

Abstract
Background
This study aims to provide genetic diagnoses for 30 cases of fetal skeletal dysplasia, and a molecular basis for the future prenatal diagnosis of fetal skeletal dysplasia.
Methods
A total of 30 cases of fetal skeletal dysplasia detected with ultrasound between January 2014 and June 2017 were analyzed. Among these fetuses, 15 fetuses had local skeletal malformations, while 15 fetuses had short limb malformations. Samples of fetal umbilical cord blood, amniotic fluid, and/or aborted tissue were collected from all cases. Karyotyping, whole genome sequencing, and targeted next-generation sequencing of skeletal disease-related pathogenic genes were performed, as needed. Blood samples were taken from the parents for verification using Sanger sequencing.
Results
Among the 30 cases of fetal skeletal dysplasia, two cases were diagnosed with trisomy 18. However, none of these cases were identified with any microdeletions or microreplications associated with skeletal dysplasia. Among the 28 chromosomally normal cases with fetal skeletal dysplasia, 21 cases were detected with mutations in genes related to skeletal diseases. Furthermore, collagen gene mutations were detected in six fetuses with short limb malformations, while heterozygous disease-causing mutations in the fibroblast growth factor receptor 3 (FGFR3) gene were detected in seven fetuses. The remaining fetuses carried mutations in other various genes, including tumor protein p63 (TP63), cholestenol delta-isomerase (EBP), cholinergic receptor nicotinic gamma subunit (CHRNG), filamin B (FLNB), and SRY-box 9 (SOX9). Three compound heterozygous mutations in CHRNG, COL11A2 and SOX9 were carried by phenotypically healthy parents.
Conclusion
Targeted next-generation sequencing can significantly improve the prenatal diagnoses of fetal skeletal dysplasia, providing parents with more precision medicine, and improved genetic counseling.
Keywords: Fetal skeletal dysplasia, Chromosomes, Targeted next-generation sequencing, Whole genome sequencing, Prenatal diagnosis
Background
Fetal skeletal dysplasia is an osteochondroblastic disease that has strong clinical heterogeneity, affecting approximately 2.4–4.5 of 10,000 births [1–4]. Although fetal skeletal dysplasia is associated with few chromosomal abnormalities, this disease is mostly associated with mutations in genes that regulate bone formation [5, 6]. At present, the prenatal diagnosis of fetal skeletal dysplasia mostly relies on ultrasound, X-ray and magnetic resonance imaging [7–9]. In 40–49% of cases with fetal skeletal dysplasia, ultrasound cannot differentiate among the different types of skeletal dysplasia. Hence, this has been merely used to identify severe lethal skeletal dysplasia [10–12]. In the 2010 revision of the Nosology and Classification of Genetic Skeletal Disorders, 456 conditions were classified into 40 groups defined by molecular, biochemical and radiographic criteria [13]. Among these conditions, 316 conditions were associated with mutations in one or more of 226 different genes, providing a basis for the molecular genetic diagnosis of fetal skeletal dysplasia.
Most previous studies have focused on specific genetic diagnoses for dyschondroplasia, osteogenesis imperfecta, and simple limb deformities [2, 14, 15]. The present study analyzed 30 cases of fetal skeletal dysplasia. The aim of the present study was to increase the scope of prenatal diagnoses and improve the genetic counseling offered to parents. In addition, the present study aimed to provide a theoretical basis for the early implementation of birth defect intervention and reproductive risk assessment.
Methods
Demographic features of cases
The present study comprised of 30 cases of fetuses diagnosed with skeletal abnormalities via ultrasound at the Obstetrics and Gynecology Hospital of Capital Medical University, Beijing, China between January 2014 and June 2017. Among these 30 cases, 15 cases were fetal skeletal malformations, while 15 cases were systemic skeletal dysplasias, which were characterized as short limb deformities. For the diagnosis of short limb deformity, two criteria must be simultaneously satisfied. First, the long bones of the extremities (i.e. the femur and humerus) must be shorter than the 5th percentile for fetuses of the same gestational age. In addition, the femur length (FL) to abdominal circumference (AC) ratio must be ≤1.6. Second, there must be ultrasonographic manifestations of abnormal bone morphology, such as long bone bending, angulation, fractures, “telephone receiver-shaped” changes, thoracic dysplasia, or changes in bone mineral density [10]. The present study was approved by the ethics committee of our hospital, and all parents of the fetuses provided a signed informed consent prior to prenatal diagnosis and sample collection.
Sample collection
After routine disinfection, fetal umbilical cord blood puncture was performed, and 2 mL of blood was drawn from the umbilical vein into a vacuum blood collection tube containing ethylenediaminetetraacetic acid (EDTA). If no fetal blood sample could be obtained, two pieces of fetal muscle tissue (3 × 3 cm) containing the skin were removed after abortion. For all cases, 5 mL of venous blood was collected from both parents in vacuum blood collection tubes containing EDTA. All specimens were treated and stored at − 80 °C until use. Both the sources of the samples and methods of blood sampling were approved by the Institutional Ethics Committee of the Obstetrics and Gynecology Hospital of Capital Medical University. The informed consent forms were completed by the parents.
Detection of fetal chromosomal abnormalities
Fetal amniotic fluid or umbilical cord blood samples were taken for chromosome G band karyotype analysis.
Whole genome sequencing (WGS) to detect fetal microdeletions/microduplications
Umbilical cord blood punctures were performed on fetuses with abnormal skeletal abnormalities. Complete genomic DNA was extracted using a commercial DNA extraction kit (Puregene; Qiagen, Hilden, Germany) from either the umbilical cord blood, or muscle tissue samples. WGS with 20–30× coverage, combined with bioinformatics analysis, accurately localize microdeletions and microduplications ≥100 kb long. If ≥100 kb disease-related microdeletions are detected in fetal tissues, these results were verified in the parent samples using Sanger sequencing.
Detection of variants in known genes related to congenital skeletal anomalies
If neither chromosomal abnormalities, nor disease-related microdeletions/microduplications were detected, the protein-coding regions and adjacent regions of 30 bp of known genes related to congenital skeletal anomalies were deeply sequenced using the targeted gene sequencing method. A special capture array (BGI, China) was used to detect the variants of the 363 genes involved in congenital skeletal anomalies. The overall sequencing coverage of the target regions was 97.81% for 20× depth of coverage in each of the chromosomes. After filtration by SAMtools (version 1.4), the sequencing data were mapped to the human genome (NCBI37/hg19) using the Burrows Wheeler Aligner software, and single nucleotide variants were identified using the SOAPsnp software (version 2.0). Then, the biological information was compared to databases, including ExAC, dbSNP, HapMap, 1,000 Genomes Asian, ESP6500, Cosmic, and HGMD. All variants were classified according to American College of Medical Genetics and Genomics recommended standards. After matching the inherent patterns of the disease, the pathogenicity of these loci were further determined by assessing the clinical symptoms and genetic data.
Verification of gene mutations
If pathogenic mutations were detected in the fetal samples, these mutations were further verified in the parents by Sanger sequencing.
Results
Clinical features of cases
Among the 30 cases in the present study, 15 fetuses had local deformities (including varus deformities, finger/toe deformities, missing fingers/toes, and/or absence of upper/lower limbs), while 15 fetuses had systemic general skeletal dysplasia characterized by short limb deformities. All deformities were confirmed by postpartum clinical and pathological analysis (Table 1).
Table 1.
