Oxidation (nM) |
Ki value S-Tetralol Oxidation (nM) |
Ki value Δ4-AD Reduction (μM) |
Ki Ratios Δ4-AD : S-Tetralol Inhibition |
IC50 AR- Binding Displacement of R1881 |
Ratio IC50 AR Binding: Ki Δ4-AD Reduction |
IC50 Luciferase Activity With DHT |
Ratio IC50 Luciferase: Ki Δ4-AD Reduction |
|---|---|---|---|---|---|---|---|---|
| R1881 | ND | ND | ND | ND | 0.42 ± 0.05 nM | ND | ND | ND |
| Enzalutamide | 12300 ± 880 | ND | ND | ND | 162 ± 3 nM | ND | 311 ± 70 nM | ND |
| R-Bicalutamide | 10000 ± 100 | ND | ND | ND | 160 ± 3 nM | ND | 937 ± 69 nM | ND |
| GTX-560 | 35 ± 2 | 13 ± 2.9 | 0.54 ± 0.7 | 42 | 80.0 ± 4 μM | 148 | 2.1 ± 0.58 μM | 3.88 |
| Compound 1 | 86 ± 11 | 52.9 ± 4.8 | 2.4 ± 0.27 | 45 | 8.0 ± 2 μM | 3.3 | 21 ± 6 μM | 8.75 |
| Compound 3 | 53 ± 2 | 40.6 ± 3.6 | 0.87 ± 0.14 | 21 | >100 μM | ≫115 | ND | ND |
| Compound 4 | 82 ± 6 | 75 ± 6.7 | 1.6 ± 0.46 | 21 | >100 μM | ≫ 63 | ND | ND |
| ASP9521 | ND | ND | 0.29 ± 0.05 | ND | >100 μM | ≫345 | 300 ± 132 μM | 1034 |
| Abiraterone | 3000 | ND | ND | ND | >100 μM | ND | 2.6 ± 0.27 μM | ND |
The mean ± S.E mean is shown.
3. 5. Comparison of Lead NAB analogs to other AKR1C3 inhibitors in advanced development.
We compared the inhibitory potency of the lead compounds with two AKR1C3 inhibitors in advanced development GTx-560 (GTx-Therapeutics)[26] and ASP9521 (Astellas) [24, 34]. GTx-560 gave a Ki value of 13 nM for the competitive inhibition of S-tetralol assay and Ki value of 0.54 μM for the competitive inhibition of 4-AD reduction. It was not possible to determine an IC50 value for ASP9521 in the S-tetraol assay due to interfering fluorescence from the inhibitor. However, it gave a Ki value of 0.29 μM for the competitive inhibition of 4-AD reduction, Table 2. The Ki value for the reduction of 4-AD observed for ASP9521 was only modestly lower than that observed for compounds 1, 3 and 4, which ranged from 0.87 μM to 2.4 μM. Abiraterone gave an IC50 value of 3.0 μM for the inhibition of S-tetralol oxidation. Knowing that the Ki value for Δ4-AD reduction is routinely 40-fold higher than the Ki value for S-tetralol oxidation due to the preference for the inhibitor to bind the E.NADP+ complex, abiraterone was not pursued further as a AKR1C3 inhibitor.
3. 6. NAB-analogs inhibit Androgen Biosynthesis in AR Dependent Prostate Cancer Cell Lines.
We also compared compounds 1, 3, 4 with GTx-560 and ASP9521 to inhibit the conversion of 4-AD to testosterone in LNCaP-AKR1C3 cells. In this assay, free testosterone is detected after treating the cell-media with β-glucuronidase. Percent inhibition was compared to the inhibition observed with 30 μM indomethacin (84% inhibition). Compounds 1, 3 and 4 at 30 μM inhibited testosterone production by 78%, 36% and 84%, respectively. By contrast GTx-560 inhibited 85% of testosterone production and ASP9521 inhibited 100% of testosterone production at 30 μM, (see Table 3.
Table 3.
