

Abstract
Metabolism is a continuous source of acids. To keep up with a desired metabolic rate, tumors must establish an adequate means of clearing their acidic end-products. This homeostatic priority is achieved by various buffers, enzymes, and transporters connected through the common denominator of H+ ions. Whilst this complexity is proportionate to the importance of adequate pH control, it is problematic for developing an intuition for tracking the route taken by acids, assessing the relative importance of various acid-handling proteins, and predicting the outcomes of pharmacological inhibition or genetic alteration. Here, with the help of a simplified mathematical framework, the genesis of cancer pH regulation is explained in terms of the obstacles to efficient acid venting and how these are overcome by specific molecules, often associated with cancer. Ultimately, the pH regulatory apparatus in tumors must (i) provide adequate lactic acid permeability through membranes, (ii) facilitate CO2/HCO3−/H+ diffusivity across the interstitium, (iii) invest in a form of active transport that strikes a favorable balance between intracellular pH and intracellular lactate retention under the energetic constraints of a cell, and (iv) enable the necessary feedback to complete the homeostatic loop. A more informed and quantitative approach to understanding acid-handling in cancer is mandatory for identifying vulnerabilities, which could be exploited as therapeutic targets.
Keywords: Tumors, Active transport, Carbonic anhydrase, Monocarboxylate transport, Set point, Lactate
Introduction
Tissue compartments will invariably contain H+ ions from the ionization of water and a myriad of biochemical substances. The concentration of these ions, commonly expressed on a pH scale [1], influences the activity of all proteins that undergo protonation: the most rapid and reversible post-translational modification [2–4]. The activity of enzymes, for instance, is strongly influenced by changes in pH, which is one reason why certain types of enzymes are grouped together in sub-cellular compartments of distinct pH, such as proteolytic enzymes inside acidic lysosomes [5]. A collection of enzymes can be ascribed an optimal pH; for example, the ensemble of cytoplasmic enzymes, including those involved in glycolysis, is predicted to operate optimally near pH 7.3 [5], and it should be in the interest of cells to maintain cytoplasmic pH near to this level.
If there was no net production of acids (or bases) in cells, tissue pH could remain constant, even in the absence of a dedicated regulatory system. However, essentially all tissues, including tumors, are net-producers of acid because mitochondrial respiration and fermentative metabolism generate large flows of CO2 and lactic acid, respectively [6, 7]. Genetic and epigenetic changes [8], as well as oxygen depletion, reprogram cancer metabolism towards a more glycolytic phenotype [9], but in order to adequately supply ATP, this low-yielding energy pipeline must be upregulated, resulting in an exacerbated output of lactic acid [6, 10]. Aberrant blood perfusion, which is a characteristic of many tumors, erects a barrier to the efficient venting of this acidic burden [11, 12]. A consequence of these circumstances is low extracellular pH (pHe), a chemical signature of the tumor microenvironment [13–16].
Microenvironmental acidity is not merely a collateral waste product of tumor biology, but a valuable source of feedback that controls various processes [17–20], including metabolic rate [21]. The sum of the effects of pH on cell biology is powerful enough to influence survival, which has been likened to a selection process favoring a particular phenotype of cancer cell among a genetically diverse population [6, 22, 23]. In order for acid-driven somatic evolution to take place, there must be a means by which the successful (and presumably more aggressive) subpopulations have adapted to microenvironmental acidity. Such a survival advantage can take one of two forms, which are not mutually exclusive.
The first involves a re-modeling of pH sensitivity, which could be achieved through genetic mutations involving titratable residues, such as histidines [4, 24–26]. The protonation state of histidine changes dramatically over the expanded physiological range, bestowing proteins with exquisite pH-dependence [27–29]. A shift in the pH sensitivity curve may, for example, allow mutant proteins to remain active even at an abnormal level of pH [4]. The scope of this effect on cell biology is, however, restricted to the functional remit of the mutated protein.
