

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects motor neurons in the brainstem, spinal cord and motor cortex. ALS is characterized by genetic and clinical heterogeneity, suggesting the existence of genetic factors that modify the phenotypic expression of the disease. We previously identified the axonal guidance EphA4 receptor, member of the Eph-ephrin system, as an ALS disease-modifying factor. EphA4 genetic inhibition rescued the motor neuron phenotype in zebrafish and a rodent model of ALS. Preventing ligands from binding to the EphA4 receptor also successfully improved disease, suggesting a role for EphA4 ligands in ALS. One particular ligand, ephrin-A5, is upregulated in reactive astrocytes after acute neuronal injury and inhibits axonal regeneration. Moreover, it plays a role during development in the correct pathfinding of motor axons towards their target limb muscles. We hypothesized that a constitutive reduction of ephrin-A5 signalling would benefit disease progression in a rodent model for ALS. We discovered that in the spinal cord of control and symptomatic ALS mice ephrin-A5 was predominantly expressed in neurons. Surprisingly, reduction of ephrin-A5 levels in SOD1G93A mice accelerated disease progression and reduced survival without affecting disease onset, motor neuron numbers or innervated neuromuscular junctions in symptomatic mice. These findings suggest ephrin-A5 as a modifier of disease progression that might play a role in the later stages of the disease. Similarly, we identified a more aggressive disease progression in patients with lower ephrin-A5 protein levels in the cerebrospinal fluid without modifying disease onset. In summary, we identified reduced expression of ephrin-A5 to accelerate disease progression in a mouse model of ALS as well as in humans. Combined with our previous findings on the role of EphA4 in ALS our current data suggests different contribution for various members of the Eph-ephrin system in the pathophysiology of a motor neuron disease.
Electronic supplementary material
The online version of this article (10.1186/s40478-019-0759-6) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis, Neurodegeneration, Motor neuron, SOD1G93A, EphA4, Ephrin-A5
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease in which upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord degenerate. The course of the disease is relentless and progressive and ultimately results in death 3–5 years after symptom onset [5]. A large number of mutations in a wide variety of genes can cause the disease, such as the copper–zinc superoxide dismutase (Sod1), chromosome 9 open reading frame 72 (C9orf72), fused in sarcoma (Fus) and TAR DNA-binding protein 43 (Tardbp) genes [39]. In ALS, there is not only diversity in the number of disease-causing genes, but also large differences exist in the phenotypic presentation of the disease even in patients carrying the same mutation [12, 36]. This indicates that other factors can alter disease onset and/or progression. We have previously shown that EphA4 is a disease modifier in ALS, and that reduction of its signalling ameliorates the motor phenotype in different ALS models [42]. EphA4 is a tyrosine kinase receptor member of the Eph/ephrin family, which is composed by Eph tyrosine kinase receptors and their ephrin ligands [34]. When receptor and ligand interact, bidirectional signalling occurs downstream of both Ephs and ephrins, giving rise to attractive or repulsive stimuli of two opposing cell membranes [34]. Although the role of EphA4 ligands in ALS is unknown, an EphA4 agonist that impedes EphA4 binding to its ligands, and an EphA4-Fc recombinant protein that reduces binding of EphA4 ligands to the receptor beneficially affected the phenotypes in mouse models for ALS [44, 47].
One particular EphA4 ligand, ephrin-A5 (efnA5), is of specific interest since its expression is increased in reactive astrocytes after stroke, resulting in reduced growth of cortical axonal projections during recovery [6, 23, 33]. Blocking efnA5 signalling improved the recovery of mice after stroke injury [33]. This upregulation of efnA5 in reactive astrocytes was not only induced after experimental stroke, but also by physical stress as shown in vitro [33], suggesting that efnA5 might be upregulated in astrocytes under stressful conditions. Since astrocytes, microglia and oligodendrocytes contribute to the selective vulnerability of motor neurons in ALS, the study of the role of efnA5 in astrocytes in this disease is of interest [1, 19, 45].
In vitro studies have shown that adding recombinant efnA5 or efnA5 overexpressing cells to neuronal cultures induces growth cone collapse and neurite retraction by affecting reorganization of the actin cytoskeleton [10, 11, 28, 29, 43]. During development, efnA5 is expressed in the ventral limb and also in developing motor axons that project from the lateral division of the lateral motor column (LMCl) in the spinal cord towards the limb. EfnA5 present in both compartments works simultaneously through forward and reverse signalling pathways that involve Eph receptors, to guide these axons so that they properly project dorsally in the limb [3, 27]. In ALS, denervation of neuromuscular junctions (NMJs) and axonal retraction occurs before motor neurons start degenerating, and compensatory re-sprouting mechanisms exist to re-innervate NMJs [14, 35]. Axonal guidance mechanisms involved during developmental stages might also play a role in disease mechanisms of de- and re-innervation.
Overall, given the role of efnA5 in astrocytes and in controlling attraction/repulsion responses, we aimed to investigate the expression and the role of efnA5 in a SOD1G93A mouse model of ALS. We hypothesized efnA5 reduction to improve disease progression in a mouse model for ALS, potentially via a role in the communication between astrocytes and motor neurons, and/or in axonal retraction and re-sprouting.
Materials and methods
Animals
All animal experiments were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments, and all animal experiments were approved by the local ethical committee of the KU Leuven (P229/2013 and P229/2017). Animals were housed under standard conditions according to the guidelines of the University of Leuven (KU Leuven), with a 12 h light-dark cycle and with access to food and water ad libitum. The human wild-type SOD1 overexpressing mouse (B6.SOD1WT; B6SJL-Tg(SOD1)2Gur/J; stock number 002297) and the human mutant SOD1 overexpressing mouse (B6.SOD1G93A; B6.Cg-Tg(SOD1*G93A)1Gur/J; stock number 004435) were purchased from the Jackson Laboratory (Ben Harbor, ME), and maintained in a C57BL/6 J genetic background. The efnA5 knockout mouse colony was generated by Dr. Jonas Frisén (Karolinska Institute, Stockholm, Sweden) [16], and was kindly provided by Dr. Renping Zhou (Rutgers University, Piscataway, NJ, USA). Mice were received and maintained under a mixed C57BL/6 J and S129 background, since EfnA5−/− mice are embryonically lethal in a pure C57BL/6 J background [38]. For ALS-related experimental purposes, EfnA5+/− mice were backcrossed for more than 10 generations to obtain EfnA5+/− mice in a pure C57BL/6 J background, which were crossed with SOD1G93A mice to obtain the following experimental groups: SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice. Only the mice whose father had a lifespan shorter than 165 days, were used for breeding and for experimental purposes. Only litters that contained the genotype of interest and the control genotype within the same gender were included in the experiments. Both males and females were used, and all experiments were conducted in a blinded manner.
Animal motor performance
Established guidelines were followed to assess ALS disease phenotype [26]. The ability of mice to hang upside down from an elevated grid (hanging wire test) and their ability to walk on the rotarod rotating at a fixed speed of 15 rpm, was measured to determine motor coordination and strength. The latency to fall was measured with cut-offs of 60 s in the hanging wire test and of 300 s in the rotarod. Motor performance on these tests and weight were assessed in experimental mice three times every week from the age of 60 days until an end-stage point of the disease. Mice reached a disease end-stage point when their righting reflex took longer than 10 s. According to humane-endpoints, mice were euthanized at this time point and the date was annotated for survival analysis. Disease onset was determined as the age at which mice could no longer perform the maximum score of 60 s in the hanging wire test, and disease duration was calculated as the time between disease onset and mouse survival.
Nerve conduction studies
Recordings of the compound muscle action potential (CMAP) were performed once a week in mice from the age of 60 days until the age of 115 days. Mice were anesthetized with 3% isoflurane/O2 gas inhalation and immobilized on a heating pad at 37 °C. Recordings were performed with subdermal needle electrodes (Technomed Europe). The stimulating electrode was placed at the level of the sciatic notch and the recording electrode was placed on the gastrocnemius muscle. CMAPs were measured by supramaximal stimulation (1 pulse per second and 0.1 ms stimulus duration) using a Medelec EMG monitor (Medelec Vickers) and Synergy software (version 20.1.0.100).