Inhibition of Testosterone Production in LNCaP-AKR1C3 Cells by AKR1C3 Inhibitors
| Cell Line | Treatment | Percent Testosterone Formed1 |
|---|---|---|
| LNCaP-AKR1C3 Control | 0.2% DMSO | 100% |
| LNCaP- parental cells | 0.2% DMSO | 0% |
| LNCaP-AKR1C3 | 30 μM Indomethacin | 16.5% |
| LNCaP-AKR1C3 | 10 μM GTx 560 | 17.23% |
| LNCaP-AKR1C3 | 30 μM GTx 560 | 15.63% |
| LNCaP-AKR1C3 | 10 μM ASP9521 | 12.24% |
| LNCaP-AKR1C3 | 30 μM ASP9521 | 0% |
| LNCaP-AKR1C3 | 10 μM Compound 1 | 65.21% |
| LNCaP-AKR1C3 | 30 μM Compound 1 | 21.75% |
| LNCaP-AKR1C3 | 10 μM Compound 3 | 76.9% |
| LNCaP-AKR1C3 | 30 μM Compound 3 | 64.23% |
| LNCaP-AKR1C3 | 10 μM Compound 4 | 54.33% |
| LNCaP-AKR1C3 | 30 μM Compound 4 | 16.62% |
LNCaP-AKR1C3 cells were incubated with 100 nM [3H]-4-androstene-3,17-dione for 48 h and the formation of testosterone as testosterone-17β-glucuronide determined in the cell culture media in presence and absence of 10 μM or 30 μM of the respective inhibitor, and percent inhibition reported. Testosterone 17β-glucuronide was analyzed as free testosterone after treatment of the aqueous-phase with β-glucuronidase and was detected by radio-chromatography.
3. 7. Lead NAB analogs as AR antagonists in competitive bindings assays.
We showed that compound 1 acted as an AR antagonist in AR-dependent luciferase assays in HeLa cells. We now report the ability of lead compounds 1, 3 and 4, GTX-560, AS9521 to competitively displace the radioactive synthetic androgen methyltrienolone ([3H]-R1881) from the AR in in-cell binding assays. Only two compounds were effective competitors, compound 1 which gave an IC50 value of 8.0 μM and GTX-560 which gave an IC50 value of 80 μM. Positive controls, unlabeled R1881, MDV3100 (enzalutamide), and R-biaclutamide which gave IC50 values of 0. 42 nM, 162 nM and 160 nM, respectively. Of the compounds tested compound 1 appeared to be almost equipotent as an AKR1C3 inhibitor (4-AD reduction) and AR antagonist, see Figure 6.
Figure 6.

Displacement of [3H]-R1881 Binding from AR in Hela Cells. Cells (4 × 105) were incubated with 0.1 nM [3H]-R1881 for 2 hr in a 96-well plate in the presence and absence of increasing concentrations of drug. Bound [3H]- R1881 from each well was measured by scintillation counting after cell lysis. Non-specific binding was defined as the amount of ligand that was bound in the presence of 10 nM DHT. Displacement by unlabeled R1181 (○); displacement by enzalutamide (◆); displacement by R-biaclutamide (∇); displacement by compound 1 (•); displacement by GTX-560 (∎); displacement by compound 3 (▼); displacement by compound 4 ( ▲ ); and displacement by ASP9521 (●). Mean + SD (n=3)
3. 8. Ability of Lead NAB compounds Inhibit Transactivation of the AR in Reporter Gene Assays.
We screened the ability of lead compounds 1, 3 and 4, GTX-560, AS9521 to inhibit AR dependent reporter gene activity in Hela Cells transfected with an AR reporter (HeLa-AR3a-PSA-(ARE)4-Luc 13) and AR. HeLa cells also endogenously express AKR1C3. In these assays we observed IC50 values of 21 μM and 2.1 μM for compound 1 and GTX-560, respectively. We attribute the lower IC50 value for GTx-560 in this assay versus its high IC50 value in competitive binding assays for the AR due to its preference to block the co-activator function of AKR1C3 rather than act as a direct AR antagonist. As positive controls we observed IC50 values of 311 nM and 940 nM for enzalutamide and R-biaclutamide, respectively, Figure 7. The inhibition of the reporter gene assay by abiraterone yielding an IC50 = 2.6 μM could be related to its metabolism to the Δ4-abiraterone metabolite [35].
Figure 7.