Since a large fraction of pH-sensitive proteins resides inside cells, another adaptation to an acidic microenvironment is for cells to defend a favorable (usually alkaline) intracellular pH (pHi), using an appropriately powered homeostatic mechanism. This adaptive strategy has the advantage of influencing all intracellular proteins collectively. A “perfect” homeostatic system would keep the pH of the internal environment constant at the set point, irrespective of the external conditions or other constraints; in achieving this, cells acquire a substantial degree of independence, which is particularly empowering for cancer cells. However, cells placed under acid stress will not universally manifest such perfect pHi homeostasis; instead, there will be variation in regulatory prowess which relates to “acid fitness” and could provide substrate for selection pressures. pH-regulatory proteins underpin this phenotype, and in recent years, much attention has been given to testing their therapeutic utility [30–33].
There is now an extensive literature about the various genes and proteins that contribute towards the pH regulatory phenotype of cancer [31, 34–36], producing ever more bewildering schematics such as the one shown in Fig. 1. It falls outside the scope of our intuition to predict, from such schematics, which is the dominant route taken by acid, or how such a system responds to modifications in one or more of its elements (e.g., inhibition by drugs). To fill this niche, mathematical models can be used to simulate complex processes, and arrive at inferences that help in formulating a more accessible narrative. Here, using conceptually simple mathematics (Table 1), I explain the genesis of pH regulation and the role played by the distinct classes of proteins involved in this process.
Fig. 1.

Schematic of a cancer cell, showing the major molecules involved in pH regulation. The complexity of the system is factually correct, but unpalatable for estimating the distribution of H+ ions fluxes through the various processes, deriving a value for the steady-state pHi, or predicting how the system would respond to changes in one or more of these processes. MCT: H+-monocarboxylate transport; CA: carbonic anhydrase; CHE: Cl−/OH− exchange; AE: anion exchange; NBCe: electrogenic Na+-HCO3− cotransport; NBCn: electroneutral Na+-HCO3− cotransport; NHE: Na+/H+ exchange; organelle: acidic lysosome/endosome with V-type ATPase
Table 1.
List of variables used in the mathematical model for simulating steady-state pH and lactate concentration under the various scenarios presented in Fig.s 2, 3 and 4
| Parameter | Definition | Fig 2 | Fig 3 | Fig 4 | Reference |
|---|---|---|---|---|---|
| r | Radius of cell | 7 μm | 7 μm | 7 μm | [37] |
| R | Distance from capillary |
(case 1) 0 μm (2–3) 150 μm |
(case 1–2) 0 μm (3) 150 μm |
150 μm | [38] |
| pHi0 | Starting intracellular pH | 7.3 | 7.3 | 7.3 | |
| pHec | Extracellular pH in capillary | 7.4 | 7.4 | 7.4 | |
