

Abstract
Hybrid type I PKS/NRPS biosynthetic pathways typically proceed in a collinear manner wherein one molecular building block is enzymatically incorporated in a sequence that corresponds with gene arrangement. Here, genome mining combined with use of a fluorogenic azide-based click probe led to the discovery and characterization of vatiamides A-F, three structurally diverse alkynylated lipopeptides and their brominated analogs, from the cyanobacterium Moorea producens ASI16Jul14-2. These derive from a unique combinatorial non-collinear PKS/NRPS system encoded by a 90 kb gene cluster in which an upstream PKS cassette interacts with three separate cognate NRPS partners. This is facilitated by a series of promiscuous inter-module PKS-NRPS docking motifs possessing identical amino acid sequences. This interaction confers a new type of combinatorial capacity for creating molecular diversity in microbial systems.
Keywords: Natural products, Combinatorial chemistry, Cyanobacteria, Click chemistry, Assembly-line biosynthesis
Graphical Abstract

Combination dock:
a unique natural example of polyketide synthase/non-ribosomal peptide synthetase combinatorial biosynthesis is reported in the discovery of the vatiamides from the cyanobacterium Moorea producens. The molecules are produced by a PKS/NRPS system encoded by a 90 kb gene cluster, whereby encoded enzymes interact via promiscuous docking domains with identical sequence to produce three distinct natural product series.
Numerous microbial species contain polyketide synthase/non ribosomal peptide synthetase (PKS/NRPS) biosynthetic gene clusters which produce diverse modified lipopeptide natural products (NP).[1] In cyanobacteria, the study of saxitoxin, cylindrospermopsin, curacin A, and apratoxin A pathways, among others, has provided much knowledge of protein-protein interaction, novel gene function, and loading module diversity in NP biosynthesis.[2–8] In typical PKS/NRPS systems, activated acyl-CoAs and amino acids are sequentially coupled by individual PKS or NRPS modules. This occurs collinearly such that the order in which their genes are transcribed is coincident with the functioning of proteins that selectively interact to elongate NPs with high fidelity. The interface between modules occurs by interacting type I or II docking domains (dd) between PKSs and communication (COM) domains between some NRPS modules.[9] A third type of structurally distinct docking interaction between combinations of PKS and/or NRPS modules was characterized in tubulysin and rhabdopeptide/xenorpeptide (RXP) biosynthesis, referred to here as (C-terminus-N-terminus) β–αββαα type domains.[10–12] We used a bioactivity and reactivity-guided approach to characterize six new lipopeptides and their biosynthetic pathway from the benthic cyanobacterium Moorea producens ASI6Jul14-2. This pathway features a unique nonselective combinatorial dd interaction that is antithetical to collinearity and represents a significant new mechanism by which prokaryotes may generate NP diversity.
An environmentally-derived culture designated ASI16Jul14-2 “ASI” from Vatia Bay, American Samoa, yielded a single non-axenic filamentous cyanobacterium after successive propagation and isolation of individual filaments in seawater BG-11 media. By 16S rRNA sequence comparison and morphology characteristics, it was identified as Moorea producens (Figure S1, Table S12).[13] Brine shrimp toxicity and NCI-H460 human lung cancer bioassay of fractionated extracts (Figure S2) from cultures of ASI led to the discovery and structure characterization of major compounds 1-4 by NMR and MS-based analysis methods (Figure 1, Table S2–S6, Figures S5–S25). Each molecule features an identical fatty-acid tail that possesses a terminal alkyne or bromoalkyne, a secondary methyl group and a vinyl chloride moiety 7/8, identical to that found in the jamaicamides 9/10.[14] This ‘tail’ is extended with an N,O,O-trimethyl-l-tyrosine in 1/2 whereas in 3/4, it is appended by a glycine-derived amide and an extended bicyclic C10 polyketide that contains a hemiketal ring, secondary methyl group, methoxy group, and a terminating unsaturated δ-lactone. Amino acid chirality of compound 1 was determined to be l (S) by Marfey’s analysis compared to an FDAA-derived standard of N,O,O-Me-l-tyrosine (Figure S35). In compound 3, stereocenters C16, C17, C18, C20 and C22 were assigned as S*,S*,R*,S*,R*. 1H-NMR J-coupling values were key to assigning the relative configuration of the lactol ring. NOESY correlations from the lactol ring to the lactone, as well as molecular modeling, were used to infer the relative configuration at C22 (Figures S38–S40). The configuration of the C9 branching methyl group was predicted by sequence analysis of the VatK KR domain. This motif possessed a LDD loop, although it was modified to LSD in VatK, and the catalytic region site 5 lacked a P residue. Thus, the VatK KR was determined to be a “B1”-type (Figure S37);[15] therefore, C9 was assigned to be R in 1-6. Compound 2 was established by HR-ESI-MS and MS2-based comparison to 1 (Figure S11), while 4 was established using a combination of MS and 1H and 13C NMR data in comparison to 3. (Table S4, Figures S23–S25).