Inhibition of AR Reporter Gene Assays by Lead Compounds. The ability of compounds to inhibit AR transcriptional activity induced by 0.1 nM DHT in HeLa cells cells that are stably transfected with the AR and a luciferase reporter gene (HeLa-AR3A-PSA-(ARE)4-Luc13 cell, Hela-13) were used for this assay. The luciferase activity was measured in (1.5 × 104) cells incubated in the presence of 0.1 nM DHT in the presence and absence of increasing concentrations of the drug shown. Enzalutamide (◆); R-biaclutamide ( ∇ ); compound 1 (•); GTX-560 (∎) ); ASP9521 (●) and Abiraterone (X). Mean + SD (n=3)
3. 9. Examination of R-Bicalutamide and Enzalutamide as Inhibitors of AKR1C3.
As compound 1 acts as an AR antagonist, R-biaclutamide and enzalutamide were also screened as inhibitors of S-tetralol oxidation. These compounds were shown to yield IC50 values in the 10 μM range confirming a shared pharmacophore for both AR antagonism and AKR1C3 inhibition, Figure 8. By contrast these AR antagonists gave limited inhibition of AKR1C2.
Figure 8.

Inhibition of the NADP+ dependent oxidation of S-tetralol catalyzed by AKR1C3 and AKR1C2 by R-biaclutamide and Enzalutamide.
3. 10. Compound 1 Blocks PSA Production and Nuclear Translocation of AR mediated by DHT.
To further explore the ability of compound 1 to act as both a competitive inhibitor of AKR1C3 and act as an AR antagonist we examined its ability to block the 4-AD and DHT dependent expression of PSA in LNCaP-AKR1C3 cells. Compound 1 blocked PSA expression driven by the AKR1C3 substrate (4-AD) and by the AR ligand (DHT), indicating the bifunctionality of the compound, Figure 9.
Figure 9.

Compound 1 (10 – 40 μM) inhibits PSA expression in LNCaP-AKR1C3 cells treated with either 100 nM 4-AD (AKR1C3 substrate) or by 1.0 nM by DHT (AR agonist). PSA expression, AR expression were measured by immunoblot analysis in cell lysates where β-tubulin was used as a loading control.
Compound 1 also blocked nuclear translocation of AR at concentrations 10-40 μM, by contrast enzalutamide blocked nuclear translocation at comparable levels at a concentration of 12.5 μM, Figure 10a. Compound 1 also caused the disappearance of AR from whole cell lysates in the presence and absence of DHT, Figure 10b.
Figure 10.

Compound 1 inhibits the translocation of the AR to the nucleus stimulated by 0.1 nM DHT. AR expression was measured by immunoblot analysis in the nuclear fraction of HeLa cells and normalized to β-tubulin (A). Compound 1 inhibits the expression of AR in LNCaP cells in the presence and absence of 0.1 nM DHT (B).
4. Discussion
We previously reported that compound 1 was a unique potent and selective AKR1C3 inhibitor and AR antagonist [27]. To explore the structure-activity relationships responsible for this bifunctionality we synthesized four classes of N-(naphthen-1-yl)amino-benzoates. Structure-activity relationships were consistent with those previously reported for the N-phenyl-amino benzoates [22, 23]. Compounds in class 1, which contain the carboxylic acid in the meta-position of the N-benzoic acid and had an EWG on the para-position of the B-naphthyl ring were the most potent and selective AKR1C3 inhibitors. Three lead compounds 1, 3 and 4 were shown to competitively inhibit the NADP+-dependent oxidation of S-tetralol and the NADPH-dependent reduction of 4-AD catalyzed by AKR1C3. These compounds gave 20-40 fold lower Ki values in the oxidation direction than in the reduction direction. This difference in Ki values has been noted previously and is due to the formation of two different abortive complexes, E•NADP+•I (in the oxidation direction) and E•NADPH•I (in the reduction direction) [21]. Formation of these complexes to cause competitive inhibition is entirely consistent with an ordered bi bi mechanism where the enzyme is either in the E.NADP+ form (oxidation direction) or the E,NADPH.I from (reduction direction). These compounds also showed greater than a 100-fold selectivity for AKR1C3 over other human AKR1C isoforms.