| β int | Intrinsic buffering capacity | 15 mM/pH | 15 mM/pH | 15 mM/pH | [39] |
| β e | Extracellular buffering capacity | 3 mM/pH | 3 mM/pH | 3 mM/pH | [40, 41] |
| D H | Interstitial H+ diffusion coefficient | 12,000 μm2/s | 12,000 μm2/s | 12,000 μm2/s | [40, 41] |
| τ e | Tortuosity in extracellular space | 0.5 | 0.5 | 0.5 | [40, 41] |
| J CO2 | CO2 production rate | 0–15 mM/min | 0 | 0 | [42] |
| K CO2 | CO2 dissociation constant | 10–6.15 M | 10–6.15 M | 10–6.15 M | |
| k h | Spontaneous CO2 hydration constant | 0.16 s−1 | 0.16 s−1 | 0.16 s−1 | [38, 40, 41] |
| CAe | Extracellular CA activity |
(case 1–2) 1 (3) 1000 |
1000 | 1000 | [38, 40, 41] |
| [CO2]ec | Extracellular CO2 concentration in capillary | 1.2 mM | 1.2 mM | 1.2 mM | |
| P CO2 | CO2 membrane permeability | 1000 μm2/s | 1000 μm2/s | 1000 μm2/s | [40] |
| D CO2 | Interstitial CO2 diffusion coefficient | 2400 μm2/s | 2400 μm2/s | 2400 μm2/s | [40] |
| D HCO3 | Interstitial HCO3− diffusion coefficient | 1800 μm2/s | 1800 μm2/s | 1800 μm2/s | [40] |
| J HLa | Lactic acid production rate | 0 | 0–20 mM/min | 0–20 mM/min | [43–48]. |
| K HLa | Lactic acid dissociation constant | – | 10–3.86 M | 10–3.86 M | |
| [HLa]ec | Extracellular lactic acid concentration in capillary | – | 0 mM | 0 mM | |
| P HLa | Apparent lactic acid membrane permeability | – |
(case 1) 10 μm/s (2–3) 1000 μm/s |
1000 μm/s | |
| D La | Interstitial lactate diffusion coefficient | – | 1300 μm2/s | 1300 μm2/s | [40] |
| D HLa | Interstitial lactic acid diffusion coefficient | – | 1300 μm2/s | 1300 μm2/s | [40] |
| V max | Maximum flux generated by active transporter | – | – |
(case 1) 0 mM/min (2) 1 mM/min (3–4) 10 mM/min |
|
| K a | Apparent H+ binding constant of active transporter | – | – |
(1–3) 10–7.0 M (4) 10–6.7 M |
[31, 39] |
| n | Hill cooperativity of active transporter | – | – | 2 | [31, 39] |
| J loading | Regulated acid-loading flux |
(case 1) 0 mM/min (2) 0.2 mM/min (3–4) 2 mM/min |
|||
| pHsetpoint | Intracellular pH setpoint | – | – |
(case 1–3) 7.3 (4) 7.0 |
Diffusion and chemical equilibration
For the many cells in the body that are juxtaposed to functional capillaries, the supply of oxygen is adequate for aerobic respiration. Such cells, particularly in a differentiated state, would be expected to opt for oxidative phosphorylation as a rich source of ATP [21]. The acidic end-product, CO2, is a gas which freely permeates lipid bilayers and possibly also through gas channels [49], although the significance of this facilitated route is debated [40, 50]. CO2 production rate can be estimated from measurements of oxygen consumption, which can be as high as 15 mM per minute [42]. Even at these high production rates, biological membranes cannot support gradients of a highly permeant gas, therefore the intra- and extracellular partial pressures of CO2 must equalize. Blood capillaries are designed to remove CO2 efficiently, and since there are no