Figure 1.
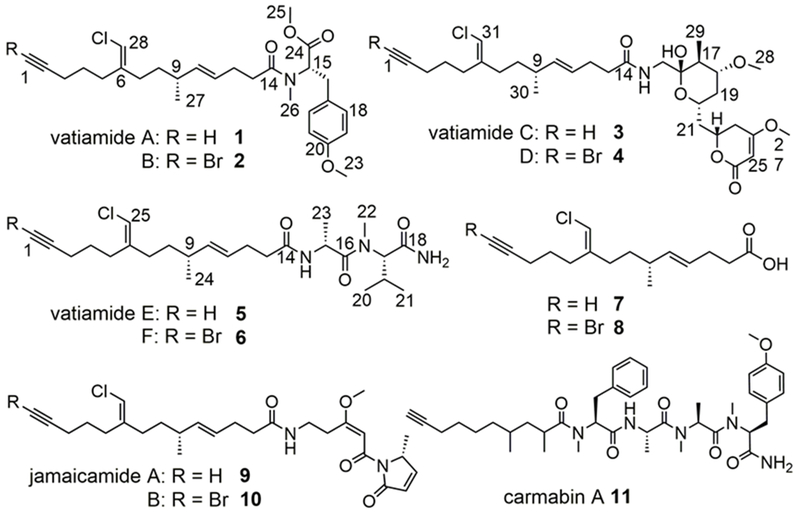
Vatiamides (1-6) from Moorea producens ASI16Jul14-2 “ASI” isolated in this study, jamaicamides (9,10) from Moorea producens JHB, and carmabin (11) from Moorea producens 3L. Compound 7 is produced by the organism in small quantity (Figure 4, S63). Free compound 8 was not observed.
Bioinformatic analysis of the ASI genome derived from short-read sequence data enabled assignment of the biosynthesis of 1/2, but not 3/4 due to the fragmented nature of the assembly. The 1/2 biosynthetic pathway was evident in a 50 kb PKS/NRPS BGC vatA-M which contained the biosynthetic machinery homologous to that of the chloro fatty-acid moiety of 9/10, but diverging at VatN, an NRPS module with an adenylation (A) domain predicted to activate a tyrosine residue. This is followed by an in-module N-methyltransferase (MT) and O-MT, peptidyl carrier protein (PCP), thioesterase (TE), and standalone O-MT enzyme, VatO, in accordance with the structure of 1/2. However, no biosynthetic genes in an orientation or domain structure matching that of 3/4 were found. Nevertheless, a second sequencing and assembly effort combining long and short reads illuminated the complete 90 kb vat pathway, with the original 50 kb pathway extended by 40 kb of NRPS and PKS modules immediately downstream of VatO (Figure 2, Table S11). VatP, a standalone ACP with high homology to the ACP of VatM, may play a role in transfer of 7/8 to subsequent NRPS modules. VatQ is an NRPS module predicted to incorporate Ala, and contains an epimerase domain (E). Module VatR is an NRPS domain with A(Val), N-MT, and PCP motifs, and a terminal domain bearing homology to an NADPH-binding motif and putative amidotransferase of uncharacterized function. COM dds mediate interaction of VatQ/VatR. The VatS NRPS module contains an A(Gly), which is then followed by five PKS modules, VatT-W, and contains reduction and methylation domains required to biosynthesize 3/4. Importantly, VatT contains an inactive KR0 by active site prediction which likely enables spontaneous heterocyclization of the C20 hydroxy group with the C16 VatS glycine-derived carbonyl, forming the lactol. A thioesterase (TE) embedded in VatW is believed to participate in an intramolecular cyclization to create the lactone observed in 3/4 by facilitating esterase activity on the terminal thioester C26. However, an additional uncharacterized gene, Orf1, possesses homology to various hydrolases including cholylglycine hydrolase, penicillin V acylase, and C-N amide hydrolase, all of which catalyze hydrolytic attack of a carbonyl; thus, Orf1 may be responsible for one or more of the heterocyclization events. Finally, the O-MT VatX is predicted to methylate the enol functionality on the terminal δ-lactone. Owing to the exactly synchronous domain-to-structure accord between predicted and observed functionalities, we propose a non-collinear transfer of 7/8 from VatM to VatS.