General data for 30 cases of fetal skeletal dysplasia, including chromosome information and microdeletion/microduplication test results
| No. | Ultrasound results | Gestation (weeks) | Chromosome | Micro-deletion/duplication results and significance |
|---|---|---|---|---|
| 1 | Right choroid plexus cyst and left foot inversion in the fetus | 21 | Trisomy 18 | |
| 2 | Absence of radius in the upper limbs of the fetus, abnormal posture of both hands | 20 | Trisomy 18 | |
| 3 | Continuously interrupted left upper lip, broken upper alveolar bone, possible small jaw, and absent fetal femur | 25 | 46,XN | No abnormalities |
| 4 | Spinal fissure (isopathology), gastroschisis, double lung dysplasia, bipedal varus, left foot polydactyly | 25 | 46,XN |
arr Xq26.2(133,527,188-133,533,879)×1 There is a 6.6 Kb deletion in the Xq26.2 segment of the fetal X chromosome. This fragment spans exon 4 and exon 5 of the PHF6 gene and does not correlate well with the patient’s clinical phenotype. |
| 5 | Bending bilateral femur, tibia and fibula, fixed knee, internal crossed flexion, and fixed foot position | 23 | 46,XN | No abnormalities |
| 6 | Cleft lip and palate, right kidney cystic dysplasia, bilateral foot fissure, and syndactyly, partially absent fingers of both hands | 25 | 46,XN | No abnormalities |
| 7 | Fetal bilateral femoral angulation deformity, fetal heart ventricular septal defect | 24 | 46,XN |
46,XN,dup(7q11.21)(64,635,655-64,947,696) × 3,46,XN,del(11p11.12)(49,009,009-49,120,197) × 1,46,XN,del(17p12)(14,099,119-15,464,828) × 1 No clear pathogenic >100Kb microdeletions/microreplications |
| 8 | Right foot inversion, bilateral rocker bottom feet, scoliosis, spina bifida occulta | 30 | 46,XN | No abnormalities |
| 9 | Fetal scoliosis, fetal bipedal varus, ventricular septal defect | 26 | 46,XN | No abnormalities |
| 10 | Absence of bilateral ulna and radius, absence of bilateral humerus, foot inversion, abnormal wrist joints | 24 | 46,XN |
arr 2q24.3(166,914,464-166,920,459)×1 There is a 5.9Kb deletion in chromosome 2q24.3 on chromosome 2, which is not associated with a clinical phenotype. |
| 11 | Absence of bilateral humerus and left foot | 26 | 46,XN | No abnormalities |
| 12 | Only two visible metacarpal bones on the left hand and part of distal phalanx on the lateral and medial sides | 26 | 46,XN | No abnormalities |
| 13 | Left foot inversion, absence of right lower limb | 26 | 46,XN,21cenh+ |
arr 5q35.1(170,405,440-171,071,061)×3 There is a repeat of 665 Kb fragment in the 5q35.1 segment of fetus chromosome 5, which includes 4 OMIM genes such as NPM1. The correlation with clinical phenotype is not high. |
| 14 | Absence of fetal fibula, foot inversion, partial absence of phalanges, absence of fingers, hand cleft deformity | 22 | 46,XN | No abnormalities |
| 15 | Fetal sirenomelia | 17 | 46,XN | No abnormalities |
| 16 | Short limbs - incomplete osteogenesis? | 23 | 46,XN | No abnormalities |
| 17 | Short limbs - incomplete osteogenesis | 22 | 46,XN | No abnormalities |
| 18 | Short limbs | 22 | 46,XN | No abnormalities |
| 19 | Short fetus limbs: cartilage hypoplasia? | 22 | 46,XN | No abnormalities |
| 20 | Short limbs, spine and vertebral ossification are not obvious: cartilage hypoplasia? | 16 | 46,XN | No abnormalities |
| 21 | Short limbs, ventricular septal defect | 15 | 46,XN | No abnormalities |
| 22 | Abnormal long bones in fetal limbs, narrow chest | 24 | 46,XN | No abnormalities |
| 23 | Short fetal limbs (chronic dysgenesis) | 25 | 46,XN | No abnormalities |
| 24 | Short limbs (cartilage hypoplasia) | 25 | 46,XN | arr 19p13.2(11,135,293-11,139,948)x, There is a deletion of 4.6 Kb fragment in the 19p13.2 segment of chromosome 19 of the fetus and there is no correlation with the patient’s clinical phenotype. |
| 25 | Short limbs | 21 | 46,XN | No abnormalities |
| 26 | Short limbs | 21 | 46,XN | No abnormalities |
| 27 | Short limbs | 27 | 46,XN | 46,XN,dup(2p11.2)(87,384,213-87,862,105) × 3, Polymorphism |
| 28 | Short limbs | 25 | 46,XN | No abnormalities |
| 29 | Achondroplasia | 15 | 46,XN | No abnormalities |
| 30 | Uneven arrangement of fetal spine, short limbs, bilateral foot inversion, small mandibular, left ventricular punctate strong echo, ventricular septal defect | 23 | 46,XN |
Both chromosomes 1 and 16 have microduplications. Dup(Xq27.1)(139,911,843-140,072,771) × (2~3) Polymorphism |
arr microarray, dup duplication, del deletion
Skeletal chromosomal abnormalities and microdeletions/microduplications
All 30 cases of fetal skeletal dysplasia were tested for chromosomal abnormalities. Trisomy 18 was detected in two cases of fetal local skeletal malformation (cases 1–2, Table 1). However, neither chromosomal abnormalities, nor pathological microdeletions/microduplications related to skeletal dysplasia were identified in the remaining 28 cases.
Sequencing and verification of hereditary bone disease in 13 cases of fetal local skeletal malformation
Among the 13 cases of fetal local skeletal malformation, in which no chromosomal abnormalities were identified, seven cases (cases 9–15) had no pathogenic mutations, while six cases (cases 3–8) had mutations in known genes associated with bone diseases (Table 1). In three of these cases (cases 3–5), the mutations were of unknown clinical significance, while in the remaining three cases (cases 6–8), the mutations were considered as causative.
Detection of hereditary bone disease mutations using targeted gene sequencing and validation using sanger sequencing in 15 cases of systemic skeletal dysplasia (short extremities)
Collagen gene mutations were detected in six cases (cases 16–21). The clinically unexplained mutation was only identified in case 20: c.2419G > A (p.Gly807Arg) in the COL2A1 gene. For the remaining cases, the mutations were known to cause the disease.
FGFR3 mutations were detected in seven cases diagnosed with short limbs (cases 23–29). Four of these cases (cases 23–26) carried the same mutation: c.742C > T (p.Arg248Cys) in the FGFR3 gene. Each of the remaining three cases carried a single unique mutation: c.1144G > A (p.Gly382Arg), c.1124A > G (p.Tyr375Cys), or c.2426G > C (p.X809S,101). Sanger sequencing confirmed that none of these mutations were carried by any parent.
In case 22, a heterozygous mutation in the emopamil binding protein (EBP) gene (NM_006579.2) was detected (C.440G > A, p.Arg147His). In case 30, a clinically significant heterozygous mutation in the filamin B (FLNB) gene was detected (c.475A > C, p.Thr159Pro). For both cases, the parents were not the carriers, suggesting that these were new fetal mutations. The mother of case 22 has subsequently given birth to a healthy baby boy (Table 2).
Table 2.