Motor neuron counts and neuromuscular junctions in ALS mice
ALS mice were followed with hanging wire test, and a whole litter was sacrificed as soon as one mouse of the litter could no longer hang from the wire for 2 s, which was considered as a late-symptomatic stage of the disease [42]. At that point, an overdose of Dolethal (20 mg/ml; Vetoquinol) was used to sacrifice the litter. Gastrocnemius muscles were dissected and snap-frozen in ice-cold isopentane, and then mice were transcardially perfused with 4% paraformaldehyde (PFA). Lumbar spinal cords were dissected, post-fixed overnight with 4% PFA, cryoprotected with 30% sucrose and embedded in OCT embedding matrix (CellPath) and stored at − 80 °C for later use. For motor neuron counts, spinal cords were cryosected on a CryoStar NX70 Cryostat (ThermoFischer Scientific). Every sixth 20 μm-thick cryosection was stained with Neurotrace 500/525 (1:100; N21480; ThermoFischer) or cresyl violet (Sigma) and used for quantitative analysis of the number of neurons. Images of ten sections in total per spinal cord were obtained with a Zeiss Axioimager M1 epifluorescence and brightfield upright microscope with a Zeiss A-Plan 10X/0.25 objective (Carl Zeiss, Germany), and an Axiocam MRm monochrome digital camera and the Zen 2.3 software (Carl Zeiss, Germany). Only neurons within one ventral horn of each spinal cord were counted using ImageJ v1.51u by Wayne Rasband (National Institutes of Health) and the number of neurons in different size groups was determined. To quantify the innervation of NMJs, longitudinal 20 μm-thick gastrocnemius cryosections were stained overnight at 4 °C with Alexa-488-conjugated neurofilament-L antibody (1:500; Cell Signaling Technologies) and 1 h at RT with Alexa-555-conjugated α-bungarotoxin (Invitrogen; 1:1000). Slides were mounted with ProLong Gold antifade reagent (Life Technologies), and visualized with a Zeiss Axioimager M1 epifluorescence upright microscope with a Zeiss A-Plan 40X/0.65 ∞/0.17 objective (Carl Zeiss). At least 100 NMJs per muscle were analysed for innervation, as determined by the co-localization of the neurofilament-L and α-bungarotoxin labelling. Analyses were performed in a blinded manner. Representative Z-stack confocal images were obtained on a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems Heidelberg GmbH) with an HC PL APO CS2 40x/0.85 dry lens with a pinhole of 0.5 Airy Units. Sum projections of the Z-stacks was done with the freeware ImageJ v1.51u by Wayne Rasband (National Institutes of Health).
Sciatic nerve crush surgery
Four months-old EfnA5+/+ and EfnA5−/− mice maintained under a mixed C57BL/6 J and S129 background were deeply anesthetized for surgery with 3% isoflurane/O2 gas inhalation. Vetergesic (15 mg/kg; Ecuphar) was administered subcutaneously to minimize post-operative pain. The fascial plane between the gluteus maximus and the anterior head of the biceps femoris was opened in both hind limbs to expose the sciatic nerves, but while the left one was crushed, the right one was left untouched. The crush was performed at 42 mm from the most distal toe three times during 15 s with Dumont #5/45 Forceps (F.S.T.). Graphite powder (Sigma-Aldrich) was used to mark the crush site at the third crush. Finally, musculature and skin were sutured with non-absorbable ethicon-coated vicryl 6–0 sutures (Johnson & Johnson) and mice were allowed to recover under a heating lamp. Mice were sacrificed with an overdose of Dolethal (20 mg/ml; Vetoquinol) 11 days after the surgery. Gastrocnemius and tibialis anterior muscles were dissected and snap-frozen in ice-cold isopentane. Innervation of NMJs was assessed as for the ALS mice.
RNAscope in situ hybridization
An overdose of Dolethal (20 mg/ml; Vetoquinol) was used to sacrifice ALS mice at 135 days of age. Mice were transcardially perfused with 4% paraformaldehyde (PFA). Spinal cords were then dissected, post-fixed overnight with 4% PFA, cryoprotected with 30% sucrose and embedded in OCT embedding matrix (CellPath) and stored at − 80 °C for later use. A CryoStar NX70 Cryostat (ThermoFischer Scientific) was used to cut 20 μm-thick cryosections, which were mounted on Superfrost Plus slides (ThermoFischer Scientific). To improve spinal cord slice attachment on the slides, slices were post-fixed during 30 min with cold 4% PFA (Life Technologies) at 4 °C, rinsed with PBS (Sigma-Aldrich), dehydrated with sequential incubation steps of 50, 70 and 100% ethanol, and baked for 30 min at 60 °C. RNAscope in situ hybridization was performed as indicated by the manufacturer with the RNAscope Multiplex Fluorescent Reagent Kit v2 (ACD Diagnostics). In brief, slides underwent an antigen retrieval step of 5 min at 98-104 °C with a Braun Multiquick FS-3000 Steamer (Braun) and a Protease III incubation step of 30 min at 40 °C in a HybEZ™ Oven (ACD Diagnostics). RNAscope probes against Efna5 (RNAscope Probe - Mm-Efna5-C1; ACD Diagnostics), Synaptophysin (Syp; 1:100; RNAscope Probe - Mm-Syp-C3; ACD Diagnostics) and Slc1a3 (1:50; RNAscope Probe - Mm-Slc1a3-C2) were used. Signal amplification was performed as stated in the manufacturer’s instructions with TSA Plus Fluorescein (1:750; Perkin Elmer), TSA Plus Cyanine 3 (1:500; Perkin Elmer) and TSA Plus Cyanine 5 (1:3000; Perkin Elmer). Nuclear counterstain was performed with Hoechst 33342 (5 μg/ml; Sigma) and slides were finally mounted with ProLong Gold antifade reagent (Life Technologies). Images of the ventral horns of every spinal cord section were obtained with a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems Heidelberg GmbH, Manheim, Germany) with an HC PL APO CS2 20x/0.75 dry lens and a pinhole of 0.5 Airy Units. Sum projection of 2 Z-stacks separated 2 μm from each other was done with the freeware ImageJ v1.51u by Wayne Rasband (National Institutes of Health) and images were next automatically quantified with the NIS-Elements Microscope Imaging Software (Nikon). Cells positive for Syp were detected based on intensity and designated as neurons. All detected nuclei that did not colocalize with Syp-positive cells were considered as glial nuclei, and a small perimeter (NIS-Elements thickening command, three iterations) around each nucleus was also selected to detect part of the cytoplasm of these cells. Glial nuclei with the small perimeter around them were considered as glial cells. Neurons were subdivided into large and small neurons according to the Syp-positive area: > 400 μm2 and 150–400 μm2, respectively, as previously described [15, 22, 42]. In accordance with Lalancette-Herbert et al., the cut-off of 400 μm2 used in our study differentiates alpha-motor neurons from gamma-motor neurons [22]. Although atrophy of alpha-motor neurons is present in SOD1G93A mice, such cut-off still allows differentiating both motor neuron subtypes in this mouse model [22]. Slc1a3 was used as an astrocyte marker. Neurons and glial cells containing at least two EfnA5 puncta or three Slc1a3 puncta were considered EfnA5 and/or Slc1a3 positive. Single-cell EfnA5 expression levels, were calculated as dot density in every cell analysed.