Figure 2.
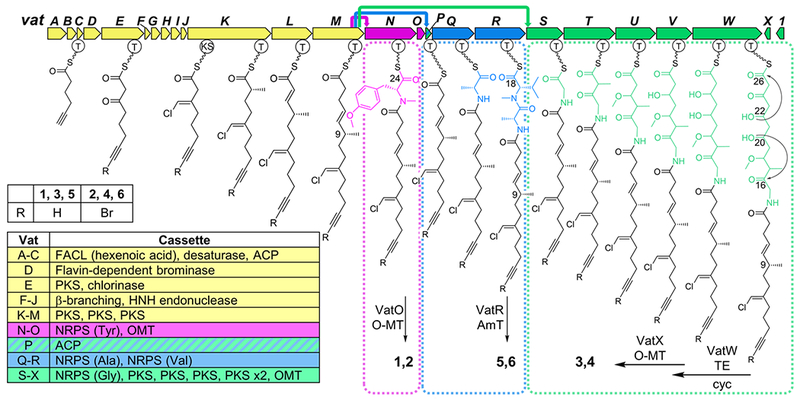
Vatiamide biosynthetic pathway vat in Moorea producens ASI16Jul14-2; biosynthetic genes for 7/8 in yellow. Three VatM/VatP cognate NRPS module partners and their subsequent PKS or NRPS cassettes highlighted as follows: pink 1/2, green 3/4, and blue 5/6, with colored arrows at top indicating transmittal of 7/8 to VatN, VatP, and VatS, respectively. ACP; acyl carrier protein, AmT; amidotransferase, FACL; fatty-acid CoA ligase, OMT; O-methyltransferase, T; thiolase, TE; thioesterase (Table S11).
An alignment of the intermodule docking motifs revealed that the first 45 amino acids of the N-terminal NRPS docking motif (Ndd) on VatN, VatQ, and VatS were 100% identical, and that the final six residues of the C-terminal dd (Cdd) on VatM and VatP were identical as well. Sequence alignment of these dds matches previously structurally characterized β–αββαα type dds.[10,11] Generally, as residues 24-29 on the β2 sheet of the N-terminal NRPS dd confer interaction with the terminal 5-6 residues of the “β3” sheet of the upstream C-terminal dd, we theorized that there was no selective preference by the upstream C-terminal VatM/P dds to downstream N-terminal VatN/Q/S dds. Thus, VatM or VatP thioester-bound 7/8 may interact with either the VatN, VatQ, or VatS Ndd stochastically. This was validated by the observed product composition wherein 7/8 appear in 1-4 but the ‘head’ group of 1/2 (VatM → VatN transfer) differs from that of 3/4 (VatM → VatS transfer).