The AKR1C3 inhibition potency of the lead compounds were slightly less to those determined for two AKR1C3 inhibitors developed by industry GTx-560 and ASP9521, Table 2. Compound 1, GTx-560 and ASP9521 all inhibited the conversion of Δ4-AD to testosterone in LNCaP-AKR1C3 cells consistent with the competitive inhibition observed with these agents. However, there is a difference in potency of the compounds based on the in vitro enzyme assays and the assays performd in LNCaP-AKR1C3 cells, which may be related to cell permeability issues. All the inhibitors are carboxylic acids and would likely require transport by OATPs for cell entry.
We now report the binding of compounds 1, 3 and 4 as well as GTx-560 and ASP9521 to the AR by measuring the displacement of [3H]-R1881 in a competitive in-cell binding assay. Of these compounds, only compound 1 and GTx-560 antagonized ligand binding to the AR yielding IC50 values of 8 and 80 μM, respectively. In the same assay R-bicalutamide and enzalutamide gave IC50 values of 162 and 160 nM, respectively. The ratio of IC50 R1881 displacement (AR binding) versus Ki 4-AD reduction (AKR1C3 reaction) for compound 1 was 3.3 and that for GTx-560 was 148 making compound 1 a bifunctional AR antagonist and AKR1C3 inhibitor whereas GTx-560 does not act as a potent ligand for AR. By contrast ASP9521 had no effect on AR ligand binding.
Consistent with the AR ligand binding assay compound 1 blocked AR transcriptional activation with an IC50 value = 21 μM. The ratios of IC50 luciferase activity (AR transactivation) and Ki 4-AD reduction (AKR1C3 reaction) were 8.75 and 3.88 for compound 1 and GTx560, respectively. The low IC50 value for GTx-560 in this assay is consistent with its ability to block the coactivator function of AKR1C3, instead of acting as a direct AR antagonist. By contrast ASP9521 had no effect on AR transactivation. Thus, in every assay ASP9521 which was taken into phase1/1b clinical trial acts only as a monofunctional AKR1C3 inhibitor [24, 36].
To further explore the bifunctionality of compound 1, we were able to show that it blocked PSA production mediated by both an AKR1C3 substrate and an AR agonist, it also caused AR disappearance from cell lysates. The bifunctionality of compound 1 and the reliance on the presence of an EWG, suggested that we examine R-biaclutamide and enzalutamide as AKR1C3 inhibitors. We found that both compounds blocked the oxidation of S-tetralol catalyzed by AKR1C3 with IC50 values = 10 μM. The ratio of IC50 values for inhibition of S-tetralol oxidation catalyzed by AKR1C3 versus IC50 values to block [3H]-R1881 binding to the AR were 76 and 63 for enzalutamide and R-biaclutamide, respectively.
The AKR1C3•NADP+• 1 complex shows that compound 1 adopts a similar pose to that of flufenamic acid (PDB ID: 1S2C). The carboxylate group of the N-benzoic acid ring interacts with the oxyanion site through hydrogen bonds to Tyr 55 and His 117 (Figure 11) and the phenylamino ring extends into the SP1 subpocket (Ser 118, Asn 167, Phe 306, Phe 311, and Tyr 319). Substitution of the EWG in the para-position allows the functional groups to penetrate deeper into the SP1 pocket, increasing the observed selectivity over other AKR1C isoforms which contain shallower SP1 pockets. Molecular docking of compounds 3 and 4 into AKR1C3, generated binding poses similar to compound 1. The N-benzoic acid rings extend into the oxyanion site, while the naphthylamino ring interacts with residues within SP1. The remainder of the molecule interacts with active site hydrophobic amino-acids between the two sites.
Figure 11.

Molecular Modeling Experiments: Binding to AKR1C3 and AR. Predicted binding poses for Compound 4 in the AKR1C3 active site. AKR1C3 subpocket 1 (cyan), AKR1C3 subpocket 3 (green), Compound 1 (purple), Compound 4 (white), oxyanion site (residues highlighted in magenta). The structures are based on the crystal structure of the AKR1C3•NADP+ and 3’-[4-nitronaphthalen-1-yl)amino]benzoic acid complexes (PDB code: 4DBS), (A). Comparison of the binding of compound 1 in the AR-LBD site to S-1 and R-bicalutamide in AR-LBD and AR-LBD W741L (B). Left panel, red bonds in compound 1 shows similarity with the AR ligands. Right panel, AR-LBD (green), nonsteroidal agonist modulator S-1 (magenta), AR-LBD W741L (cyan), R-bicalutamide (yellow) and compound 1 (white). Structures of AR-LBD W741L in complex with R-bicalutamide (1Z95) The figure was generated with Pymol (Delano Scientific).