other barriers to CO2 movement, blood perfusion will seamlessly drive CO2 out of cells. Under these circumstances, pHi remains constant, as there is no meaningful buildup of CO2 (Fig. 2a(1)). For the simulations shown in Fig. 2, starting pHi was set at 7.3, the predicted optimal for cytoplasmic enzymes. Whilst efficient CO2 removal ensures the constancy of pHi, it cannot influence the level at which pHi is kept. Offsetting pHi relative to pHe ultimately requires an input of energy, whereas the process of CO2 venting is purely dissipative.
Fig. 2.
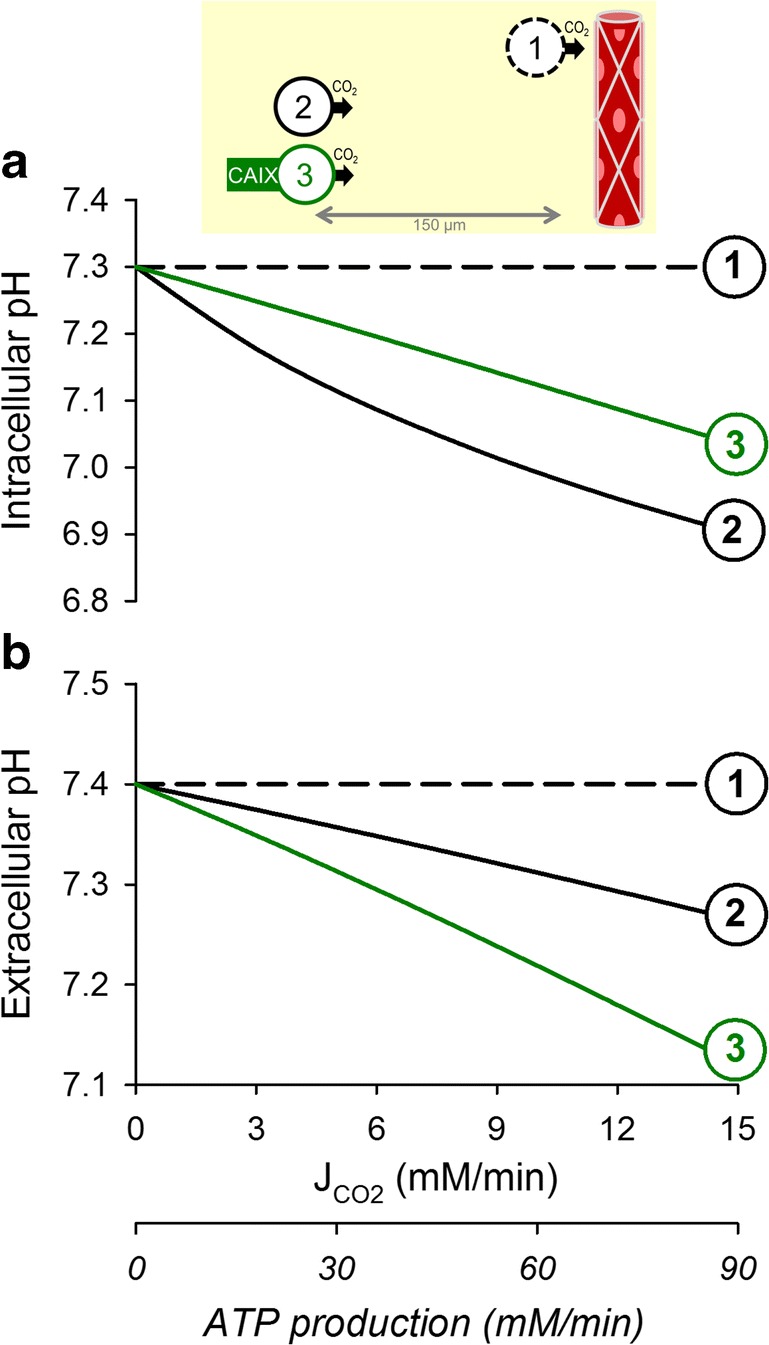
The simulated relationship between aerobically-produced CO2 and (a) intracellular pH and (b) extracellular pH over the range of metabolic rates reported in cells. (1) Cell juxtaposed to a capillary, i.e., absence of a meaningful barrier to CO2 diffusion in the extracellular space. Rapid permeation of CO2 across the surface membrane results in a tight coupling between intra- and extracellular CO2 partial pressures, and hence rapid clearance of CO2 from the cell. (2) Distance between the cell and its capillary expanded to 150 μm, a commonly accepted hypoxic limit. This barrier to the flow of CO2 produces an intracellular buildup of CO2 and establishes a radial gradient of extracellular CO2 partial pressure, which is responsible for driving CO2 venting. Consequently, both the intra- and extracellular compartments of the tissue acidify. (3) Under adequate CA catalysis (e.g. by CAIX), the magnitude of extracellular CO2 venting is enhanced by means of facilitated diffusion (transport in the form of HCO3− and H+ ions), a consequence of which is a further degree of extracellular acidification. More efficient CO2 venting reduces the degree of intracellular CO2 buildup and intracellular acidification
In poorly perfused tissues, such as tumors, the distance to the nearest capillary can become substantial. This constitutes a barrier to CO2 movement, which requires an adequately steep gradient of CO2 partial pressure to drive the flow of gas: invariably, cells will accumulate CO2 and acidify (Fig. 2a(2)). This scenario also leads to an undesirable coupling between pHi, diffusion distance, and metabolic rate, which greatly limits the scope of cancer cell behaviors. A way of improving CO2 venting is to increase its effective diffusivity by enabling a parallel transport of H+ and HCO3− ions. The necessary chemical conversion is normally very slow, but can be catalyzed enzymatically by exofacial isoforms of carbonic anhydrase, such as CAIX and CAXII (coded by genes CA9, CA12) [38, 51–53]. Faster CO2 clearance reduces the extent of intracellular acidification (Fig. 2a(3)), but also leads to a more pronounced extracellular acidification (Fig. 2b(3)). This latter effect has been documented in 3D spheroids of cancer cells in vitro [54] and in xenografts in vivo [55], and is believed to be important in the acid-selection process in cancer [6, 22].