Identification of genes associated with skeletal diseases in 30 cases of fetal skeletal dysplasia
| No. | Ultrasound results | Inheritance | Bone gene-encoded | Reference sequence | Nucleotide change/mutation | Amino-acid change | Genetic subregion | Heterogeneity | Chromosomal Loci | Mutation type | Sanger verification | Next pregnancy/child | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Paternal | Maternal | ||||||||||||
| 1 | Right choroid plexus cyst and left foot inversion in the fetus | Trisomy 18 | Delivered a healthy child | ||||||||||
| 2 | Absence of radius in the upper limbs of the fetus, abnormal posture of both hands | Trisomy 18 | Delivered a healthy child | ||||||||||
| 3 | Continuously interrupted left upper lip, broken upper alveolar bone, possible small jaw, and absent fetal femur | AD | TBX4 | NM_018488.2 | c.1200G > T | p.Glu400Asp | EX8E | Het | Chr17:59560439 | VUS | N | Het | |
| 4 | Spinal fissure (isopathology), gastroschisis, double lung dysplasia, bipedal varus, left foot polydactyly | AD | TNNT3 | NM_006757.3 | c.88G > A | p.Ala30Thr | EX7 | Het | Chr 11:1950355 | VUS | Het | N | Delivered a healthy child |
| 5 | Bending bilateral femur, tibia and fibula, fixed knee, internal crossed flexion, and fixed foot position | AD | SOX9 | NM_000346.3 | c.344G > C | p.Trp115Ser | EX1 | Het | Chr17:70117876 | VUS | N | N | Delivered a healthy child |
| 6 | Cleft lip and palate, right kidney cystic dysplasia, bilateral foot fissure, and syndactyly, partially absent fingers of both hands | AD | TP63 | NM_003722.4 | c.952C > T | p.Arg318Cys | EX7 | Het | Chr3:189585691 | Pathogenic | N | N | |
| 7 | Fetal bilateral femoral angulation deformity, fetal heart ventricular septal defect | AR | POR | NM_000941.2 | c.1370G > A | p.Arg457His | EX12 | Het | Chr7: 75614497 | Pathogenic | N | Het | Angular femoral deformity found at the 15th week of pregnancy, pregnancy terminated |
| c.744C > G | p.Tyr248* | EX8 | Het | Chr7: 75611554 | Pathogenic | Het | N | ||||||
| 8 | Right foot inversion, bilateral rocker bottom feet, scoliosis, spina bifida occulta | AR | CHRNG | NM_005199 | C.13C > T | p.Q5X,513 | EX1 | Het | Chr2 | Pathogenic | N | Het | Same abnormality as the previous pregnancy, undergoing PGD |
| C.202C > T | p.R68X,450 | EX3 | Het | Chr2 | Pathogenic | Het | N | ||||||
| 9 | Fetal scoliosis, fetal bipedal varus, ventricular septal defect | No abnormalities | |||||||||||
| 10 | Absence of bilateral ulna and radius, absence of bilateral humerus, foot inversion, abnormal wrist joints | No abnormalities | |||||||||||
| 11 | Absence of bilateral humerus and left foot | No abnormalities | |||||||||||
| 12 | Only two visible metacarpal bones on the left hand and part of distal phalanx on the lateral and medial sides | No abnormalities | |||||||||||
| 13 | Left foot inversion, absence of right lower limb | No abnormalities | |||||||||||
| 14 | Absence of fetal fibula, foot inversion, partial absence of phalanges, absence of fingers, hand cleft deformity | No abnormalities | Delivered a healthy child | ||||||||||
| 15 | Fetal sirenomelia | No abnormalities | Delivered a healthy child | ||||||||||
| 16 | Short limbs - incomplete osteogenesis? | AD | COL1A1 | NM_000088.3 | c.1678G > A | p.Gly560Ser | EX25 | Het | Chr17: 48268739 | Pathogenic | N | N | |
| 17 | Short limbs - incomplete osteogenesis | AD | COL1A2 | NM_000089.3 | c.1774G > A | p.Gly592Ser | EX31 | Het | Chr7: 94045726 | Pathogenic | N | N | |
| 18 | Short limbs | AD | COL1A2 | NM_000089.3 | c.1072G > A | p.Gly358Ser | EX20 | Het | Chr7: 94039590 | Pathogenic | N | N | Delivered a healthy child |
| 19 | Short fetus limbs: cartilage hypoplasia? | AD | COL2A1 | NM_001844.4 | c.3013G > A | p.Gly1005Ser | CDS44 | Het | Chr12:48371891 | Pathogenic | N | N | |
| 20 | Short limbs, spine and vertebral ossification are not obvious: cartilage hypoplasia? | AD | COL2A1 | NM_001844.4 | c.2419G > A | p.Gly807Arg | EX37 | Het | Chr12:48375170 | VUS | N | N | |
| 21 | Short limbs, ventricular septal defect |
AD/ AR |
COL11A2 | NM_080680.2 | c.966dupC | p.Thr323Hisfis*19 | EX8 | Het | Chr6:33152074 | Pathogenic | Het | N | Undergoing PGD |
| c.1773 + 8 T>A | – | IVS19 | Het | Chr6:33146204 | VUS | N | Het | ||||||
| 22 | Abnormal long bones in fetal limbs, narrow chest | XD | EBP | NM_006579.2 | c.440G>A | p.Arg147His | EX4 | Het | ChrX:48385644 | Pathogenic | N | N | Delivered a healthy boy |
| 23 | Short fetal limbs (chronic dysgenesis) | AD | FGFR3 | NM_001163213.1 | c.742C > T | p.Arg248Cys | EX7 | Het | Chr4:1803564 | Pathogenic | N | N | Delivered a healthy child |
| 24 | Short limbs (cartilage hypoplasia) | AD | FGFR3 | NM_001163213.1 | c.742C > T | p.Arg248Cys | EX7 | Het | Chr4:1803564 | Pathogenic | N | N | |
| 25 | Short limbs | AD | FGFR3 | NM_001163213.1 | c.742C > T | p.Arg248Cys | EX7 | Het | Chr4:1803564 | Pathogenic | N | N | |
| 26 | Short limbs | AD | FGFR3 | NM_001163213.1 | c.742C > T | p.Arg248Cys | EX7 | Het | Chr4:1803564 | Pathogenic | N | N | |
| 27 | Short limbs | AD | FGFR3 | NM_001163213.1 | c.1144G > A | p.Gly382Arg | EX9 | Het | Chr4:1806119 | Pathogenic | N | N | |
| 28 | Short limbs | AD | FGFR3 | NM_001163213.1 | c.1124A > G | p.Tyr375Cys | EX9 | Het | Chr4:1806099 | Pathogenic | N | N | |
| 29 | Achondroplasia | AD | FGFR3 | NM_001163213.1 | c.2426G > C | p.X809S,101 | EX18 | Het | Chr4:1806099 | Pathogenic | N | N | |
| 30 | Uneven arrangement of fetal spine, short limbs, bilateral foot inversion, small mandibular, left ventricular punctate strong echo, ventricular septal defect | AD | FLNB | NM_001164317.1 | c.475A > C | p.Thr159Pro | EX2 | Het | Chr3:58062955 | VUS | N | N | |
Het heterogeneity, Pathogenic known disease-causing mutation, VUS Mutations of unknown clinical significance, N normal, AD autosomal dominant, AR autosomal recessive, PGD preimplantation genetic diagnosis
Discussion
Prenatal diagnosis of local skeletal dysplasia
Different types of chromosomal abnormalities complicate the wide, diverse variety of skeletal abnormalities [16]. For example, fetuses carrying trisomy 13, trisomy 18, or even trisomy 21 (Down’s syndrome) may have abnormal skeletal development. In the present study, two fetuses with trisomy 18 (cases 1 and 2) exhibited chromosomal abnormalities in local bone lesions, suggesting that the screening for chromosomal abnormalities remains vital when skeletal lesions are observed (Fig. 1).