RNA extraction, quantitative PCR and digital droplet PCR
ALS mice were sacrificed at 130 days of age or at end-stage point of the disease with cervical dislocation and lumbar spinal cords were dissected and homogenized in TRIzol (ThermoFischer Scientific) with Lysing Matrix D beads (MP Biomedicals) and a MagNa Lyser oscillator (Roche) at 6500 rpm for 30 s thrice with a 1 min interval on ice. Total RNA was precipitated with isopropanol. cDNA was prepared with the SuperScript III First-Strand Synthesis System (ThermoFischer Scientific). Quantitative PCRs were performed with the TaqMan Fast Universal PCR Master Mix 2X (Life Technologies) and the following Taqman assays: Chat (Mm01221882_m1), Rbfox3 (Mm01248771_m1) and Aif (Mm00479862_g1) from Life technologies, and Efna5 (Mm.PT.58.28681125), Gfap (Mm.PT.58.10570926), Vimentin (Mm.PT.58.8720419), Polr2a (Mm.PT.58.13811327) and Gapdh (Mm.PT.39a.1) from IDT. Reaction was performed in a StepOnePlus instrument (Life Technologies) and relative gene expression was analysed with the Qbase+ software (Biogazelle). Digital droplet PCR was performed following Bio-Rad guidelines. PCR was done with ddPCR Supermix for Probes (No dUTPs; Bio-Rad) and with Efna5-FAM Taqman assays Mm.PT.58.28681125 (exon 1–2) and Mm.PT.58.29725953 (exon 3–5) from IDT, multiplexing each of them with Polr2a-HEX labelled Taqman assay Mm.PT.58.13811327 (Integrated DNA Technologies). Droplets were generated in a QX200 Droplet Generator and scanned with a QX200 Droplet Reader (Bio-Rad). Results were analysed with QuantaSoft software from Bio-Rad.
Label-free proteomic analysis of tissue samples
Lumbar spinal cord samples were obtained from SOD1WT, SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice at 130 days of age after cervical dislocation, and were lysed by addition of lysis buffer containing 6 M guanidine hydrochloride, 10 mM Tris (2-carboxyethyl)phosphine hydrochloride (TCEP), 40 mM 2-chloroacetamide and 100 mM triethylammonium bicarbonate (TEAB). Samples were then sonicated, heated at 95 °C for 10 min and centrifuged at 20000 g for 30 min at 4 °C, and the supernatant was collected for further analysis. Protein concentration was determined using the Qubit Fluorometric Quantification Kit (Thermo Scientific). Protein digestion was performed with a filter-aided sample preparation (FASP) protocol and analysed by LC-MS/MS as previously described [4]. Proteins were identified using MaxQuant 1.5.2.8 and the Mus musculus reference proteome from UniProt (downloaded 12th March 2018). A false discovery rate (FDR) of 1% was used for peptide and protein identification (total protein IDs after exclusion of contaminants: 5525) and protein quantification was performed with the MaxLFQ algorithm [8]. Quantitative analysis of the data was performed with Perseus 1.5.2.6 [40]. Proteins with valid values in at least three samples per group were used for the quantitative comparison (4065 proteins).
Patients and cerebrospinal fluid collection
Patients (Table 1) were enrolled at the Ulm University Hospital, Department of Neurology, where cerebrospinal fluid (CSF) was collected by lumbar puncture at diagnostic follow-up (except asymptomatic ALS gene carriers and non-carriers, see below). Samples were centrifuged and stored at − 80 °C within 2 h according to standard operating procedures. Control patients had no neurodegenerative disease and CSF was collected to rule out acute or chronic inflammation of the brain. Asymptomatic ALS gene carriers and their relatives without known ALS mutation were recruited via the German Presymptomatic-ALS cohort [32]. ALS was diagnosed according to Ludolph et al. [25] and disease onset was determined by interviewing of the patients. All patients or their relatives gave written informed consent to participate in the study, and the collection and analysis of CSF samples during the diagnostic pathway was approved by the Ethics Committee of Ulm University.
Table 1.
Table showing the characteristics of the humansamples analysed in the present study. The total number (N) ofsamples and the median age with its interquartile range areshown for every group: Young and aged controls (Con),asymptomatic ALS patients (Asympt.), patients carrying a knowngenetic mutation (gALS) and patients without any identifiedgenetic mutation (sALS)
| Con (young) | Con (aged) | Asympt. ALS | gALS | sALS | |
|---|---|---|---|---|---|
| N (f/m) | 24 (14/10) | 32 (13/19) | 21 (14/7) | 38 (14/24) | 70 (25/45) |
| Age (yr) | 42 (31-50) | 63 (57-72) | 45 (35-51) | 59 (51-67) | 65 (56-70) |
Multiple reaction monitoring of CSF samples
For multiple reaction monitoring (MRM) analysis of efnA5 in CSF samples, 200 μL of CSF were spiked with TEAB buffer and a quantitative protein epitope signature tag (QPrEST, Atlas Antibodies AB, #QPrEST25042) of efnA5 as internal standard. Samples were reduced and alkylated with 1 mM TCEP and 1 mM CAA at 95 °C for 10 min. Proteins were then digested for 16 h at 37 °C by adding 1.2 μg trypsin/LysC (Promega). Digestion was stopped by addition of 800 μL 1.25% TFA and peptides were transferred to strong cation exchange STAGE-Tips by centrifugation as previously described [31]. Peptides were washed with 0.2% TFA and fractionated with 75, 125, 200, 300, 450 mM ammonium acetate in 20% acetonitrile/0.5% formic acid (fraction 1–5) and 5% ammonium hydroxide/80% acetonitrile (fraction 6). After vacuum drying, peptides were dissolved in 27.5 μL 6% acetonitrile/0.1%TFA and analysed by LC-MRM.
Analysis of efnA5 was performed with a QTRAP6500 mass spectrometer (AB Sciex), Eksigent MicroLC200 and Agilent 1260 HPLC pump. The fractions were loaded on a C18 PepMap100, 5 μm, 0.3 × 5 mm trap column (Thermo Fisher Scientific). Separation was performed on an Eksigent HALO Fused-core C18, 2.7 μm, 0.5 × 100 mm column at 40 °C with mobile phase A: 4% DMSO/0.1% formic acid, and mobile phase B: 4% DMSO/96% acetonitrile/0.1% formic acid and a linear gradient from 1 to 30% B within 9.85 min. The following transitions of efnA5 (peptide TIGVHDR) were measured: 399.2–583.3 (y5), 399.2–427.2 (y3) (light peptide); 404.2–593.3 (y5), 404.2–437.2 (y3) (heavy peptide). For relative quantification, the light-to-heavy (L/H) peptide ratio (mean of the two transitions) was calculated using Skyline v4.2. CSF quality control (QC) samples were included in each run.
Statistics
Statistics were performed using Graphpad prism 7.01 software (Graphad Software), except for the analysis of the proteomic data (see below). Unpaired two-tailed t-test, one-way ANOVA followed by the Dunnett’s multiple comparisons posthoc test, two-way ANOVA with repeated measurements followed by the Sidak’s multiple comparisons posthoc test and two-way ANOVA followed by the Tukey’s multiple comparisons posthoc test, were used to determine differences among conditions as stated in the figure legends. Survival and disease onset were analysed using the Log-rank test if data showed a parametric distribution. For non-parametric data, a two-tailed Mann-Whitney test was performed. Statistical analysis of proteomic data was performed with Perseus 1.5.2.6. Protein levels between groups were compared by two-tailed Student’s t-test applying an FDR of 5% and S0 = 0.1 to correct for multiple testing. Data are visualized by Volcano plots. For efnA5 protein determination in CSF samples, groups were compared by Kruskal-Wallis test and Dunn’s post hoc test. A p-value < 0.05 was regarded as significant. For all the tests, significance level was considered for p values lower than 0.05.
Based on previous experiments in SOD1G93A mice, we estimated that a sample size of 32 mice in each group would provide 80% power at an α = 0.05 to detect an extended survival of 11 days, as previously found for EphA4+/− SOD1G93A mice [42]. For the sciatic nerve crush experiments, we estimated a sample size of 4–6 mice per group to have an 80% power at α = 0.05 to detect a difference of 24% in re-innervation as previously observed for EphA4−/− mice [42]. For these experiments we used 5 mice per group.