Facilitated membrane permeation
In underperfused tissues, the diffusion path that restricts the outflow of CO2 will also restrict the counterflux of oxygen. With reduced O2 penetration, tumor cells must rely on glycolysis. Intriguingly, cancer cells typically manifest a glycolytic phenotype even in the presence of oxygen, a phenomenon known as the Warburg Effect [56]. The rates of lactic acid production by cancer cells are in the low mM/min range [43–47], but some cancer cells can attain rates as high as 20 mM/min [48]. Compared to CO2, lactic acid ionizes more completely, which reduces the availability of its uncharged lipid-soluble form. Thus, lactic acid permeability across lipid bilayers is low in relation to the venting demand placed by glycolysis. Without any form of facilitated permeation, a substantial transmembrane gradient of lactic acid would be necessary to drive an adequate efflux: consequently, cells would accumulate high levels of lactic acid and lactate (Fig. 3b(1)). A solution to this conundrum is in the form of H+-monocarboxylate transporters of the SLC16 gene family [57], such as the ubiquitously expressed MCT1 (SLC16A1). By shuttling H+ and lactate ions across membranes, MCTs greatly increase the apparent membrane permeability to lactic acid; consequently, a much smaller concentration gradient is necessary to drive an adequate lactic acid efflux (Fig. 3a/b(2)). In the case of well-perfused cells expressing MCT isoforms, intracellular lactate accumulation and acidification are minimal and compatible with pHi constancy. However, this system is unable to offset pHi to a desired set point because protein-assisted permeation is solely dissipative.
Fig. 3.
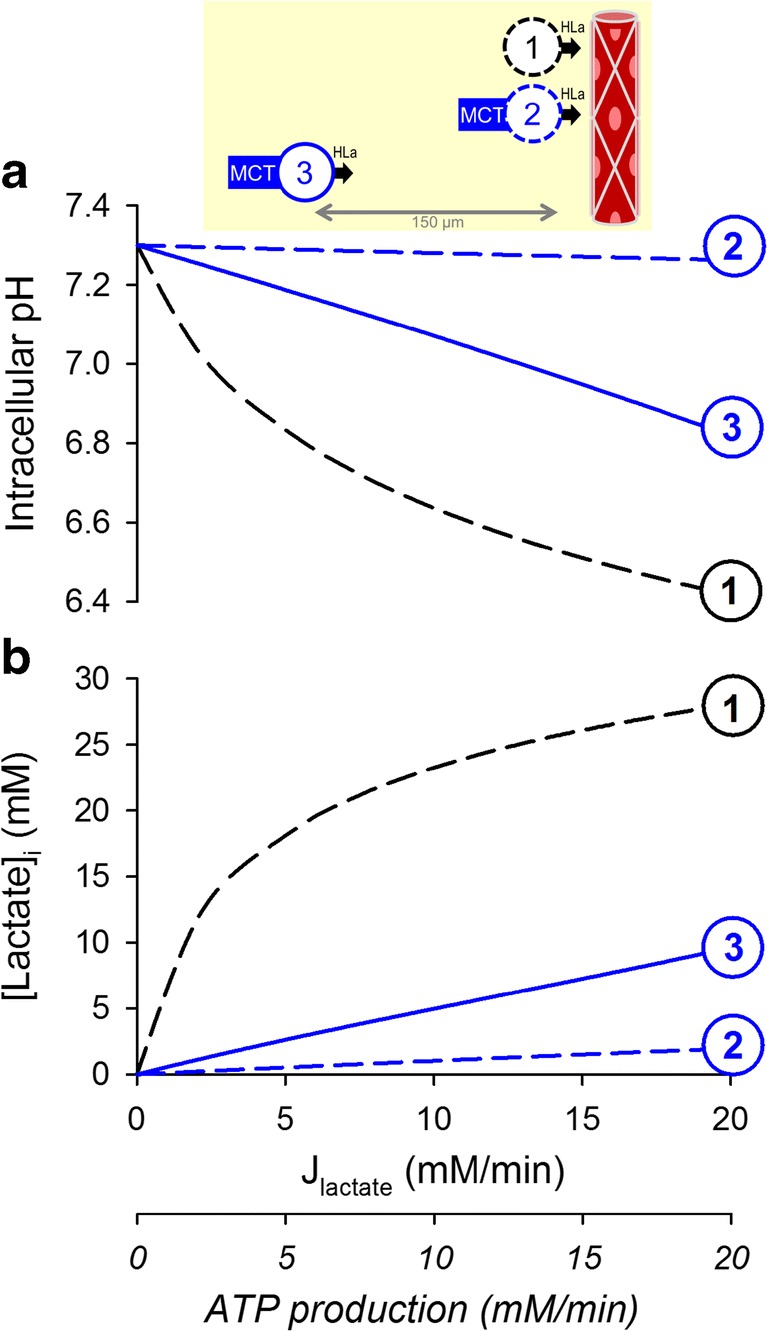
The simulated relationship between glycolytic lactic acid production and (a) intracellular pH and (b) intracellular lactate retention over the range of metabolic rates reported in cancer cells. (1) Cell lacking protein-facilitated permeability to lactic acid, juxtaposed to a capillary, i.e., absence of a meaningful barrier to lactic acid diffusion in the extracellular space. Since lactic acid is only poorly permeant across lipid bilayers, its venting is severely restricted by the cell membrane, resulting in an intracellular buildup of lactate and H+ ions. (2) Cell with high lactic acid permeability attained with MCT isoforms (e.g., MCT1 and MCT4), juxtaposed to a capillary. With higher permeability, a much smaller gradient is required to drive lactic acid efflux, resulting in a considerably diminished intracellular buildup of lactate and H+ ions. (3) Distance between the cell and its capillary expanded to 150 μm, a commonly accepted hypoxic limit. CO2/HCO3− equilibration is ensured by high CA activity. As a consequence of the extracellular diffusional barrier to lactic acid movement, a substantial gradient of lactic acid is required to drive venting. This results in a greater intracellular retention of H+ and lactate ions. Thus, steady-state pHi becomes subservient to both metabolic rate and distance from capillary, i.e., is not independently regulated