N-naphthylamino-benzoates which contain a p- carboxylates in the A-ring have reasonable potency as AKR1C3 inhibitors as they inhibit in the low micromolar range but this is not comparable with that observed for compounds 1, 3 and 4 which have inhibitory potency in the mid-nanomolar range. The p-carboxylates have also lost their selectivity over AKR1C3 and this is a more important issue. We know from crystal structures that the B’-rings of the naphthyl moiety bind into the SP1 pocket which is absent in AKR1C2 [27]. Thus, the loss of selectivity is due to the possibility that the naphthyl ring can no longer enter into the SP1 pocket when the p-carboxyl group anchors to the catalytic tyrosine.
No crystal structure exists of the AR-LBD with antagonist-bound in the antagonist conformation, and thus understanding the structural basis for compound 1 antagonism of AR is difficult to address. We docked compound 1, into the AR-LBD that is bound by the selective androgen receptor modulator S-1, a structural analog of R-biaclutamide. In addition, the mutant AR-LBD W741L binds R-bicalutamide in an agonist conformation [37, 38] and this structure was superimposed on the docked ligands to compare binding interactions. Compound 1 is predicted to interact with AR-LBD and AR-LBD W741L through hydrophobic interactions similar to that observed with S-1 and R-bicalutamide (Figure 11). Key hydrophobic interactions were predicted to be with Met 745, Met 787, Leu 704 and Trp 741. The meta-carboxylic acid is located near Arg 752, while a water molecule in proximity to the EWG groups could form hydrogen bonds with Glu 738, Tyr 739, and Leu 741 to strengthen the interaction.
5. Conclusion
We conducted SAR on (3-((4-nitronaphthalen-1-yl)amino)benzoic acid 1 which acts as a potent and selective AKR1C3 inhibitor and AR antagonist. Compound 1 efficiently blocked the formation of testosterone in LNCaP-AKR1C3 cells, which are a model for CRPC cells. The compound blocked ligand binding to the AR, and AR transactivation. A head-to-head comparison with two AKR1C3 inhibitors in advanced development e.g. GTx-560 and ASP9521, demonstrate that (3-((4-nitronaphthalen-1-yl)amino)benzoic acid is a “first-in-class” bifunctional AKR1C3 inhibitor and AR antagonist.
Supplementary Material
Table 1S. Gridbox parameters for docking experiments
Highlights.
AKR1C3 plays a pivotal role in androgen biosynthesis in CRPC
Compound 1 is a “first-in-class” AKR1C3 competitive inhibitor and AR antagonist
Compound 1 was compared with advanced AKR1C3 inhibitors GTx560 and ASP9521
Only compound 1 showed equal potency as an AKR1C3 inhibitor and AR antagonist
ACKNOWLEDGMENT.
This work was supported by P30-ES013508 awarded to TMP.