Results
Ephrin-A5 is mainly expressed in neurons of the ventral horn
We first determined EfnA5 gene expression in whole lysates of adult mice lumbar spinal cord in age-matched SOD1WT and mutant SOD1G93A mice, at different disease stages (pre-symptomatic, onset, and early and late-symptomatic). EfnA5 mRNA levels were lower in mutant SOD1G93A mice compared to SOD1WT (Fig. 1a). We also quantified the amount of efnA5 protein with MRM in CSF from ALS patients and controls. No difference in the protein amount was detected when we compared aged and young control samples, and no differences were observed when we compared control, non-symptomatic and symptomatic ALS samples (Fig. 1b).
Fig. 1.

EfnA5 gene expression levels are reduced in SOD1G93A mice whereas efnA5 protein is not altered in ALS patients CSF. a EfnA5 gene expression was determined in the lumbar spinal cord of SOD1WT and SOD1G93A overexpressing mice (N = 4–9). Data represents mean ± SEM, and different conditions were compared with a two-way ANOVA with Tukey’s multiple comparisons test: ** P < 0.01 as compared with SOD1WT. b EfnA5 protein relative quantification in the CSF of patients carrying an ALS genetic known mutation (gALS) and patients without any known mutation (sALS) were compared to the levels found in the CSF of young and aged controls (Con) and asymptomatic ALS patients carrying a known mutation for the disease (asympt ALS). Boxes show median and interquartile range, and whiskers are minimum and maximum. The light-to-heavy (L/H) peptide ratio was used as relative quantification of EfnA5 protein
To determine cell type specific expression of efnA5 and potential single-cell changes related to ALS, we performed RNAscope in situ hybridizations in lumbar spinal cord slices of SOD1WT and SOD1G93A symptomatic mice at 135 days of age and examined EfnA5 gene expression in the ventral horn in neurons and in glial cells (Fig. 2a). Since in ALS different vulnerability has been observed in neurons of different soma size [42], EfnA5 expression was evaluated in the most vulnerable, large, (> 400 μm2) and small (150–400 μm2) neurons. In SOD1WT spinal cords, more than 92% of the neurons (Syp positive cells) expressed EfnA5 (Fig. 2b). In contrast, only 8% of glial cells (Syp negative cells) and 4% of astrocytes (Syp negative, Slc1a3 positive cells) expressed EfnA5, indicating that in the spinal cord EfnA5 is mainly expressed in neurons and minimally in astrocytes (Fig. 2c, d). In the SOD1G93A spinal cord the percentage of EfnA5 positive large and small neurons and astrocytes was similar to SOD1WT, but we observed a reduction of 2% of total glial cells expressing EfnA5 (Fig. 2b-d). We next determined differential expression of EfnA5 in neurons and glia at a single cell level. EfnA5 expression was higher in the large compared to the small neurons in SOD1WT mice (Fig. 2e), and we observed a reduction of EfnA5 in individual large neurons of SOD1G93A compared to SOD1WT mice (Fig. 2e), as well as in glial cells including astrocytes (Fig. 2f, g).
Fig. 2.

In the ventral horn of the spinal cord EfnA5 is mainly expressed in neurons. RNAscope in situ hybridization was performed to determine cell type-specific expression of EfnA5. A total of 15–18 images of different lumbar spinal cord ventral horns from 3 different mice were analysed for every condition. a Representative images show lumbar spinal cord ventral horns of SOD1WT and SOD1G93A mice at 135 days of age that were stained with probes against EfnA5, Syp and Slc1a3. Hoechst was used as a counter stain for nuclei. Scale bar = 50 μm. b Percentage of neurons (Syp + cells) that stained positive for EfnA5 were quantified and shown as percentage of the total Syp + cells. c Glial cells (Syp- cells) and d astrocytes (Syp-, Slc1a3+ cells) positive for EfnA5 were quantified and represented as percentage of the total amount of glial cells. Single-cell EfnA5 expression was quantified by measuring EfnA5 puncta density (dots/μm2) in e neurons, f glial cells and g astrocytes. Data represents mean ± SEM, and different conditions were compared with a two-tailed t-test: * P < 0.05; ** P < 0.01 as compared to SOD1WT. b and e Data represents mean ± SEM, and different conditions were compared with a two-way ANOVA with Tukey’s multiple comparisons test: * P < 0.05 as compared with SOD1WT; ## P < 0.01 as compared with 150–400 μm2 neurons
Reduction of ephrin-A5 levels accelerates disease progression in SOD1G93A mice
In contrast to a stroke model [33], EfnA5 was minimally expressed in astrocytes. Similarly to EphA4 [42], EfnA5 expression was higher in the large and most vulnerable motor neurons, and therefore, we hypothesized that this higher expression could contribute to mechanisms that drive to the selective retraction and vulnerability of large motor neurons in ALS [14, 35]. To study disease progression in SOD1G93A mice with lower efnA5, we reduced EfnA5 expression by crossbreeding SOD1G93A mice with a constitutive EfnA5 knockout mouse [16]. Neurodevelopmental axon miswiring alterations have been reported in EfnA5−/− mice [2, 9, 16, 21], but EfnA5+/− and EfnA5−/− adult mice had similar amounts of motor neurons in the ventral horn of the lumbar spinal cord and innervated NMJs in the gastrocnemius muscle as compared to EfnA5+/+ mice (Additional file 1: Figure S1). Since EfnA5−/− mice present with embryonic lethality when they are bred in a C57BL/6 J background [38], we crossbred SOD1G93A mice to EfnA5+/− mice and thus reduced efnA5 levels to 50%. SOD1G93A EfnA5+/− mice had comparable body weight, as assessed from 60 days until mice reached an end-stage point of the disease (Fig. 3a). Disease onset was unaltered and motor performance in both hanging wire and rotarod tests of SOD1G93A EfnA5+/− mice progressively declined in a similar manner as SOD1G93A EfnA5+/+ mice (Fig. 3b-d). However, mice reached disease end-stage faster than their littermate controls, with a median survival reduction of 17 days (Fig. 3e), which was also reflected in a shorter disease duration (25 days in SOD1G93A EfnA5+/− compared to 40 days in SOD1G93A EfnA5+/+) (Fig. 3f). We excluded compensatory mechanisms potentially resulting in upregulation of EfnA5 gene expression by confirming a 50% reduction in EfnA5 expression levels at end-stage in SOD1G93A EfnA5+/− compared to SOD1G93A EfnA5+/+ (Fig. 3g). Since we observed an accelerated disease progression instead of an extended survival, we stopped the experiment with 26 and 28 mice per group.
Fig. 3.

Reduction of efnA5 levels in SOD1G93A mice is detrimental and shortens survival and disease duration. Disease progression was closely monitored in a-f SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice from the age of 60 days onwards (N = 26–28). a Weight and b and c motor performance decline over time on b the hanging wire (HW) and c rotarod tests is shown. Data is represented as mean ± SEM and two-way ANOVA test with repeated measurements was used to determine differences among genotypes. d Disease onset differences were compared between the two genotypes with the Log-rank test whereas e survival was analysed with the two-tailed Mann-Whitney test for non-parametric data: * P < 0.05. f Disease duration was analysed with a two-tailed student t-test: * P < 0.05. g EfnA5 expression levels in the lumbar spinal cord of end-stage mice were determined by qPCR and data is represented as mean ± SEM (N = 7–9). Different genotypes were compared with a one-way ANOVA with Tukey’s multiple comparisons test: *** P < 0.001
Reduction of ephrin-A5 levels does not alter electrophysiological and histological findings at a symptomatic stage of the disease
We next examined electrophysiological and histological alterations. For this purpose, a group of SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice were followed from 60 days until 115 days for electrophysiological recordings. When mice became symptomatic, we analysed the number of motor neurons in the spinal cord and the percentage of innervated NMJs in the gastrocnemius muscle. The decrease of the CMAP amplitudes progressed similarly in both experimental groups until the last time point measured (Fig. 4a). The number of remaining motor neurons in the ventral horn of the lumbar spinal cord was comparable in both SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice (Fig. 4b). In agreement with this result a similar reduction could be observed in the expression levels of the neuron and motor neuron marker genes Rbfox3 and Chat in the lumbar spinal cord of SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice as compared to SOD1WT mice at the age of 130 days (Fig. 4c, d). The percentage of innervated NMJs in the gastrocnemius muscle was also similar in both genotypes (Fig. 4e).