Fig. 1.
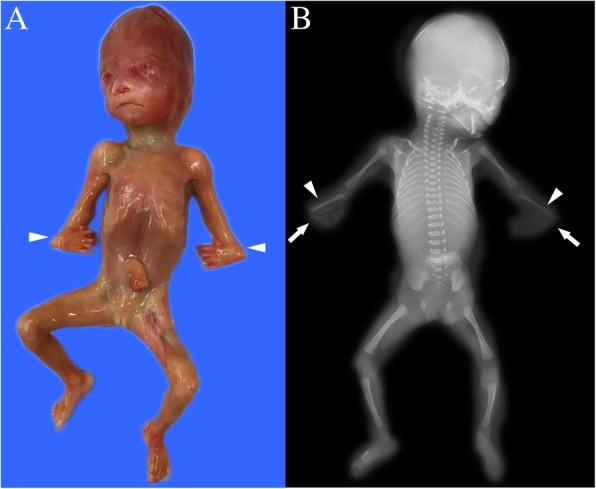
Case 2: The fetus with trisomy 18 and the absence of the radius. a Gross anatomy of the aborted fetus, showing the bilateral wrist flexion (arrowheads). b X-ray image of the aborted fetus, showing the bilateral bone defects (arrowheads) and bilateral wrist flexion (arrows)
The WGS revealed no significant copy number variations in the 13 cases of local fetal skeletal abnormalities (cases 3–15, Table 1). In addition, no mutations of skeletal pathogenic genes were identified in the seven cases (cases 9–15, Table 2). In contrast to osteodystrophy, local skeletal abnormalities are regulated by other factors in addition to hereditary genes. However, these additional factors require further study.
In the six fetuses with skeletal lesions (cases 3–8), skeletal gene mutations were detected with targeted gene sequencing (Table 2). In three of these cases (cases 3–5), the fetuses carried heterozygous mutations of unclear clinical significance. Both case 3 (femoral absence, micrognathia, and cleft lip and palate) and case 4 (spinal fracture, ventral fissure, introversion, and left foot polydactyly) carried clinically unexplained heterozygous mutations. The fetus in case 3 carried TBX4-induced small zygomatic complexes, while the fetus in case 4 carried TNNT3-related distal type 2B. Both of these are autosomal dominant genetic diseases. However, the Sanger sequencing suggested that one of the healthy parents carried the mutation. Hence, these two mutations could not explain the reason of case 3 and 4.
In case 5, the fetus carried a clinically unexplained heterozygous mutation in the SOX9 gene. SOX9-related trunk dysplasia is an autosomal dominant genetic disease, and in such diseases, heterozygous mutations can induce a disease phenotype. In this case, the Sanger sequencing indicated that neither of the parents were mutation carriers. Hence, this mutation was considered de novo and likely pathologic. Notably, the mother became naturally pregnant again and gave birth to a healthy baby boy.
In the remaining three cases (cases 6–8), known disease-causing mutations were detected in the fetuses. The available information about these mutations was useful in fetal disease diagnosis and in the subsequent genetic counseling for the parents.
In case 6 (fetal cleft lip and palate, right kidney cystic hypoplasia, bilateral foot fissure, lateral toe, and abnormal hand development), the fetus carried a mutation in the TP63 gene, which was correlated to ectrodactyly, ectoderm dysplasia, and cleft lip/palate syndrome-3 (EEC3). EEC3 is an autosomal dominant genetic disease, and in such diseases, heterozygous mutations can cause the disease. Alves et al. [17], Hydern et al. [18] and Clements et al. [19] all reported several mutations of the TP63 gene in families of EEC3. The Sanger sequencing revealed negative results for both parents of case 6, and this mutation was thereby considered novel in the fetus. The subsequent genetic counseling advised the parents to continue to conceive naturally.
In case 7 (angular femoral deformity and ventricular septal defect), two compound heterozygous causative mutations in the POR gene were detected: c.1370G > A (p.Arg457His) and c.744C > G (p.Tyr248*), which were associated with the Antley-Bixler syndrome. The Antley-Bixler syndrome is an autosomal recessive disease, and this type of homozygous or compound heterozygous mutation can induce a disease phenotype. The mutation c.1370G > A (p.Arg457His) has been detected in patients diagnosed with Antley-Bixler syndrome [20, 21]. The other mutations have also been reported in recent years [22, 23]. The mutation c.744C > G (p.Tyr248*) is a nonsense mutation that prematurely terminates the encoding of the POR protein at amino acid 248 (the unmutated protein is 680 amino acids long). Although the specific mutation identified in the present study has not yet been reported in literature, previously reported nonsense mutations after position 248 were deleterious. POR gene mutations c.1370G > A and c.744C > G have been considered disease-causing in their compound heterozygous forms. In the present study, the Sanger sequencing verified that both parents were carriers of this disease-causing mutation. The parents chose to conceive naturally at 6 months after pregnancy termination. However, the pregnancy was terminated at the 15th week of the second pregnancy, because the ultrasound examination revealed bilateral femoral angulation again in the fetus. Compound heterozygous POR gene mutations c.1370G > A and c.744C > G were verified on the analysis of the aborted tissues. The parents conceived naturally for the third time. The ultrasound results were normal, and the amniotic fluid analysis indicated that the POR gene mutation was absent. The mother eventually delivered a healthy baby girl.
In case 8 (two-sided rocker bottom feet, scoliosis, and spina bifida occulta), compound heterozygous mutations were detected in the CHRNG gene. The fetus was diagnosed with both double-curve scoliosis and varus, and both parents were carriers of this disease-causing mutation. These mutations had an autosomal recessive inheritance pattern. Hence, both parents presented with a normal phenotype. The final fetal diagnosis was Escobar variant of multiple pterygium syndrome (EVMPS). The parents are presently trying to conceive with PGD.
Prenatal diagnosis of systemic skeletal dysplasia
In the present study, no chromosomal abnormalities were identified in any of the cases of systemic skeletal dysplasia that manifested in short limb deformities (Table 1). The targeted gene sequencing identified several disease-causative mutations in known genes related to the disease (Table 2).
Collagen is an indispensable component of bone tissue found in the extracellular matrix. Mutations in the collagen gene may lead to insufficient collagen production. There are substantial differences in the severity of skeletal abnormalities caused by different types of collagen mutations. Mutations in collagen genes were detected in cases 16–21 in the present study. Three fetuses (cases 16–18) were diagnosed with osteogenesis imperfecta based on prenatal ultrasound, gross postnatal pathology, and X-ray examination (Fig. 2). Heterozygous mutations in COL2A1 were found in the other two fetuses (cases 19 and 20) diagnosed with achondrogenesis type II.
Fig. 2.