ABBREVIATIONS
- AR
androgen receptor
- CRPC
castration resistant prostate cancer
- CYP17A1
cytochrome P45017A1, 17α-hydroxylase/17,20-lyase
- DHEA
epiandrosterone
- DHEA-S
dehydroepiandrosterone-SO4
- DHT
5alpha-dihydrotestosterone
- NAB
nitronaphthen-1-yl)aminobenzoates
- SAR
structure-activity relationships
Footnotes
See e-Component supporting information synthesis:
3-((4-nitronaphthalen-1-yl)amino)benzoic acid (1)
3-(naphthalen-1-ylamino)benzoic acid (2)
3-((4,5-dinitronaphthalen-1-yl)amino)benzoic acid (3)
3-((4-acetylnaphthalen-1-yl)amino)benzoic acid (4)
5-((4-acetylnaphthalen-1-yl)amino)isophthalic acid (5)
3-((4-cyanonaphthalen-1-yl)amino)benzoic acid (6)
3-((4-(trifluoromethyl)naphthalen-1-yl)amino)benzoic acid (7)
3-((4-fluoronaphthalen-1-yl)amino)benzoic acid (8)
3-(quinolin-4-ylamino)benzoic acid (9)
4-(naphthalen-1-ylamino)benzoic acid (10)
4-((4-acetylnaphthalen-1-yl)amino)benzoic acid (11)
2-(naphthalen-1-ylamino)benzoic acid (12)
4-methoxy-2-(naphthalen-1-ylamino)benzoic acid (13)
5-acetyl-2-(naphthalen-1-ylamino)benzoic acid (14)
5-acetyl-2-((4-nitronaphthalen-1-yl)amino)benzoic acid (15)
5,5-dimethyl-3-(4-nitronaphthalen-1-yl)imidazolidine-2,4-dione (16)
5,5-dimethyl-3-(4-nitronaphthalen-1-yl)-2-thioxoimidazolidin-4-one (17)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- 1.Huggins CB, and Hodges CV, Studies on prostatic cancer 1. Effect of castration, estrogen and androgen injection on serum phosphatases in metatstatic carcinoma of the prostate. Cancer Res., 1941. 1: p. 293–397. [Google Scholar]
- 2.Huggins CB, Two principles in endocrine therapy of cancers: Hormone deprival and hormone interference. Cancer Res., 1965. 25: p. 1163–67. [PubMed] [Google Scholar]
- 3.Sharifi R, Bruskewitz RC, Gittleman MC, Graham SD Jr., Hudson PB and Stein B, Leuprolide acetate 22.5 mg 12-week depot formulation in the treatment of patients with advanced prostate cancer. Clin. Ther, 1996. 18(4): p. 647–57. [DOI] [PubMed] [Google Scholar]
- 4.SEER, Surveilance, Epidemiology, and End Results Program. 2014: http://seer.cancer.gov/statfacts/html/prost.html.
- 5.Knudsen K, and Scher HI, Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res, 2009. 15: p. 4792–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knudsen K, and Penning TM, Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab, 2010. 21: p. 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martinins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, and de Bono JS, Phase 1 clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol, 2008. 28: p. 4563–4571. [DOI] [PubMed] [Google Scholar]
- 8.Attard G, Reid AH, A'Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, and de Bono JS, Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J. Clin. Oncol, 2009. 27: p. 3742–37482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, Taplin ME, Bubley GJ, Kheoh T, Haqq C, Molina A, Anand A, Koscuiszka M, Larson SM, Schwartz LH, Fleisher M and Scher HI, Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J. Clin. Oncol, 2010. 28(9): p. 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, and Sawyers CL Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium., Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet, 2010. 375: p. 1437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, and Sawyers CL, Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science, 2009. 324: p. 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS and Montgomery RB, Resistance to CYP17A1 inhibition with abiraterone in castrate-resistance prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res, 2011. 17: p. 5913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penning TM, Mechanisms of drug resistance that target the androgen axis in castration resistant prostate cancer (CRPC). J. Steroid Biochem. & Mol. Biol, 2015. 153: p. 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu C, A.C., Lou W, Lombard A, Evans CP, Gao AC, Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther, 2017. 16: p. 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, L.W., Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC, Intracrine androgens and AKR1C3 activation confer resistance to Enzalutamide in prostate cancer. Cancer Res., 2015. 75: p. 1413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byrns MC, Mindnich R, Duan L, and Penning TM, Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5α-reductase inhibitor finasteride. J. Steroid Biochem. Mol. Biol. , 2012. 130: p. 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penning TM, Burczynski ME, Jez JM, Hung C-F, Lin H-K, Ma H Moore M, Palackal N, and Ratnam K, Human 3a-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J, 2000. 351: p. 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin H-K, Jez JM, Schlegel BP, Peehl DM, Pachter JA and Penning TM, Expression and characterization of recombinant type 2 3a-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3a 17b-HSD activity and cellular distribution. Mol. Endocrinol, 1997. 11: p. 1971–1984. [DOI] [PubMed] [Google Scholar]