Fig. 4.
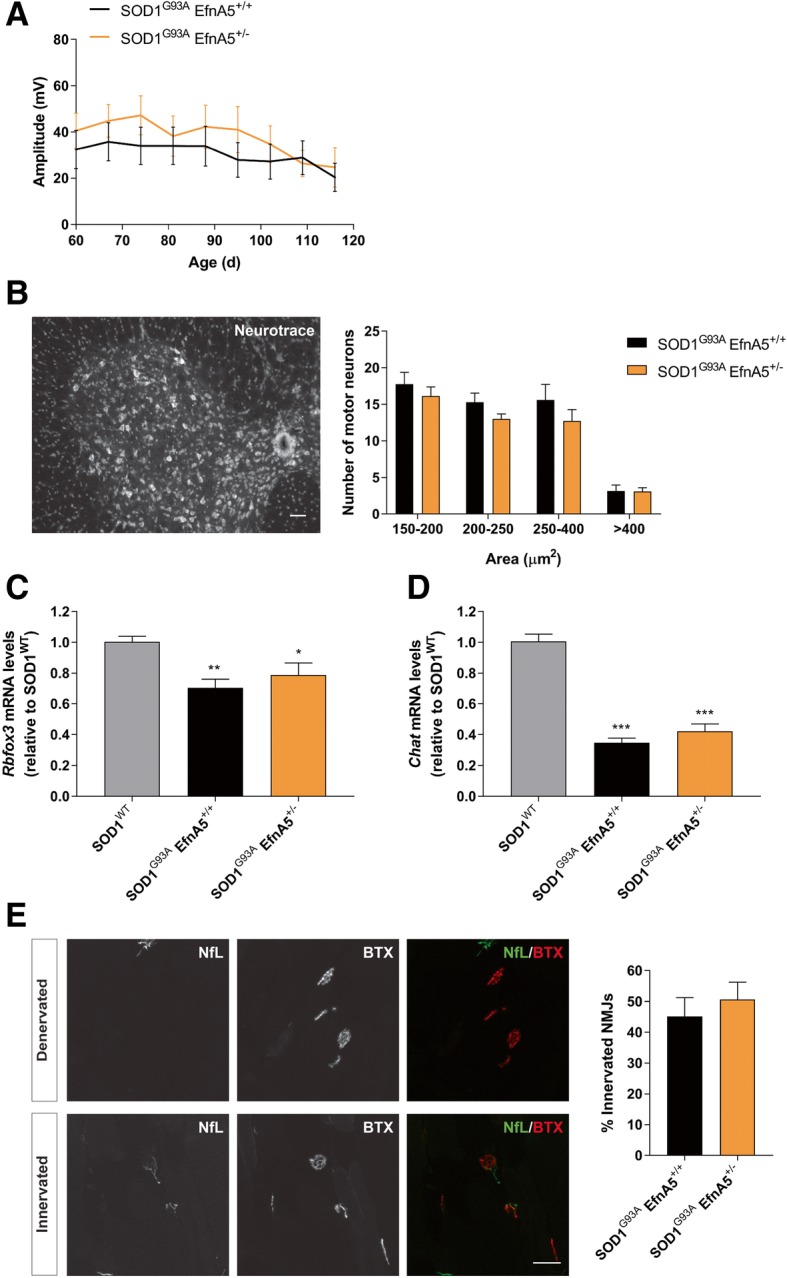
Electromyography decline, neuronal loss and neuromuscular junction denervation in SOD1G93A mice is not affected in SOD1G93A mice with reduced efnA5 levels. a Compound muscle action potential amplitude (expressed in mV) was measured in SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice from the age of 60 days (d) onwards. Data is represented as as mean ± SEM (N = 7–8). b The number of motor neurons of different soma area was counted in the ventral horn of the lumbar spinal cord. Data is represented as mean ± SEM (N = 6–7) and it was analysed with a two-way ANOVA. Representative image shows Neurotrace staining of a spinal cord slice. Scale bar = 50 μm. c and d Expression levels of c Rbfox3 and d Chat were unaltered after efnA5 knockdown in mice at 130 days of age. Data represents mean ± SEM (N = 6–7), and different conditions were compared with a one-way ANOVA with Tukey’s multiple comparisons test: * P < 0.05; ** P < 0.01; *** P < 0.001 as compared with SOD1WT mice. e Neuromuscular junctions (NMJs) innervation was scored in the gastrocnemius muscle. Data represents mean ± SEM (N = 5–6) and it was analysed with a two-tailed t-test. Representative images are shown of denervated and innervated NMJs in the gastrocnemius muscle that were stained with neurofilament-L (NfL) and α-bungarotoxin (BTX). Scale bar = 50 μm
Ephrin-A5 reduction does not affect gene expression of glial markers nor the overall proteome
In ALS, non-cell autonomous mechanisms contribute to the selective motor neuron death [1, 19, 45]. Therefore, we analysed the expression in spinal cord tissue from mice at 130 days of age of several genes involved in glial function, which are altered in ALS, and compared their expression between SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice, to find a potential mechanism for the worsening of disease progression. The increase in the microglial marker Aif and the reactive astrocyte markers Gfap and Vimentin was analogous in SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice compared to SOD1WT mice (Fig. 5a-c). Next, we performed a proteomic analysis in spinal cord samples from the same mice to detect alterations in any neuronal or glial molecular pathway in an unbiased way. We detected a total of 4065 proteins for the comparative analysis between the groups. Alterations in 817 proteins were altered in SOD1G93A mice compared to SOD1WT mice, but no differences could be detected between SOD1G93A EfnA5+/− and SOD1G93A EfnA5+/+ mice (Fig. 6a, b).
Fig. 5.

Reduction of efnA5 levels does not alter glial gene expression in symptomatic mice. Gene expression of several glial genes was quantified in the lumbar spinal cord of SOD1WT, SOD1G93A EfnA5+/+ and SOD1G93A EfnA5+/− mice. a Aif, b Gfap, and c Vimentin levels were unaltered in SOD1G93A EfnA5+/− mice. Data represents mean ± SEM (N = 6–7), and different conditions were compared with a one-way ANOVA with Tukey’s multiple comparisons test: * P < 0.05; ** P < 0.01; *** P < 0.001 as compared with SOD1WT mice
Fig. 6.

Whole proteome is not altered in the lumbar spinal cord of SOD1G93A EfnA5+/− compared to SOD1G93A EfnA5+/+ mice at 130 days of age. Differences at a proteomic level were determined for a SOD1G93A EfnA5+/+ versus SOD1WT, and for b SOD1G93A EfnA5+/− versus SOD1G93A EfnA5+/+ mice. Data is represented in Volcano plots and the black lines indicate the significance level based on an FDR of 5% and S0 = 0.1 to correct for multiple testing
Similar re-innervation status after sciatic nerve injury upon efnA5 reduction
Although we did not find evidence for an increase in re-sprouting upon a 50% reduction of efnA5 expression in ALS mice, we examined whether a further reduction of efnA5 levels would affect the re-innervation potential in an acute nerve injury model. We crushed sciatic nerves of EfnA5+/+ and EfnA5−/− mice (bred in a mixed background to prevent embryonic lethality) and evaluated re-innervation. We assessed re-innervation of NMJs 11 days after the surgery in two different muscles of the lower limb, the gastrocnemius and the tibialis anterior muscles. The ipsilateral limb showed a profound denervation and we did not observe any difference between EfnA5+/+ and EfnA5−/− mice in the percentage of re-innervated NMJs (Fig. 7a, b), suggesting that efnA5 is not involved in axonal regeneration in acute neuronal crush injury.