In under-perfused tumors, the diffusion distance across the interstitium is an additional “resistance” to the flow of lactic acid, which mostly takes the form of lactate and H+ ions. Cells in such niches may induce hypoxia-upregulated MCT4 to minimize the permeability barrier at their surface membrane [58], but this response cannot address the problem of diffusion across the interstitial space. Of the two chemical species released by glycolytic cells, the diffusive flux of H+ ions is likely to be rate-limiting because it is dramatically restricted by reversible binding to buffers [59–61] in an environment that does not support fast transport involving proton wires (Grotthuss mechanism) [62]. H+ ion diffusion can be facilitated by the mobile CO2/HCO3− buffer with adequate levels of exofacial CA activity; however, even with maximal enzymatic facilitation, the diffusional barrier cannot be collapsed. In glycolytic tissues, the diffusional delay across the interstitium will result in an intracellular retention of lactate and H+ ions, reaching levels that may become physiologically untenable (Fig. 3a/b(3)). These circumstances would justify the implementation of additional homeostatic measures, ultimately resorting to uphill (active) transport.
Active transport and the pH set point
The components of pH regulation described thus far address the issue of slow diffusion of the CO2/HCO3−/H+ system across extracellular spaces (CA) and inadequate lactic acid permeation across membranes (MCT). These protein-assisted processes are passive: they do not consume energy but, instead, hasten equilibration. It would be thermodynamically implausible for these processes, alone, to maintain tumor pHi at a certain set point under continuous metabolic acid loading. Any departure from the “passive” pHi and pHe curves plotted in Figs 2 and 3 would require a form of active transport, which historically has been at the center of research into pH regulation. There are many types of transporters that engage in active transport, and these can be classified as being either primary active (V-type H+ ATPase, P-type H+/K+ ATPase) or secondary active (e.g., Na+/H+ exchangers of the SLC9 gene family [63]) [31, 34–36]. The latter class also includes transporters that carry HCO3− ions, which is chemically equivalent to a counterflux of H+ ions (e.g., Na+-HCO3− cotransporters of the SLC4 gene family, see Bødtkjer, this volume) [64]. Whilst HCO3−-importing pHi regulators can be distinguished from H+-exporting counterparts by experimental maneuvers (e.g., the system’s response to the removal of CO2/HCO3− buffer) [65], their physiological outcomes are equivalent: both produce an equimolar intracellular alkalinization.