However, a conundrum resulted from the lack of an observed product from vatQ and vatR. To sensitively probe for these predicted products, we employed a reactivity-guided approach.[16,17] We synthesized fluorogenic “click”-based probe 14, which undergoes a copper-catalyzed azide-alkyne cycloaddition (CuAAC) with terminal alkynes (Figure 3A, Scheme S1–S2, Table S9, Figures S43–S50).[18–20] Incorporation of a bromine atom on the coumarin ring endows the probe with a characteristic isotopic pattern, aiding in identification of tagged compounds during LC/MS analysis of probed extracts.[21] In addition, the probe exhibits “turn-on” fluorescence at 490 nm upon formation of a triazole adduct, enabling benchtop screening for terminal alkyne-bearing NPs in crude extracts. Proof of concept was demonstrated by a model click reaction with alkyne propargyl benzoate at 90% yield, and subsequently reaction of 14 with purified 1. (Figure 3B–3C, Scheme S3, Table S10, Figures S51–S60). Probing the extracts of the Moorea producens strains ASI, JHB (9-10 producer),[14] 3L (11 producer),[22,23] PAL15Aug08-1 (no known alkynes), and Moorea bouillonii PNG5-198 (no known alkynes) clearly indicated the presence of alkynes in ASI, JHB, and 3L but not PNG5-198 or PAL15Aug08-1, which showed near baseline fluorescence in comparison to the alkyne producers (Figure 3D).
Figure 3.
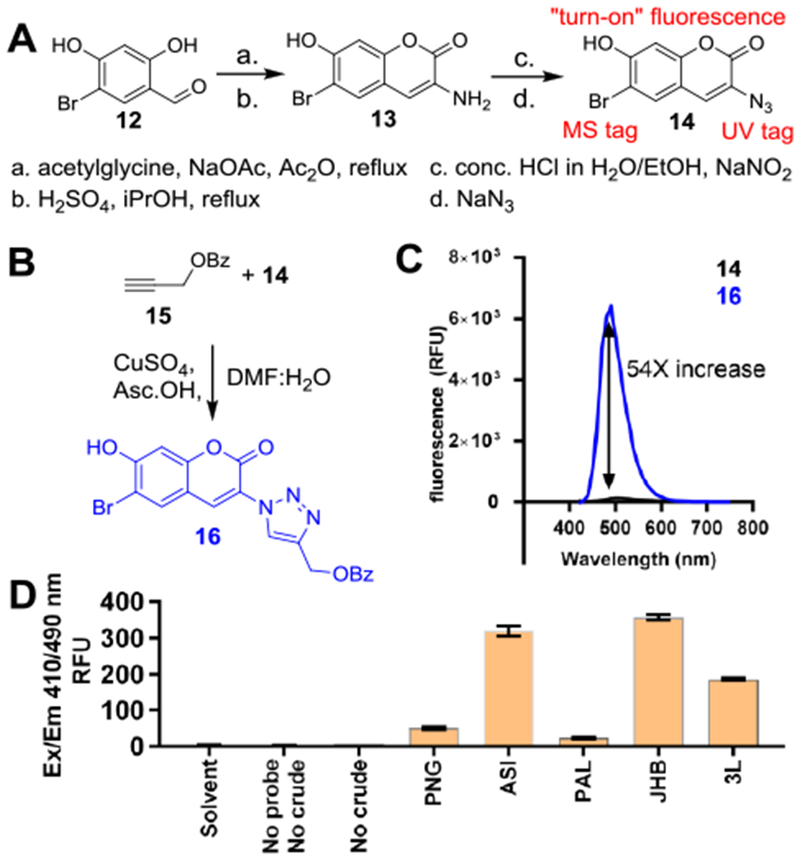
Synthesis and testing of bromoazidocoumarin click probe. A) Synthetic route of 14 from 12. B) Synthesis of propargyl benzoate-14 adduct. Asc.OH; ascorbic acid. C) Fluorescence increase upon formation of 16 from 15+14. D) Screening Moorea extracts for alkynes with probe 14.