Fig. 7.

Axonal regeneration is not enhanced in EfnA5 knockout mice after sciatic nerve crush. Percentage of re-innervated neuromuscular junctions (NMJs) was determined in the a gastrocnemius and in the b tibialis anterior muscles in the ipsilateral and contralateral limbs of EfnA5+/+ and EfnA5−/− mice 11 days after the crush (N = 5–5). Data represents mean ± SEM, and different conditions were compared with a two-tailed t-test. Representative images of ipsilateral and contralateral gastrocnemius and tibialis anterior muscles that were stained with neurofilament-L (NfL) and α-bungarotoxin (BTX) are shown. Scale bar = 50 μm
Lower levels of efnA5 in the cerebrospinal fluid of patients associates with faster disease progression
To evaluate if the findings observed in the mouse model for ALS would translate to the phenotypic expression of the disease in humans we studied efnA5 protein in the CSF of ALS patients. We correlated efnA5 levels in the CSF with disease characteristics in ALS, by comparing patients with efnA5 protein expression below and above the median value in the ALS cohort. Disease onset was similar in these two subgroups, comparable to our observations in the mouse model. However, disease progression was more severe in patients with lower levels of efnA5 as reflected by shorter disease duration, also analogous to our results in the mouse model (Fig. 8a, b).
Fig. 8.

Disease progression is more severe in patients with lower CSF efnA5 protein levels. a Disease onset and b disease progression were determined in patients with CSF efnA5 levels above the median and in patients with CSF efnA5 levels below the median (N = 32). Differences were compared between the two genotypes with the Log-rank test: ** p < 0.01
Discussion
Previously we identified EphA4 as a disease modifier of ALS [42]. Here we studied the contribution of one EphA4 ligand, efnA5, in the disease progression of a mouse model of ALS. We observed that in the ventral horn of SOD1G93A mouse spinal cord, efnA5 was predominantly present in almost all neurons and not in glial cells. Heterozygous deletion of efnA5 aggravated disease progression without affecting onset nor disease-specific glial and neuronal pathways. In an acute neuronal injury model the sprouting capacity of neurons did not improve in the absence of efnA5. Finally, patients with lower efnA5 levels had faster disease progression. Our findings indicate that reduced levels of efnA5 aggravate the clinical presentation of ALS and add to the evidence that altering ephrin signalling modifies the disease course in both mouse models and humans.
We selected efnA5 as target ligand, since in another neuronal injury model, stroke, evidence exists for upregulation of efnA5 in astrocytes, which reduced axonal regeneration after injury [33]. Moreover, the efnA5 gene contains conserved NF-κB binding sites, which makes it a direct downstream target gene of the pro-inflammatory NF-κB pathway [17]. The NF- κB pathway is activated in glial cells isolated from ALS patients and SOD1G93A mice [13, 18], and there is substantial evidence on the contributing role for astrocytes, microglia and oligodendrocytes in the selective vulnerability of motor neurons in ALS [1, 19, 45]. We revealed predominant expression of efnA5 in neurons and minimally in glial cells in the adult mouse spinal cord, with no upregulation in mice overexpressing SOD1G93A. Several mechanisms could account for this lack of efnA5 expression in astrocytes. Despite the activation of the NF-κB pathway in astrocytes and other glial cells [13, 18], various gene expression regulatory elements in those specific cell-types could interfere with efnA5 upregulation. In addition, reactive astrocytes are also very heterogeneous, and depending on the insult, their transcriptional profile differs [24, 46]. As an example, stroke-induced reactive astrocytes upregulate neurotrophic factors and seem to acquire neuroprotective properties, whereas astrocytes activated by lipopolysaccharide enhance complement cascade genes, which can be harmful for synapses [24, 46]. ALS reactive astrocytes acquire a phenotype more similar to that upon induction by lipopolysaccharide, which may not be accompanied with efnA5 upregulation [18, 24].
Within the spinal cord different types of lower motor neurons exhibit diverse vulnerability. The largest fast-twitch fast-fatigable (FF) alpha-motor neurons are the first to degenerate in ALS mouse models and patients, and they are progressively followed by the smaller fast-twitch fatigue-resistant (FR) and later on by the slow (S) motor neurons [14, 35, 37]. In contrast, gamma-motor neurons, which are the smallest in size, are relatively preserved during the progression of the disease [22]. We previously identified EphA4 as a susceptibility factor contributing to the specific motor neuron vulnerability [42]. Similarly, we found in control mice the highest levels of efnA5 in the large motor neurons, and therefore we assessed the role of efnA5 levels in motor neuron vulnerability. At 135 days efnA5 expression in SOD1G93A mice was reduced in these motor neurons compared to SOD1WT mice, and by lowering efnA5 levels in the SOD1G93A EfnA5+/− mice disease progression was aggravated. Combining these findings we hypothesize that in contrast to EphA4, the vulnerability of motor neurons increases upon reduction of efnA5. However, the number of motor neurons did not differ in symptomatic SOD1G93A mice with normal or reduced efnA5 levels. Moreover, we could not observe any difference in motor neuron molecular pathways that would indicate further neuronal dysfunction in mice with reduced efnA5 levels. Since onset was not altered in these mice but disease progression and survival were aggravated, reduced levels of efnA5 may disrupt neuronal integrity at very late stages of the disease. In addition, during ALS disease progression, remaining intact motor neurons have the capacity to re-sprout to compensate for the denervation of NMJs [14]. Interestingly, during development, neuronal efnA5 promotes the growth of LMCl motor axons that project to the dorsal limb [3, 23]. Therefore, we studied the sprouting capacity in an acute model for neuronal injury by crushing the sciatic nerve in control mice and in those lacking efnA5. However, we did not find differences in re-innervation in EfnA5−/− mice. The recovery after the injury was similar in both genotypes, indicating that even a complete absence of efnA5 did not alter neuronal re-sprouting. In addition, innervation of NMJs at the gastrocnemius muscle at least just after onset was also not altered in SOD1G93A mice with reduced efnA5 levels. These results suggest that, in contrast to developmental stages when efnA5 plays a role in axonal sprouting, in an adult-onset disease, efnA5 reduction does not alter retraction of motor axons from the muscles.
We initially considered efnA5 localized in motor neuron neighbouring cells such as astrocytes as a possible binding ligand of EphA4. However, we observed predominant neuronal efnA5 expression. Reducing efnA5 expression with 50% accelerated disease progression, whereas heterozygous deletion of EphA4 gene increased survival in SOD1G93A mice [42]. Although highly speculative efnA5 and EphA4 expressed in the same motor neurons could establish inhibitory cis-binding. The absence of efnA5 may increase EphA4 signalling, resulting in shorter survival in mice. Inhibitory cis-binding has already been reported for efnA5 in retinal axons, where cis-binding of efnA5 with EphA3 desensitizes EphA3-mediated signalling [7]. Future research could be focussed on further elucidating this hypothesis and considering the implications for other ephrin members of the family. In addition, efnA5 is a ligand of most EphA receptors [30]. Since the interaction of Ephs and ephrins mediate intercellular communication [20, 34], another hypothesis would be that efnA5 mediates neuroprotection by binding in trans to other EphA receptors expressed in other cell types surrounding neurons. Further work will address whether these other Eph receptors might have a role in ALS. Interestingly, a neuroprotective response has been suggested for the neuronal EphB1, which interacts with the astrocytic ephrin-B1 to induce a neuroprotective phenotype in astrocytes [41].