In homeostatic terms, a more relevant characterization of pHi-regulating proteins relates to their kinetics, rather than the chemical identity of the transport substrate. The maximal transport rate (Vmax) describes the capacity for surface-expressed transporters to produce a flux of H+ ions or their chemical equivalents. A powerful pHi-regulatory system is expected to produce fluxes that comfortably exceed the sum of disturbances, such as glycolysis. However, for such a system to be efficient, its energetic footprint must not be excessive to avoid an unwarranted depletion of ATP. pHi regulators must also receive feedback that gauges the progress of their actions: as pHi rises, the acid-extrusion process should slow. The relationship between flux and pHi can be described in terms of an apparent affinity constant (Ka) and cooperativity (a measure of steepness). Although high pHi can allosterically inhibit acid-extrusion, it cannot block this efflux completely within the physiological pHi range. Consequently, a regulatory system comprising only of acid-extruders would manifest an upwardly drifting pHi rather than stabilize at a steady-state pHi. To ensure that the steady-state condition is met, acid-extrusion at the desired set point pHi must be matched by an equal acid-loading flux, such as that generated by the activity of various Cl−-coupled transporters belonging to the SLC4 or SLC26 families of genes [66–68]. The magnitude of these equal but opposite acid-fluxes determines the robustness of the system’s response to acid-base disturbances, in addition to its baseline energy consumption. For example, higher fluxes make the system better at defending pHi during transient challenges, such as bursts of metabolic activity, but these require higher ATP production to power the apparently futile cycle of Na+-dependent acid-extrusion and Cl−-dependent acid-loading. The compromise that a cell strikes between these conflicting interests influences its survival in acidic niches.
To explore how the various parameters relating to active transport influence steady-state pHi, a simplified kinetic representation of acid-extrusion, designed to defend a set point pHi of 7.3, was included in the model. The transporter’s pHi-sensitivity was modeled with a pKa that was 0.3 units lower than the set point pHi, and a cooperativity of 2. These values are within the range reported for Na+/H+ exchangers expressed in cancer cells [31, 37, 39]. For a maximal flux (Vmax) set to 1 mM/min, the balancing acid-loading flux would need to be 0.2 mM/min, i.e., an ATP consumption of 0.07 mM/min (calculated on the basis that the Na+/K+ pump which ultimately drives secondary-active transport has a stoichiometry of 3Na+/ATP). This relatively low flux is inadequate to defend pHi in highly glycolytic and diffusively-restricted tumors (Fig. 4a(2)). Raising Vmax to 10 mM/min produces a system that is able to maintain pHi at the set point, even under high glycolytic rates, but its higher ATP demand (0.7 mM/min) is the price the cell must pay for the improvement in pHi control (Fig. 4a(3)).
Fig. 4.
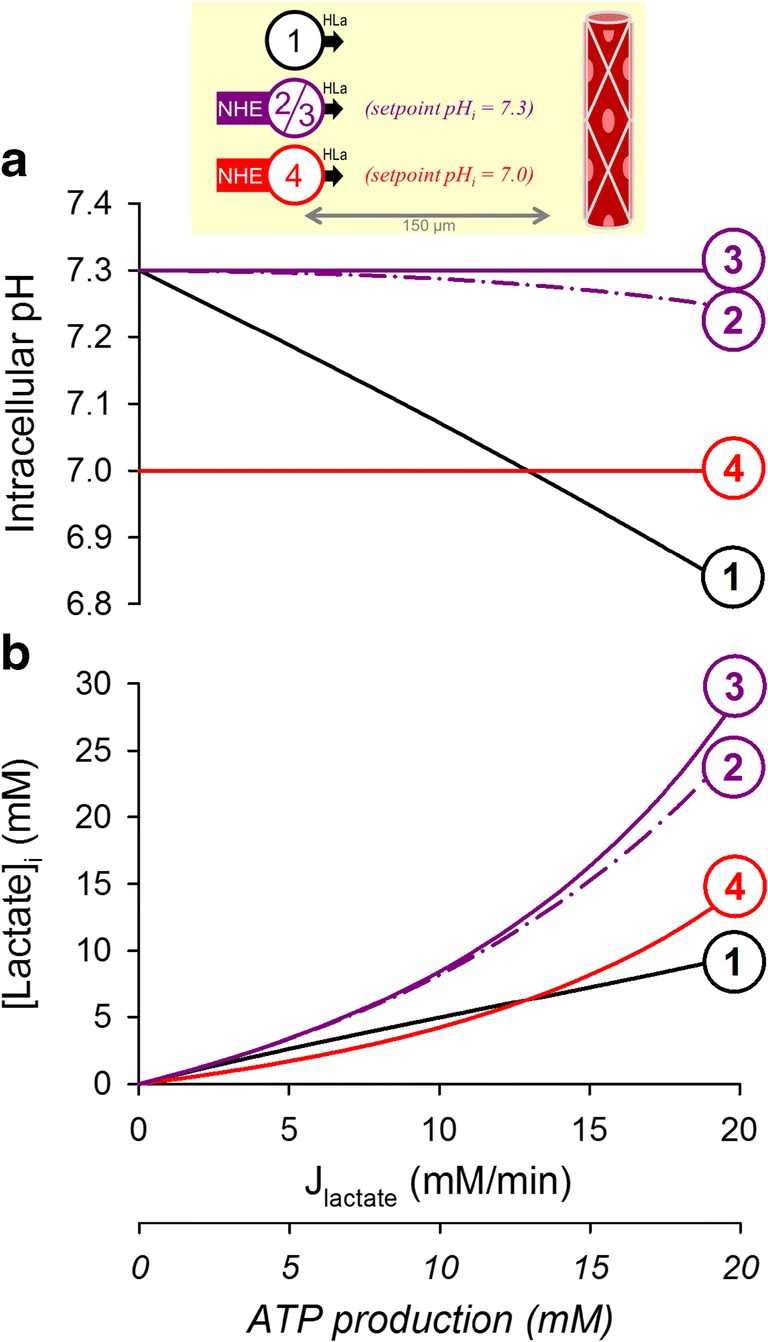