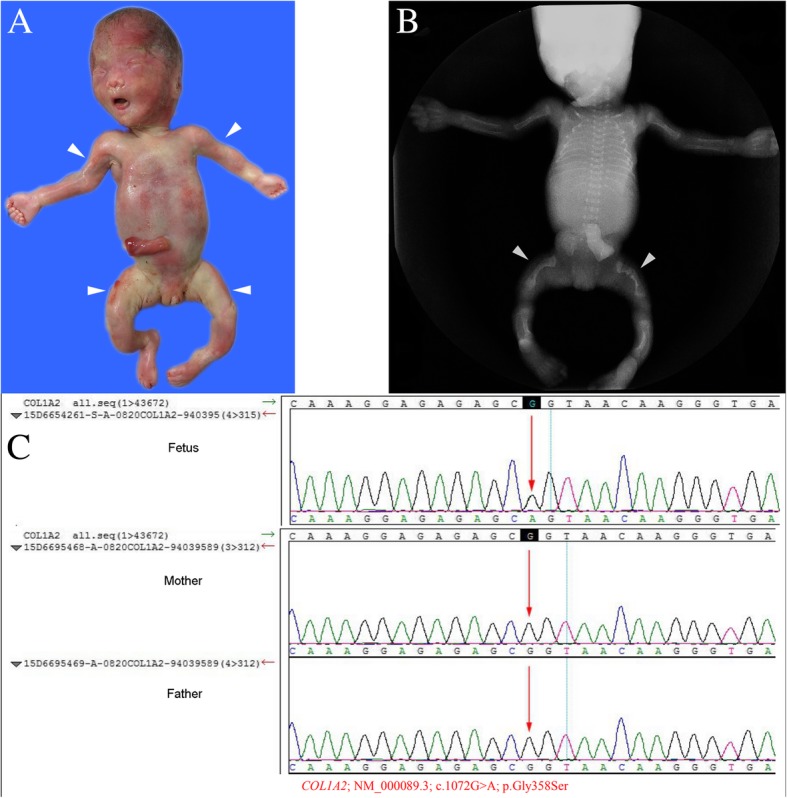
Case 18: The fetus with osteogenesis imperfecta type II with collagen gene mutation (COL1A2). a Gross anatomy of the aborted fetus, showing the short arms and bending short legs (arrowheads). b X-ray images of the aborted fetus, showing the short and bent femurs, both with fractures (arrowheads). c The fetus carried the c.1072G > A (p.Gly358Ser) mutation in the COL1A2 gene. The Sanger verification revealed that this mutation was new, and was not carried by the parents
In case 16, a heterozygous causative mutation (c.1678G > A, p.Gly560Ser) in COL1A1 was detected in the fetus. In cases 17 and 18, two known pathogenic mutations were detected in the COL1A2 gene: c.1774G > A (p.Gly592Ser) and c.1072G > A (p.Gly358Ser). These mutations have been reported to be pathogenic mutations associated with osteogenesis imperfecta [24–28]. However, none of these mutations were detected in the parents, suggesting that these mutations are novel fetal mutations.
Approximately 90% of osteogenesis imperfecta cases are due to causative variants in the COL1A1 and COL1A2 genes, which result in abnormal collagen I fibrils formation, while the remaining 10% of cases are associated with recessive variants of known or yet to be discovered genes [24, 25].
Heterozygous mutations in COL2A1 were found in two fetuses (cases 19 and 20) diagnosed with achondrogenesis type II (Fig. 3). Mutations in COL2A1 disrupt the Gly-XY motif necessary for the formation of a triple helix structure, resulting in type II collagen over-modification, cellular retention and decreased secretion [29, 30]. All these collagen-related mutations were new in the fetuses, and the parents were not mutation carriers. The parents were advised to continue natural conception.
Fig. 3.
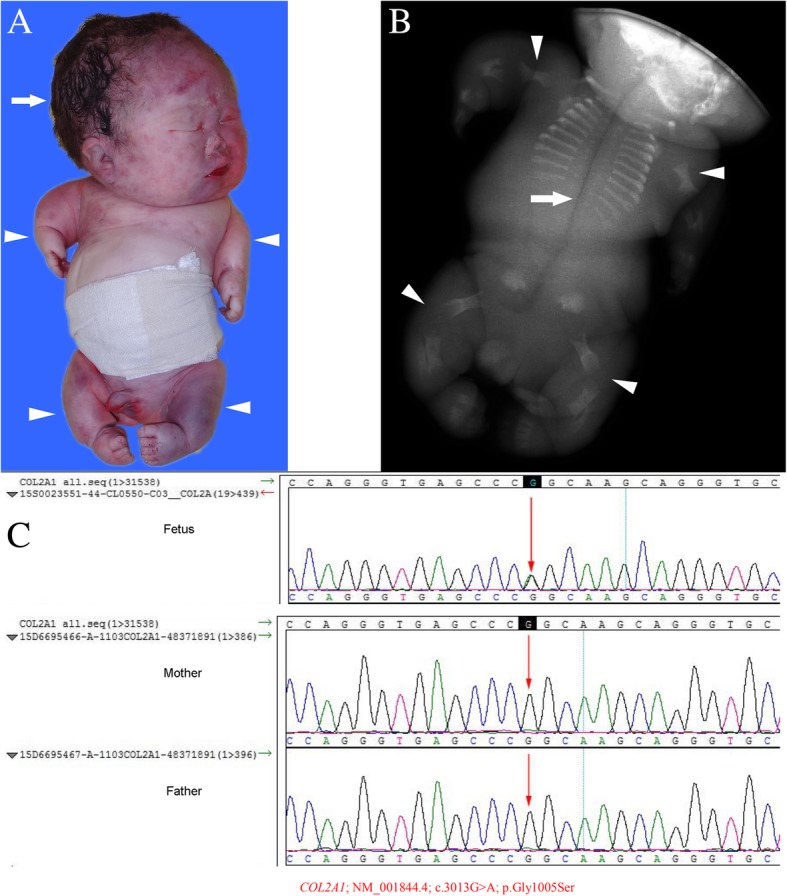
Case 19: The fetus with achondrogenesis. a Gross anatomy of the aborted fetus, showing the markedly short arms and legs (arrowheads), with a giant skull (arrow). b X-ray images of the aborted fetus, showing the severe short limb (arrowheads), and the defective ossification of the spine, sciatic, pubic and iliac bone (arrow). c The fetus carried the c.3013G > A (p.Gly1005Ser) mutation in the COL2A1 gene. The Sanger verification revealed that this mutation was new, and was not carried by the parents
Case 21 was eventually diagnosed with fibrocartilage hyperplasia type II, which was caused by mutations in the COL11A2 gene. The two COL11A2 mutations identified in the present study have only been previously identified once [31]. These mutations revealed autosomal recessive/dominant inheritance. The Sanger sequencing verified that these phenotypically normal parents were mutation carriers. These parents were trying to conceive again with PGD.
Fibroblast growth factors (FGFs) play an important role in endochondral osteogenesis and intramembranous osteogenesis. Cells generally aggregate in several areas on osteophyte growth plates, and in the proximal dormant area, chondrocytes proliferate. Then, the chondrocytes differentiate into primary hypertrophic chondrocytes, and gradually become mature hypertrophic chondrocytes. The proliferation, differentiation and apoptosis of chondrocytes are regulated by FGF/FGFR signaling. For example, the interaction between FGF18 and FGFR3 inhibits chondrocyte proliferation. Mutations in the FGFR3 gene can increase the extracellular/tyrosine kinase domain activity of the receptor, stimulating the signaling pathways that induce the expression of extracellular signal-regulated kinase 1/2, and the signal transducers and activators of transcription protein 1 (STAT1), which leads to the arrest of chondrocyte proliferation and chondrocyte apoptosis .