In the pathophysiology of ALS various cell types acting in concert results in motor neuron death. Therefore, we studied alterations in various neuronal and glial markers in ALS mice with reduced efnA5 levels. However, we did not detect alterations in glial cell markers in the lumbar spinal cord of SOD1G93A EfnA5+/− mice when compared to SOD1G93A EfnA5+/+ mice. In an unbiased proteomics approach, we found several up- and down-regulated proteins in late-symptomatic SOD1G93A mice when compared to age-matched SOD1WT mice, but we failed to detect any protein or molecular pathway in the spinal cord that was altered in SOD1G93A EfnA5+/− mice when compared to SOD1G93A EfnA5+/+ mice, that could explain the observed modifying effect of efnA5. A limitation of our study is the fact that we performed a general efnA5 knockdown, instead of a nervous system specific knockdown. Further work and the use of conditional strategies to knockdown efnA5 in specific cell types and tissues will allow determination of cell types involved in the modifying role of efnA5 in ALS.
Conclusions
Reducing levels of efnA5 aggravates disease progression without modifying the disease onset in both an ALS mouse model as well as humans. Since efnA5 expression is mainly neuronal we hypothesize a decrease in neuronal integrity in the later stages of the disease. Our future understanding of the exact mechanism that drives this accelerated disease phenotype, may gain more insights in the complexity of Eph-ephrin binding in motor neuron diseases.
Additional file
{kind=link}
Figure S1. Reduction of efnA5 levels does not alter the numbers of motor neurons or innervated neuromuscular junctions in a non-ALS context. (a) Motor neurons were counted in the ventral horn of the lumbar spinal cord in EfnA5+/+, EfnA5+/− and EfnA5−/− mice. Data is represented as mean ± SEM (N = 4) and it was analysed with a two-way ANOVA. (b) Innervated neuromuscular junctions (NMJs) were scored in the gastrocnemius muscle of the same mice. Data represents mean ± SEM (N = 4) and it was analysed with a one-way ANOVA. (JPG 1900 kb)
Acknowledgements
The authors thank Dr. Renping Zhou (Rutgers University, Piscataway, NJ, USA) and Dr. Jonas Frisén (Karolinska Institute, Stockholm, Sweden) for providing the ephrin-A5 knockout mice, Begga Schevenels and Séraphina Penninckx for contributing in the maintenance of the mouse colonies and in genotyping and Kathrin Müller and Antje Kneer for performing genetic testing of patients.
Abbreviations
- ALS
Amyotrophic Lateral Sclerosis
- BTX
α-bungarotoxin
- CMAP
Compound muscle action potential
- CSF
Cerebrospinal fluid
- efnA5
Ephrin-A5
- FF
Fast-twitch fast-fatigable
- FR
Fast-twitch fatigue resistant
- MRM
Multiple reaction monitoring
- NfL
Neurofilament-L
- NMJ
Neuromuscular junction
- PFA
Paraformaldehyde
- S
Slow
- SOD1
Copper-zinc superoxide dismutase
Authors’ contributions
LR performed and coordinated all experiments, analysed the data and wrote the manuscript. PÖ performed multiple reaction monitoring of CSF samples and proteomics in mouse spinal cord samples, and analysed the data. MT, AL, JvdV, SS, LP and AdB performed some experiments. JHW performed genetic testing of patients and ACL collected and provided CSF samples. MO supervised the multiple reaction monitoring and proteomics experiments. LVDB, PVD, WR and RL supervised the project. RL supervised and wrote the manuscript. All authors contributed to the final manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by grants from VIB, Fund for scientific research Flanders (FWO; grant number G.0996.14 N), the ALS League Belgium, the Thierry Latran Foundation (EphrALS; grant number FTL AAP2013), the ALS Association (grant number 039CYK) and the Geneeskundige Stichting Koningin Elisabeth (G.S.K.E.). This work was also supported by the European Research Council and the European’s Seventh Framework Programme (FP7/2007–2013)/ERC Grant (grant number 340429). LR was awarded with a postdoctoral fellowship by the Fund Alfonso Martín Escudero, and AdB was supported by the Fund for Scientific Research Flanders (FWO; 1136917 N). MO, PÖ, ACL were supported by grants from the German Federal Ministry of Education and Research (project: FTLDc 01GI1007A, MND-Net 01GI0704), the EU Joint Programme - Neurodegenerative Disease Research (JPND) network, PreFrontAls (01ED1512), the foundation of the state of Baden-Württemberg (D.3830), the Thierry Latran Foundation, BIU (D.5009) and the ALS association. LVDB, PVD and WR are supported by the ALS League Belgium and the Fund ‘Opening the Future’. PVD and WR are supported by the Laevers Fund for ALS Research and the Fund ‘Een Hart voor ALS’. WR is also supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders and the European Research Council under the European’s Seventh Framework Programme (FP7/2007–2013). PVD and RL hold a clinical investigatorship of FWO-Vlaanderen. Funding for the Open Access publication charges of this article was provided by the European Commission.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author upon request.
Ethics approval and consent to participate
All animal experiments were approved by the local ethical committee of the KU Leuven (P229/2013 and P229/2017), and were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments.
All patients or their relatives gave written informed consent to participate in the study, and the Ethics Committee of Ulm University gave approval for the collection and analysis of cerebrospinal fluid samples.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 2.Bolz J, Uziel D, Muhlfriedel S, Gullmar A, Peuckert C, Zarbalis K, Wurst W, Torii M, Levitt P. Multiple roles of ephrins during the formation of thalamocortical projections: maps and more. J Neurobiol. 2004;59:82–94. doi: 10.1002/neu.10346. [DOI] [PubMed] [Google Scholar]
- 3.Bonanomi D, Chivatakarn O, Bai G, Abdesselem H, Lettieri K, Marquardt T, Pierchala BA, Pfaff SL. Ret is a multifunctional coreceptor that integrates diffusible- and contact-axon guidance signals. Cell. 2012;148:568–582. doi: 10.1016/j.cell.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockmann SJ, Freischmidt A, Oeckl P, Muller K, Ponna SK, Helferich AM, Paone C, Reinders J, Kojer K, Orth M, Jokela M, Auranen M, Udd B, Hermann A, Danzer KM, Lichtner P, Walther P, Ludolph AC, Andersen PM, Otto M, Kursula P, Just S, Weishaupt JH. CHCHD10 mutations p.R15L and p.G66V cause motoneuron disease by haploinsufficiency. Hum Mol Genet. 2018;27:706–715. doi: 10.1093/hmg/ddx436. [DOI] [PubMed] [Google Scholar]
- 5.Brown RH, Jr, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602. doi: 10.1056/NEJMc1710379. [DOI] [PubMed] [Google Scholar]
- 6.Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193:291–311. doi: 10.1016/j.expneurol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Carvalho RF, Beutler M, Marler KJ, Knoll B, Becker-Barroso E, Heintzmann R, Ng T, Drescher U. Silencing of EphA3 through a cis interaction with ephrinA5. Nat Neurosci. 2006;9:322–330. doi: 10.1038/nn1655. [DOI] [PubMed] [Google Scholar]
- 8.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Defourny J, Poirrier AL, Lallemend F, Mateo Sanchez S, Neef J, Vanderhaeghen P, Soriano E, Peuckert C, Kullander K, Fritzsch B, Nguyen L, Moonen G, Moser T, Malgrange B. Ephrin-A5/EphA4 signalling controls specific afferent targeting to cochlear hair cells. Nat Commun. 2013;4:1438. doi: 10.1038/ncomms2445. [DOI] [PubMed] [Google Scholar]
- 10.Donoghue MJ, Lewis RM, Merlie JP, Sanes JR. The Eph kinase ligand AL-1 is expressed by rostral muscles and inhibits outgrowth from caudal neurons. Mol Cell Neurosci. 1996;8:185–198. doi: 10.1006/mcne.1996.0056. [DOI] [PubMed] [Google Scholar]