Analysis of crude ASI-14 reaction mixture by HPLC-UV-HRESI-MS/MS showed six major peaks by UV. By HRESIMS, these peaks consisted of 13, 14, and adducts 1+14, 3+14, 7+14, and lastly an unidentified sixth peak (5+14) with an [M+H]+ = 747, or 465 amu when probe mass of 281 was subtracted (Figure 4, Figures S61–S63). Re-examination of unreacted ASI extract fractions revealed a minor peak with [M+H]+ of 544 by LC/MS, suggesting a potential brominated analog of the 465 amu compound. This bromoalkyne was not evident in initial crude MS/MS runs due to its low abundance and poor ionization compared to 1-4. Extraction of more biomass yielded sufficient material to characterize the [M+H]+ 544 compound (6), but only trace quantities of the 465 amu compound (5) were observed. 1D and 2D NMR revealed that partial structure 8 was linked via an amide to an alanyl residue followed by an N-methyl-valinamide residue, defining compound 6. By HRESIMS and MS2-based comparison, the non-brominated compound 5 was also observed, reconciling the function of VatQ-VatR (Figures S26–S31, S34). Analysis of 6 in DMSO-d6 indicated it was present in a 2:1 conformer ratio, likely due to hindered rotation about the C22 N-Me amide. It also showed split resonances for the terminal amide protons likely due to H-bonding. The conformer ratio was reduced from 2:1 to 8:1 by 1H-NMR analysis in d3-acetonitrile, while elevated temperature 1H-NMR (80°C) resolved the split NH2 resonance and conformers into single peaks (Figures S32–33). Marfey’s analysis of 6 indicated d-alanine and l-N-Me-valinamide, thus defining stereocenters 15R and 17S (Figure S36). We theorize that the terminal amide is formed by the undescribed domain embedded in VatR, downstream of the PCP. This domain bears homology to other amine-functionalizing enzymes, and is also found at the C-terminus of the carmabin (11) biosynthetic pathway in Moorea producens 3L; however, its enzymology and precise mechanism are unknown.[23,24]
Figure 4.

HPLC-UV of ASI crude extract with probe 14 added (blue) and without probe 14 (black).
The combinatorial biosynthesis of 1-6 is a unique and heretofore new method of generating chemodiversity with type I PKS/NRPS systems, and reveals that nature has selected for true combinatorial biosynthesis (Figure 5, S41). We postulate that this interaction is mediated by VatN, VatQ, and VatS N-terminal NRPS dds with identical sequence, enabling non-selective interaction with VatM and/or VatP ACP Cdd (Figure S64A). We do not anticipate module skipping to play a role, as VatO and VatR, the respective termini for the cassettes which generate 1/2 and 5/6, do not contain C-terminal dds and have fully intact active sites. Therefore, the VatM-thioester bound 7/8 may be transferred directly to either VatN, VatQ, or VatS, effectively forming three separate assembly lines upon translation and tertiary folding (Figure S64B). Alternatively, it is possible that the small standalone VatP plays an intermediary role in transfer of 7/8 to VatQ or VatS (Figure S64C). The GxDS phosphopantetheine (ppt) binding site of VatP ACP is found at the N-terminus of the protein; thus it is plausible that the VatP ppt arm could attack the VatM-thioester without an acyltransferase.
Figure 5.

Vatiamide pathway promiscuous dd interactions. A) VatM and VatP Cdd alignment. B) VatN/Q/S Ndd aligned with characterized dds. Key interaction points in red triangles. C) Depiction of “docking”; electrostatic and steric interactions of residues in blue dots.[10,11] D) Vat pathway promiscuity: T; thiolase, Cond; condensation domain.
There is evidence in multiple assembly-line biosynthetic pathways that significant evolutionary pressure has been placed on eliminating promiscuity, ensuring that biosynthetic modules interact with their cognate dd partner through unique dd sequences.[1,5] A BLAST search of the MIBiG pathway database using the VatM/P Cdd and VatN/S/R Ndd revealed several pathways with at least two β–αββαα dds between separate modules, yet no pathway features 100% homologous dds. This includes proteins other than PKSs and NRPSs, such as a standalone halogenase AerJ in aeruginosin biosynthesis which possesses a β–αββαα type docking pair at both its C and N terminus (Figure S42).
Still, examples of stuttering,[25] halting,[26] out-of-order module processing,[27] skipping,[28–32] bidirectionality,[33] and combinations thereof[26] have been reported in various NP biosynthetic pathways. We report here the first characterized example to date of a native reversal of selectivity in a PKS/NRPS context. The promiscuity enabled by identical dds infuses a new element into the combinatorial possibilities of both heterologous and native pathways. One may speculate that the origin in ASI is synergistic defensive toxicity imparted by the combination of more than one of the three head groups, or separate environmental targets for each. However we have only observed modest H-460 human lung cancer cytotoxicity with both pure compounds and combinations thereof, with the brominated analogs 2 and 4 being slightly more cytotoxic than the non-brominated versions 1 and 3 (Figures S3–S4).