In the present study, seven fetuses with achondroplasia (cases 23–29) were further examined with a combination of ultrasound, postnatal gross pathology, and X-ray. Based on these examinations, six of the fetuses were diagnosed with clinically fatal cartilage hypoplasia. The subsequent genetic analysis confirmed the fatal cartilage hypoplasia type I in six fetuses. Cases 23–26 carried an identical mutation (c.742C > T, p.Arg248Cys), which is a common pathogenic mutation associated with lethal achondroplasia (Fig. 4). In 2015, Barkova et al. [2] reported that eight of 20 patients with lethal dysplasia type I carried the c.742C > T mutation in FGFR3. Similar findings were reported in 2001 by Chen et al. [32] and in 2014 by Cho et al. [33]. The mutation c.1124A > G (p.Tyr375Cys) in the FGFR3 gene is also a common pathogenic mutation that causes lethal dwarfism type I. The study conducted by Rousseau et al. [34] revealed that eight of 26 (30.7%) cases of fatal dwarfism type I carried the c.1124A > G (i.e. c.1118A > G in the article) mutation in FGFR3. However, Xue et al. [35] reported that the frequency of these FGFR3 mutations was 23.7% (41 of 173 cases) in cases of fatal dwarfism type I. Another FGFR3 mutation, c.2426G > C (p.X809S,101), was identified in the present study, which has not previously been reported. This was a missense mutation that led to the false extension of protein translation. In case 27, the fetus was diagnosed with achondroplasia. Furthermore, 99% of all achondroplasia cases are caused by mutations in the FGFR3 gene, and c.1144G > A (p.Gly382Arg) is the most common pathogenic mutation. The missense mutation c.1144G > A (p.Gly382Arg) is identical to c.1138G > A (p.Gly380Arg) (different transcripts). In 1995, the study conducted by Bellus et al. [36] revealed that 187 of 193 (96.9%) cases of achondroplasia are caused by the mutation c.1138G > A. For case 27, since the parents did not carry the mutation, it was considered a new mutation in the fetus. After genetic counseling, the parents were advised to continue to conceive naturally.
Fig. 4.
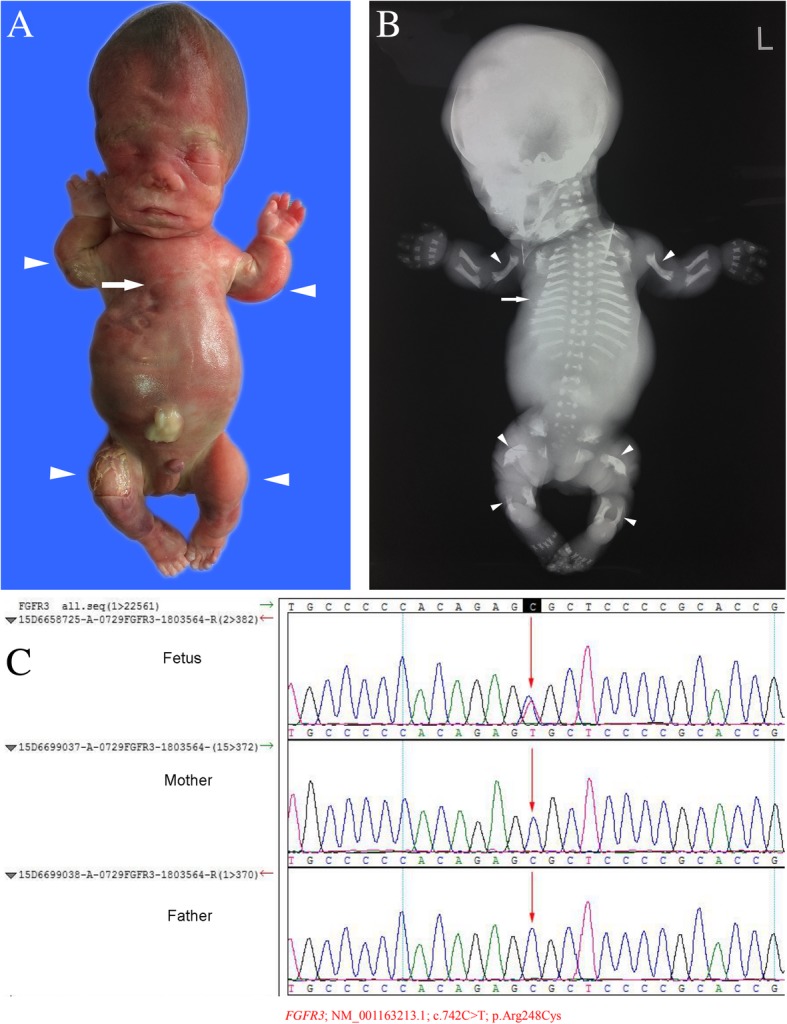
Case 26: The fetus with fatal achondroplasia and fibroblast growth factor receptor 3 (FGFR3) gene mutations. a Gross anatomy of the aborted fetus, showing the markedly short arms and legs (arrowheads), with a narrow chest (arrow). b X-ray images of the aborted fetus, showing the “telephone receiver” appearance of the femurs and humeri, the malformation of the metaphyseal (crateriform) (arrowheads), and the narrow thorax (arrow). c The fetus carried the c.742C > T (p.Arg248Cys) mutation in the FGFR3 gene. The Sanger verification revealed that this mutation was new, and was not carried by the parents
The mutation c.475A > C (p.Thr159Pro) in the FLNB gene carried by the fetus in case 30 is a missense mutation, which changes the amino acid at position 159 from threonine to proline. FLNB-related osteogenesis imperfecta type I/Larsen syndrome is an autosomal dominant disease. The sequence verification confirmed that neither of the parents carried the mutation, indicating that this was a novel mutation in the fetus. Therefore, the parents were advised to continue to conceive naturally.
In case 22, ultrasound revealed the abnormal development of long bones of the limbs (the length of the long bones was less than 1%), thick metaphysis in the right lower limb, irregular vertebral arrangement, and a narrow and small thorax in 24 weeks of gestation. The fetus carried a heterozygous mutation in the EBP gene (C.440G > A, p.Arg147His). Herman et al. investigated the mutations in 26 female patients with suspected CDPX2. Among these 26 patients, 22 had EBP mutations. Among these 22 mutations, 13 mutations were de novo [37]. The EBP gene was located on the short arm of the X chromosome (Xp11.22-p11.23), and the mutations in this gene can lead to the accumulation of 8-dehydrocholesterol and 8(9)-cholesterol in plasma, the skin and other tissues, resulting in a wide range of abnormalities [37, 38]. In the present study, a heterozygous EBP mutation (a known causative mutation; c.440G > A [p.Arg147His]) was the cause of CDPX2, and is a missense mutation (arginine to histidine). The Sanger test revealed that the patient’s parents were non-carriers, indicating that the mutation of the fetus occurred de novo. Whittock et al., Has et al. and Cañueto et al. have previously reported cases related to this mutation site. [39–41]. However, neither of the parents carried the mutation. In the present study, the 27-week-old female fetus presented with markedly short bones, ankle joint contracture, markedly asymmetric short lower limbs, and a flat face and nose bridge. The severity of the phenotype was considered to be related to X chromosome inactivation, which is also known as lyonization.
However, although targeted exome capture and sequencing have shown great advantages in disease gene identification and molecular diagnosis, some problems still needs to be immediately resolved. Fox example, exome sequencing focuses on the sequencing of exon regions. Thus, from the genome level, the information obtained was obviously incomplete. Furthermore, information for promoter regions, enhancer regions and microRNA coding regions were certainly missed. Second, a large amount of data was obtained after exome sequencing. The best method to perform an in-depth and accurate analysis of these data is the largest challenge faced at present by researchers worldwide. The deep mining of data needs to start from many aspects and perspectives, including studies at the transcription level, bioinformatics analysis, and functional genomics studies.