- 11.Drescher U, Kremoser C, Handwerker C, Loschinger J, Noda M, Bonhoeffer F. In vitro guidance of retinal ganglion cell axons by RAGS, a 25 kDa tectal protein related to ligands for Eph receptor tyrosine kinases. Cell. 1995;82:359–370. doi: 10.1016/0092-8674(95)90425-5. [DOI] [PubMed] [Google Scholar]
- 12.Fogh I, Rijsdijk F, Andersen PM, Sham PC, Knight J, Neale B, McKenna-Yasek D, Silani V, Brown RH, Jr, Powell JF, Al-Chalabi A. Age at onset in sod1-mediated amyotrophic lateral sclerosis shows familiality. Neurogenetics. 2007;8:235–236. doi: 10.1007/s10048-007-0092-2. [DOI] [PubMed] [Google Scholar]
- 13.Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi: 10.1016/j.neuron.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci U S A. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frisen J, Yates PA, McLaughlin T, Friedman GC, O'Leary DD, Barbacid M. Ephrin-A5 (AL-1/RAGS) is essential for proper retinal axon guidance and topographic mapping in the mammalian visual system. Neuron. 1998;20:235–243. doi: 10.1016/S0896-6273(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 17.Gu JM, Wang DJ, Peterson JM, Shintaku J, Liyanarachchi S, Coppola V, Frakes AE, Kaspar BK, Cornelison DD, Guttridge DC. An NF-kappaB--EphrinA5-dependent communication between NG2(+) interstitial cells and myoblasts promotes muscle growth in neonates. Dev Cell. 2016;36:215–224. doi: 10.1016/j.devcel.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–256. doi: 10.1038/nrm.2015.16. [DOI] [PubMed] [Google Scholar]
- 21.Knoll B, Zarbalis K, Wurst W, Drescher U. A role for the EphA family in the topographic targeting of vomeronasal axons. Development. 2001;128:895–906. doi: 10.1242/dev.128.6.895. [DOI] [PubMed] [Google Scholar]
- 22.Lalancette-Hebert M, Sharma A, Lyashchenko AK, Shneider NA. Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc Natl Acad Sci U S A. 2016;113:E8316–E8325. doi: 10.1073/pnas.1605210113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Carmichael ST. Growth-associated gene and protein expression in the region of axonal sprouting in the aged brain after stroke. Neurobiol Dis. 2006;23:362–373. doi: 10.1016/j.nbd.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, Shefner J, WFNRGO ALS/MND. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:291–292. doi: 10.3109/21678421.2015.1049183. [DOI] [PubMed] [Google Scholar]
- 26.Ludolph AC, Bendotti C, Blaugrund E, Chio A, Greensmith L, Loeffler JP, Mead R, Niessen HG, Petri S, Pradat PF, Robberecht W, Ruegg M, Schwalenstocker B, Stiller D, van den Berg L, Vieira F, von Horsten S. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Lateral Scler. 2010;11:38–45. doi: 10.3109/17482960903545334. [DOI] [PubMed] [Google Scholar]
- 27.Marquardt T, Shirasaki R, Ghosh S, Andrews SE, Carter N, Hunter T, Pfaff SL. Coexpressed EphA receptors and ephrin-A ligands mediate opposing actions on growth cone navigation from distinct membrane domains. Cell. 2005;121:127–139. doi: 10.1016/j.cell.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 28.Meima L, Kljavin IJ, Moran P, Shih A, Winslow JW, Caras IW. AL-1-induced growth cone collapse of rat cortical neurons is correlated with REK7 expression and rearrangement of the actin cytoskeleton. Eur J Neurosci. 1997;9:177–188. doi: 10.1111/j.1460-9568.1997.tb01365.x. [DOI] [PubMed] [Google Scholar]
- 29.Monschau B, Kremoser C, Ohta K, Tanaka H, Kaneko T, Yamada T, Handwerker C, Hornberger MR, Loschinger J, Pasquale EB, Siever DA, Verderame MF, Muller BK, Bonhoeffer F, Drescher U. Shared and distinct functions of RAGS and ELF-1 in guiding retinal axons. EMBO J. 1997;16:1258–1267. doi: 10.1093/emboj/16.6.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noberini R, Rubio de la Torre E, Pasquale EB. Profiling Eph receptor expression in cells and tissues: a targeted mass spectrometry approach. Cell Adhes Migr. 2012;6:102–112. doi: 10.4161/cam.19620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oeckl P, Metzger F, Nagl M, von Arnim CA, Halbgebauer S, Steinacker P, Ludolph AC, Otto M. Alpha-, Beta-, and gamma-synuclein quantification in cerebrospinal fluid by multiple reaction monitoring reveals increased concentrations in Alzheimer’s and Creutzfeldt-Jakob disease but no alteration in Synucleinopathies. Mol Cell Proteomics. 2016;15:3126–3138. doi: 10.1074/mcp.M116.059915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oeckl P, Weydt P, Steinacker P, Anderl-Straub S, Nordin F, Volk AE, Diehl-Schmid J, Andersen PM, Kornhuber J, Danek A, Fassbender K, Fliessbach K, German Consortium for Frontotemporal Lobar D. Jahn H, Lauer M, Muller K, Knehr A, Prudlo J, Schneider A, Thal DR, Yilmazer-Hanke D, Weishaupt JH, Ludolph AC, Otto M. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90:4–10. doi: 10.1136/jnnp-2018-318868. [DOI] [PubMed] [Google Scholar]
- 33.Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109:E2230–E2239. doi: 10.1073/pnas.1204386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- 36.Régal Luc, Vanopdenbosch Ludo, Tilkin Petra, Van Den Bosch Ludo, Thijs Vincent, Sciot Rafael, Robberecht Wim. The G93C Mutation in Superoxide Dismutase 1. Archives of Neurology. 2006;63(2):262. doi: 10.1001/archneur.63.2.262. [DOI] [PubMed] [Google Scholar]
- 37.Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 38.Sheleg M, Yochum CL, Richardson JR, Wagner GC, Zhou R. Ephrin-A5 regulates inter-male aggression in mice. Behav Brain Res. 2015;286:300–307. doi: 10.1016/j.bbr.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 41.Tyzack GE, Hall CE, Sibley CR, Cymes T, Forostyak S, Carlino G, Meyer IF, Schiavo G, Zhang SC, Gibbons GM, Newcombe J, Patani R, Lakatos A. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat Commun. 2017;8:1164. doi: 10.1038/s41467-017-01283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Hoecke A, Schoonaert L, Lemmens R, Timmers M, Staats KA, Laird AS, Peeters E, Philips T, Goris A, Dubois B, Andersen PM, Al-Chalabi A, Thijs V, Turnley AM, van Vught PW, Veldink JH, Hardiman O, Van Den Bosch L, Gonzalez-Perez P, Van Damme P, Brown RH, Jr, van den Berg LH, Robberecht W. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med. 2012;18:1418–1422. doi: 10.1038/nm.2901. [DOI] [PubMed] [Google Scholar]
- 43.Wahl S, Barth H, Ciossek T, Aktories K, Mueller BK. Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol. 2000;149:263–270. doi: 10.1083/jcb.149.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu B, De SK, Kulinich A, Salem AF, Koeppen J, Wang R, Barile E, Wang S, Zhang D, Ethell I, Pellecchia M. Potent and selective EphA4 agonists for the treatment of ALS. Cell Chem Biol. 2017;24:293–305. doi: 10.1016/j.chembiol.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao J, Cooper LT, Boyd AW, Bartlett PF. Decreased signalling of EphA4 improves functional performance and motor neuron survival in the SOD1(G93A) ALS mouse model. Sci Rep. 2018;8:11393. doi: 10.1038/s41598-018-29845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reduction of efnA5 levels does not alter the numbers of motor neurons or innervated neuromuscular junctions in a non-ALS context. (a) Motor neurons were counted in the ventral horn of the lumbar spinal cord in EfnA5+/+, EfnA5+/− and EfnA5−/− mice. Data is represented as mean ± SEM (N = 4) and it was analysed with a two-way ANOVA. (b) Innervated neuromuscular junctions (NMJs) were scored in the gastrocnemius muscle of the same mice. Data represents mean ± SEM (N = 4) and it was analysed with a one-way ANOVA. (JPG 1900 kb)
Data Availability Statement
The datasets analysed during the current study are available from the corresponding author upon request.