Additional questions arise in understanding how such a protein structure is assembled over evolutionary time: either the docking motif was replicated and inserted in front of three pre-existing NRPS modules, or a single NRPS module with this dd was duplicated twice and each evolved different amino acid specificities followed by further tailoring divergence. The high homology between the VatM ACP and VatP suggests that duplication created VatP. Discovery of this full pathway only occurred after employing long-read and short-read sequencing with hybrid assembly. Assembler graphs may not be able to reconcile three nearly identical 150-bp sequences derived from short reads in a microbial genome without breaking continuity and generating “orphan” clusters. Therefore, we predict dd-mediated combinatorial biosynthesis may be more widespread than currently recognized and may be present in numerous cyanobacterial and bacterial clades.
Experimental Section
For experimental details, supplementary tables, schemes, and figures including biosynthetic gene cluster proteins, spectra and molecule characterization, see supporting information.
vat pathway Accession Number: MK618714
Supplementary Material
Acknowledgements
We thank J. G. Sanders, G. Humphrey, J. Gaffney, R. A. Salido Benitez, J. J. Minich, C. Brennan, K. Sanders, and R. Knight for their expertise in library preparation and genome sequencing. We thank E. Glukhov, P. Kanjanakantorn, M. Kissick, B. Ni, and A. M. Hoskins for their contributions to cyanobacterial culturing, bioassay and extraction, and B. Miller for harvesting and collection. We acknowledge Y. Su, L. Gross, A. Mrse and B. Duggan for analytical expertise. We thank M. Pierce and T. Murray for bioassay testing. This work was supported by NIH GM107550, NIH GM118815 to LG and WHG. NAM was supported by NIH Training Grant in Marine Biotechnology and Microbiome and Microbial Sciences Initiative Graduate Student Fellowship.
Contributor Information
Nathan A. Moss, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA)
Grant Seiler, Department of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA).
Tiago F. Leão, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA)
Gabriel Castro-Falcón, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA).
Lena Gerwick, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA).
Chambers C. Hughes, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA)
William H. Gerwick, Center for Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA); Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA).
References
- [1].Fischbach MA, Walsh CT, Chem. Rev 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- [2].Kellmann R, Mihali TK, Young JJ, Pickford R, Pomati F, Neilan BA, Appl. Environ. Microbiol 2008, 74, 4044–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Muenchhoff J, Siddiqui KS, Poljak A, Raftery MJ, Barrow KD, Neilan BA, FEBS J 2010, 277, 3844–3860. [DOI] [PubMed] [Google Scholar]
- [4].Mihali TK, Kellmann R, Muenchhoff J, Barrow KD, Neilan BA, Appl. Environ. Microbiol 2008, 74, 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH, Smith JL, Chem. Biol 2013, 20, 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH, J. Nat. Prod 2004, 67, 1356–67. [DOI] [PubMed] [Google Scholar]
- [7].Grindberg RV, Ishoey T, Brinza D, Esquenazi E, Coates RC, Liu W, Gerwick L, Dorrestein PC, Pevzner P, Lasken R, et al. , PLoS One 2011, 6, e18565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Skiba MA, Sikkema AP, Moss NA, Lowell AN, Su M, Sturgis RM, Gerwick L, Gerwick WH, Sherman DH, Smith JL, ACS Chem. Biol 2018, 13, 1640–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miyanaga A, Kudo F, Eguchi T, Nat. Prod. Rep 2018, 35, 1185–1209. [DOI] [PubMed] [Google Scholar]