Conclusion
In summary, the results of the present study suggest that the application of targeted gene sequencing technology can significantly improve the prenatal diagnosis of systemic skeletal abnormalities, allowing for a more comprehensive and useful prenatal genetic counseling guidance for parents. Furthermore, the present study provides a theoretical basis for early intervention birth defect diagnoses and the assessment of fetal risk associated with subsequent pregnancies. In addition, the present study also provides further useful information for the continued development of skeletal dysplasia treatments based on target genes [32]. To date, mutations in 363 genes are known to be associated with more than 300 common skeletal dysplasias in humans [10]. However, genetic basis remains unknown in many additional skeletal diseases, especially local skeletal lesions, suggesting that new genes or non-genetic factors may cause these diseases.
Acknowledgements
The authors thank all the families for participation in the present study.
Abbreviations
- AC
Abdominal circumference
- CHRNG
Cholinergic receptor nicotinic gamma subunit
- EBP
Cholestenol delta-isomerase
- EDTA
Ethylenediaminetetraacetic acid
- EEC3
Ectrodactyly, ectoderm dysplasia, and cleft lip/palate syndrome-3
- EVMPS
Escobar variant of multiple pterygium syndrome
- FGFR3
Fibroblast growth factor receptor 3
- FGFs
Fibroblast growth factors
- FL
Femur length
- FLNB
Filamin B
- SOX9
SRY-box 9
- STAT1
Signal transducers and activators of transcription protein 1
- TP63
Tumor protein p63
- WGS
Whole genome sequencing
Authors’ contributions
YL been involved in drafting the manuscript and revising it critically for important intellectual content; LW and Y-KY made substantial contributions to conception and design of the work; YL, T-JZ, NL, L-MY, S-JL, DS and Q-QW made substantial contributions to the acquisition, analysis, and interpretation of data for the work; all authors given final approval of the version to be published.
Funding
This study was supported by The National Key Research and Development Program of China [No.2016YFC1000104], Beijing Municipal Administration Hospital Ascent Plan [No.DFL20151302], Beijing Municipal Science and Technology Commission [No.Z161100000116089] and The Capital Health Research and Development of Special [No.2014–2-2113].
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Ethics approval and consent to participate
The present study was approved by the ethics committee of our hospital, and all parents of the fetuses provided a signed informed consent prior to prenatal diagnosis and sample collection.
Consent for publication
We have obtained consent to publish from the parents of the fetuses to report individual patient data.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rawhani R, Abdellatif A, Abushama M, Ahmed B. Antenatal diagnosis of fetal skeletal malformation. Donald Sch J Ultrasound Obstet Gynecol. 2018;12:116–123. doi: 10.5005/jp-journals-10009-1561. [DOI] [Google Scholar]
- 2.Barkova E, Mohan U, Chitayat D, Keating S, Toi A, Frank J, Frank R, Tomlinson G, Glanc P. Fetal skeletal dysplasias in a tertiary care center: radiology, pathology, and molecular analysis of 112 cases. Clin Genet. 2015;87:330–337. doi: 10.1111/cge.12434. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson DA, Carey JC, Byrne JL, Srisukhumbowornchai S, Feldkamp ML. Analysis of skeletal dysplasias in the Utah population. Am J Med Genet A. 2012;158A:1046–1054. doi: 10.1002/ajmg.a.35327. [DOI] [PubMed] [Google Scholar]
- 4.Orioli IM, Castilla EE, Barbosa-Neto JG. The birth prevalence rates for the skeletal dysplasias. J Med Genet. 1986;23:328–332. doi: 10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, Chandler N, Deng L, Zhou J, Yuan M, Sun L. Prenatal diagnosis of skeletal dysplasias using a targeted skeletal gene panel. Prenat Diagn. 2018;38:692–699. doi: 10.1002/pd.5298. [DOI] [PubMed] [Google Scholar]
- 6.Geister KA, Camper SA. Advances in skeletal dysplasia genetics. Annu Rev Genomics Hum Genet. 2015;16:199–227. doi: 10.1146/annurev-genom-090314-045904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar M, Thakur S, Haldar A, Anand R. Approach to the diagnosis of skeletal dysplasias: experience at a center with limited resources. J Clin Ultrasound. 2016;44:529–539. doi: 10.1002/jcu.22371. [DOI] [PubMed] [Google Scholar]
- 8.Toru HS, Nur BG, Sanhal CY, Mihci E, Mendilcioğlu İ, Yilmaz E, Yilmaz GT, Ozbudak IH, Karaali K, Alper OM, Karaveli FŞ. Perinatal diagnostic approach to fetal skeletal Dysplasias: six years experience of a tertiary center. Fetal Pediatr Pathol. 2015;34:287–306. doi: 10.3109/15513815.2015.1068414. [DOI] [PubMed] [Google Scholar]
- 9.Calder AD, Offiah AC. Foetal radiography for suspected skeletal dysplasia: technique, normal appearances, diagnostic approach. Pediatr Radiol. 2015;45:536–548. doi: 10.1007/s00247-014-3130-x. [DOI] [PubMed] [Google Scholar]
- 10.Li SL. Prenatal ultrasound diagnosis and prognosis of fetal limb deformities. Chin J Practic Gynecol Obstet. 2007;23:399–400. [Google Scholar]
- 11.Milks KS, Hill LM, Hosseinzadeh K. Evaluating skeletal dysplasias on prenatal ultrasound: an emphasis on predicting lethality. Pediatr Radiol. 2017;47:134–145. doi: 10.1007/s00247-016-3725-5. [DOI] [PubMed] [Google Scholar]
- 12.Nelson DB, Dashe JS, McIntire DD, Twickler DM. Fetal skeletal dysplasias: sonographic indices associated with adverse outcomes. J Ultrasound Med. 2014;33:1085–1090. doi: 10.7863/ultra.33.6.1085. [DOI] [PubMed] [Google Scholar]
- 13.Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti-Furga A. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konstantinidou AE, Agrogiannis G, Sifakis S, Karantanas A, Harakoglou V, Kaminopetros P, Hatzaki A, Petersen MB, Karadimas C, Velissariou V, Velonis S, Papantoniou N, Antsaklis A, Patsouris E. Genetic skeletal disorders of the fetus and infant: pathologic and molecular findings in a series of 41 cases. Birth Defects Res A Clin Mol Teratol. 2009;85:811–821. doi: 10.1002/bdra.20617. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ML, Lu YP, Li RB, Ye MX, Huang K, You YQ, Wang SJ, Wang LX, Li YL. Application of targeted exome capture in identifying fetal skeletal malformation mutations. Chin J Perinat Med. 2015;18:334–338. [Google Scholar]
- 16.Xiong W, Luo H, An SY, Wu Y, Liu Y. The correlation analysis of fetal skeletal anomalies with chromosome abnormality by prenatal systematic ultrasonography examination. Chin J Med Ultrasound (Electronic Edition) 2015;12:148–151. [Google Scholar]