- 19.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ and Sharifi N, Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA, 2011. 108: p. 13728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Auchus RJ, The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab, 2004. 15: p. 432–8. [DOI] [PubMed] [Google Scholar]
- 21.Adeniji A, Uddin MJ, Zang T, Tamae D, Wangtrakuldee P, Marnett LJ, and Penning TM, Discovery of (R)-2-(6-Methoxynaphthalen-2-yl)butanoic Acid as a Potent and Selective Aldo-keto Reductase 1C3 Inhibitor. J. Med. Chem, 2016. 59: p. 7431–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, and Penning TM, Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3 ). Bioorg. Med. Chem. Lett, 2011. 21: p. 1464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, and Penning TM, Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J. Med. Chem. , 2012. 55: p. 2311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kikuchi A, Furutani T, Azami H, Watanbe K, Nimi T, Kamiyama Y, Kuromitsu S, Bakin-Bey E, Heeringa M, Uoatas T, and Enjo K, In vitro and in vivo characterization of ASP9521: a novel selective, orally bioavailable inhibitor of 17b-hydroxysteroid dehydrogenase type 5 (17b-HSD5; AKR1C3). Invest. New, Drugs, 2014. 32: p. 860–870. [DOI] [PubMed] [Google Scholar]
- 25.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ and Penning TM, Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (type 5 17b-hydroxysteroid dehydrogenase prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem, 2013. 56(6): p. 2429–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yepuru M, Wu Z, Kyulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT and Narayanan R, Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin. Cancer Res, 2013. 19: p. 5613–25. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, and Penning TM, Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg. Med. Chem. Lett. , 2012. 22: p. 3492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalton JT, Miller DD, Narayanan R, Yepuru M, Coss CC, Mohler ML, and Wu Z, Preparation of isoquinoline compounds as aldo-keto reductase subfamily 1C3 (AKR1C3) inhibitors for treating cancer and other diseases, World Intellectual Property Organization, 2013: Intermational Patent Number WO 2013/142390 A1 2013096
- 29.Watanabe K, Kakefuda A, Yasuda M, Amano Y, Enjo E, Kikuchi A, Furutani T, Naritomi Y, Otsuka Y, Okada M, Ohta M, Discovery of ASP9521, a novel, potent, selective 17β-HSD5 inhibitor. Abstract In 246th American Chemical Society meeting 2014: Indianapolis, Indiana, USA. [Google Scholar]
- 30.Trott O, and Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem, 2010. 31: p. 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burczynski ME, Harvey RG and Penning TM, Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry, 1998. 37: p. 6781–6790. [DOI] [PubMed] [Google Scholar]
- 32.Cherian MT, Wilson EM, Shapiro DJ, A competitive inhibitor that reduces recruitment of androgen receptor to androgen-responsive genes. J. Biol. Chem, 2012. 287: p. 23368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizner T, Lin H-K, Peehl DM, Steckelbroeck S, Bauman DR and Penning TM, Human type 3 3a-hydroxysteroid dehydrogenase (AKR1C2) and androgen metabolism in prostate cells. Endocrinology, 2003. 144: p. 2922–2932. [DOI] [PubMed] [Google Scholar]
- 34.Loriot Y, Fizazi K, Jones RJ, Brand, Van den J, Molife RL, Omlin A, James ND, Baskin-Bey E, Heeringa M, Baron B, Holtkamp GM, Ouatas T, and de Bono JS, Safety, tolerability and anti-tumor activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs, 2014. 32: p. 995–1004. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ and Sharifi N, Conversion of abiraterone to D4A drives antitumour activity in prostate cancer. Nature, 2015. 523: p. 347–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kikuchi A, Enjo K, Furutani T, Azami H, Nimi T, Kamiyama Y, and Kuromitsu S, ASP9521, a novel, selective, orally bioavailable AKR1C3 (type 5 17b-hydroxysteroid dehydrogenase) inhibitor: In vitro and in vivo characterization. ASCO Annual Meeting, 2013. Abstract 5046 [DOI] [PubMed] [Google Scholar]
- 37.Bohl C, Gao W, Miller DD, Bell CE, and Dalton JT, Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA, 2005. 102: p. 6201–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bohl CE, Miller DD, Chen Y, Bell CE, and Dalton JT, Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem, 2005. 280: p. 37747–37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1S. Gridbox parameters for docking experiments