- [10].Hacker C, Cai X, Kegler C, Zhao L, Weickhmann AK, Wurm JP, Bode HB, Wöhnert J, Nat. Commun 2018, 9, 4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Richter CD, Nietlispach D, Broadhurst RW, Weissman KJ, Nat. Chem. Biol 2008, 4, 75–81. [DOI] [PubMed] [Google Scholar]
- [12].O’Connor SE, Walsh CT, Liu F, Angew. Chemie - Int. Ed 2003, 42, 3917–3921. [DOI] [PubMed] [Google Scholar]
- [13].Engene N, Rottacker EC, Kaštovský J, Byrum T, Choi H, Ellisman MH, Komárek J, Gerwick WH, Int. J. Syst. Evol. Microbiol 2012, 62, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH, Chem. Biol 2004, 11, 817–833. [DOI] [PubMed] [Google Scholar]
- [15].Keatinge-Clay AT, Chem. Biol 2007, 14, 898–908. [DOI] [PubMed] [Google Scholar]
- [16].Castro-Falcón G, Millán-Aguiñaga N, Roullier C, Jensen PR, Hughes CC, ACS Chem. Biol 2018, 13, 3097–3106. [DOI] [PubMed] [Google Scholar]
- [17].Castro-Falcón G, Hahn D, Reimer D, Hughes CC, ACS Chem. Biol 2016, 11, 2328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sivakumar K, Xie F, Cash BM, Long S, Barnhill HN, Wang Q, Org. Lett 2004, 6, 4603–4606. [DOI] [PubMed] [Google Scholar]
- [19].Ross C, Scherlach K, Kloss F, Hertweck C, Angew. Chemie - Int. Ed 2014, 53, 7794–7798. [DOI] [PubMed] [Google Scholar]
- [20].Zhu X, Shieh P, Su M, Bertozzi CR, Zhang W, Chem. Commun 2016, 52, 11239–11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang L, Chumsae C, Kaplan JB, Moulton KR, Wang D, Lee DH, Zhou ZS, Bioconjug. Chem 2017, 28, 2302–2309. [DOI] [PubMed] [Google Scholar]
- [22].Hooper GJ, Orjala J, Schatzman RC, Gerwick WH, J. Nat. Prod 1998, 61, 529–533. [DOI] [PubMed] [Google Scholar]
- [23].Jones AC, Monroe EA, Podell S, Hess WR, Klages S, Esquenazi E, Niessen S, Hoover H, Rothmann M, Lasken RS, et al. , Proc. Natl. Acad. Sci 2011, 108, 8815–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Leao T, Castelão G, Korobeynikov A, Monroe EA, Podell S, Glukhov E, Allen EE, Gerwick WH, Gerwick L, Proc. Natl. Acad. Sci 2017, 114, 3198–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Olano C, Wilkinson B, Sánchez C, Moss SJ, Sheridan R, Math V, Weston AJ, Braña AF, Martin CJ, Oliynyk M, et al. , Chem. Biol 2004, 11, 87–97. [DOI] [PubMed] [Google Scholar]
- [26].Ross AC, Xu Y, Lu L, Kersten RD, Shao Z, Al-Suwailem AM, Dorrestein PC, Qian P, Moore BS, J. Am. Chem. Soc 2013, 135, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM, Chem. Biol 2003, 10, 419–430. [DOI] [PubMed] [Google Scholar]
- [28].Wenzel SC, Kunze B, Höfle G, Silakowski B, Scharfe M, Blöcker H, Müller R, ChemBioChem 2005, 6, 375–385. [DOI] [PubMed] [Google Scholar]
- [29].Ninomiya A, Katsuyama Y, Kuranaga T, Miyazaki M, Nogi Y, Okada S, Wakimoto T, Ohnishi Y, Matsunaga S, Takada K, ChemBioChem 2016, 17, 1709–1712. [DOI] [PubMed] [Google Scholar]
- [30].Beck BJ, Yoon YJ, Reynolds KA, Sherman DH, Chem. Biol 2002, 9, 575–583. [DOI] [PubMed] [Google Scholar]
- [31].Li Z-R, Li J, Gu J-P, Lai JYH, Duggan BM, Zhang W-P, Li Z-L, Li Y-X, Tong R-B, Xu Y, et al. , Nat. Chem. Biol 2016, 12, 773–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].He H-Y, Pan H-X, Wu L-F, Zhang B-B, Chai H-B, Liu W, Tang G-L, Chem. Biol 2012, 19, 1313–1323. [DOI] [PubMed] [Google Scholar]
- [33].Li S, Wu X, Zhang L, Shen Y, Du L, Org. Lett 2017, 19, 5010–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.