- 17.Alves LU, Pardono E, Otto PA, Mingroni Netto RC. A novel c.1037C > G (p.Ala346Gly) mutation in TP63 as cause of the ectrodactyly-ectodermal dysplasia and cleft lip/palate (EEC) syndrome. Genet Mol Biol. 2015;38:37–41. doi: 10.1590/S1415-475738120140125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyder Z, Beale V, O'Connor R, Clayton-Smith J. Genitourinary malformations: an under-recognized feature of ectrodactyly, ectodermal dysplasia and cleft lip/palate syndrome. Clin Dysmorphol. 2017;26:78–82. doi: 10.1097/MCD.0000000000000172. [DOI] [PubMed] [Google Scholar]
- 19.Clements SE, Techanukul T, Coman D, Mellerio JE, McGrath JA. Molecular basis of EEC (ectrodactyly, ectodermal dysplasia, clefting) syndrome: five new mutations in the DNA-binding domain of the TP63 gene and genotype-phenotype correlation. Br J Dermatol. 2010;162:201–207. doi: 10.1111/j.1365-2133.2009.09496.x. [DOI] [PubMed] [Google Scholar]
- 20.Ko JM, Cheon CK, Kim GH, Yoo HW. A case of Antley-Bixler syndrome caused by compound heterozygous mutations of the cytochrome P450 oxidoreductase gene. Eur J Pediatr. 2009;168:877–880. doi: 10.1007/s00431-008-0849-0. [DOI] [PubMed] [Google Scholar]
- 21.Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, Matsuo N, Sato S, Homma K, Nishimura G, Hasegawa T, Ogata T. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005;90:414–426. doi: 10.1210/jc.2004-0810. [DOI] [PubMed] [Google Scholar]
- 22.Oldani E, Garel C, Bucourt M, Carbillon L. Prenatal diagnosis of Antley-Bixler syndrome and POR deficiency. Am J Case Rep. 2015;16:882–885. doi: 10.12659/AJCR.895526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzetis M, Konstantinidou A, Sofocleous C, Kosma K, Mitrakos A, Tzannatos C, Kitsiou-Tzeli S. Compound heterozygosity of a paternal submicroscopic deletion and a maternal missense mutation in POR gene: Antley-bixler syndrome phenotype in three sibling fetuses. Birth Defects Res A Clin Mol Teratol. 2016;106:536–541. doi: 10.1002/bdra.23492. [DOI] [PubMed] [Google Scholar]
- 24.Zhytnik L, Maasalu K, Reimann E, Prans E, Kõks S, Märtson A. Mutational analysis of COL1A1 and COL1A2 genes among Estonian osteogenesis imperfecta patients. Hum Genomics. 2017;11:19. doi: 10.1186/s40246-017-0115-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Augusciak-Duma A, Witecka J, Sieron AL, Janeczko M, Pietrzyk JJ, Ochman K, Galicka A, Borszewska-Kornacka MK, Pilch J, Jakubowska-Pietkiewicz E. Mutations in the COL1A1 and COL1A2 genes associated with osteogenesis imperfecta (OI) types I or III. Acta Biochim Pol. 2018;65:79–86. doi: 10.18388/abp.2017_1612. [DOI] [PubMed] [Google Scholar]
- 26.Stephen J, Shukla A, Dalal A, Girisha KM, Shah H, Gupta N, Kabra M, Dabadghao P, Hasegawa K, Tanaka H, Phadke SR. Mutation spectrum of COL1A1 and COL1A2 genes in Indian patients with osteogenesis imperfecta. Am J Med Genet A. 2014;164A:1482–1489. doi: 10.1002/ajmg.a.36481. [DOI] [PubMed] [Google Scholar]
- 27.Malmgren B, Andersson K, Lindahl K, Kindmark A, Grigelioniene G, Zachariadis V, Dahllöf G, Åström E. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis. 2017;23:42–49. doi: 10.1111/odi.12568. [DOI] [PubMed] [Google Scholar]
- 28.Lee KS, Song HR, Cho TJ, Kim HJ, Lee TM, Jin HS, Park HY, Kang S, Jung SC, Koo SK. Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta. Hum Mutat. 2006;27:599. doi: 10.1002/humu.9423. [DOI] [PubMed] [Google Scholar]
- 29.Deng H, Huang X, Yuan L. Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res. 2016;768:1–13. doi: 10.1016/j.mrrev.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Mortier GR, Weis M, Nuytinck L, King LM, Wilkin DJ, De Paepe A, Lachman RS, Rimoin DL, Eyre DR, Cohn DH. Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder. J Med Genet. 2000;37:263–271. doi: 10.1136/jmg.37.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tompson SW, Faqeih EA, Ala-Kokko L, Hecht JT, Miki R, Funari T, Funari VA, Nevarez L, Krakow D, Cohn DH. Dominant and recessive forms of fibrochondrogenesis resulting from mutations at a second locus, COL11A2. Am J Med Genet A. 2012;158A:309–314. doi: 10.1002/ajmg.a.34406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CP, Chern SR, Shih JC, Wang W, Yeh LF, Chang TY, Tzen CY. Prenatal diagnosis and genetic analysis of type I and type II thanatophoric dysplasia. Prenat Diagn. 2001;21:89–95. doi: 10.1002/1097-0223(200102)21:2<89::AID-PD21>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 33.Cho I, Shim JY, Kim GH, Yoo HW, Lee EJ, Won HS, Lee PR, Kim A. Thanatophoric dysplasia in a dichorionic twin confirmed by genetic analysis at the early second trimester: a case report and literature review. Obstet Gynecol Sci. 2014;57:151–154. doi: 10.5468/ogs.2014.57.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseau F, el Ghouzzi V, Delezoide AL, Legeai-Mallet L, Le Merrer M, Munnich A, Bonaventure J. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TD1) Hum Mol Genet. 1996;5:509–512. doi: 10.1093/hmg/5.4.509. [DOI] [PubMed] [Google Scholar]
- 35.Xue Y, Sun A, Mekikian PB, Martin J, Rimoin DL, Lachman RS, Wilcox WR. FGFR3 mutation frequency in 324 cases from the international skeletal dysplasia registry. Mol Genet Genomic Med. 2014;2:497–503. doi: 10.1002/mgg3.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–373. [PMC free article] [PubMed] [Google Scholar]
- 37.Herman GE, Kelley RI, Pureza V, Smith D, Kopacz K, Pitt J, Sutphen R, Sheffield LJ, Metzenberg AB. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome) Genet Med. 2002;4:434–438. doi: 10.1097/00125817-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Braverman N, Lin P, Moebius FF, Obie C, Moser A, Glossmann H, Wilcox WR, Rimoin DL, Smith M, Kratz L, Kelley RI, Valle D. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22:291–294. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 39.Has C, Bruckner-Tuderman L, Müller D, Floeth M, Folkers E, Donnai D, Traupe H. The Conradi-Hünermann-Happle syndrome (CDPX2) and emopamil binding protein: novel mutations, and somatic and gonadal mosaicism. Hum Mol Genet. 2000;9:1951–1955. doi: 10.1093/hmg/9.13.1951. [DOI] [PubMed] [Google Scholar]
- 40.Whittock NV, Izatt L, Mann A, Homfray T, Bennett C, Mansour S, Hurst J, Fryer A, Saggar AK, Barwell JG, Ellard S, Clayton PT. Novel mutations in X-linked dominant chondrodysplasia punctata (CDPX2) J Invest Dermatol. 2003;121:939–942. doi: 10.1046/j.1523-1747.2003.12489.x. [DOI] [PubMed] [Google Scholar]
- 41.Cañueto J, Girós M, Ciria S, Pi-Castán G, Artigas M, García-Dorado J, García-Patos V, Virós A, Vendrell T, Torrelo A, Hernández-Martín A, Martín-Hernández E, Garcia-Silva MT, Fernández-Burriel M, Rosell J, Tejedor M, Martínez F, Valero J, García JL, Sánchez-Tapia EM, Unamuno P, González-Sarmiento R. Clinical, molecular and biochemical characterization of nine Spanish families with Conradi-Hünermann-Happle syndrome: new insights into X-linked dominant chondrodysplasia punctata with a comprehensive review of the literature. Br J Dermatol. 2012;166:830–838. doi: 10.1111/j.1365-2133.2011.10756.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its supplementary information files].