Simulating the effect of active transport on the relationship between glycolytic lactic acid production and (a) intracellular pH and (b) intracellular lactate retention. (1) Cell with high exofacial CA activity and high lactic acid permeability placed 150 μm away from a capillary. (2) Inclusion of an active transporter, such as Na+/H+ exchanger, with a set point at pHi = 7.3 and maximal acid extrusion rate of 1 mM/min; this relatively low corrective flux is unable to fully offset metabolic acid-loading, resulting in a modest influence of glycolytic rate on pHi. Uphill extrusion of H+ ions from the cell favors lactic acid dissociation and increases intracellular lactate retention. Active transport will reduce net ATP supply by 0.07 mM/min. (3) Raising the maximal acid-extrusion flux by 10-fold is sufficient to maintain pHi at the set point of 7.3 over a wide range of metabolic rates; this establishes a system that truly regulates pHi, independently of constraints imposed by metabolic rate or diffusion distance. However, clamping pHi to 7.3 results in substantial intracellular lactate retention. Additionally, the elevated rate of active transport reduces ATP supply by 0.7 mM/min. (4) Lowering the set point of active transport from 7.3 to 7.0 reduces the degree of lactate retention inside cells, whilst still defending constancy of pHi, albeit at a less alkaline level
A consequence of regulating pHi to an alkaline set point is that it produces a cytoplasmic milieu that favors lactic acid dissociation. Cells in diffusively-restricted tissues will thus build up lactate to levels that can be significant, reaching tens of mM, and likely exerting functional consequences, such as end-product inhibition of glycolysis. Thus, it may not necessarily be desirable for glycolytic tumors to maintain their pHi much higher than 7.0 because this invariably leads to intracellular lactate retention. Since the transmembrane distribution of lactate is set by the pHi/pHe gradient, one way of “regulating” lactate is by adjusting set point pHi towards a less alkaline level; for example, dropping this from 7.3 to 7.0 halves lactate retention (Fig. 4a(4)) without altering ATP consumption (assuming that the regulated acid-loading flux is of the same magnitude at the new steady-state pHi). To explore this further, simulations were run for a range of starting pHi and metabolic rates (Fig. 5a). The concentration of intracellular lactate attained under the simulated conditions is shown by the contour plots in Fig. 5b, and demonstrates why maintaining an invariably alkaline pHi in a milieu of low pHe may become disadvantageous for glycolytically-active tumors. Indeed, it is well-documented that even in well-perfused single cells, steady-state pHi falls modestly in response to a decrease in pHe, producing a coupling between pHe and pHi. A reason behind this seemingly imperfect homeostatic apparatus may be to strategically protect cells from excessive lactate retention, which would otherwise happen if pHi remained substantially higher than pHe. Thus, the burden of lactate retention is lessened by allowing cells to modestly acidify in niches of low pHe.
Fig. 5.
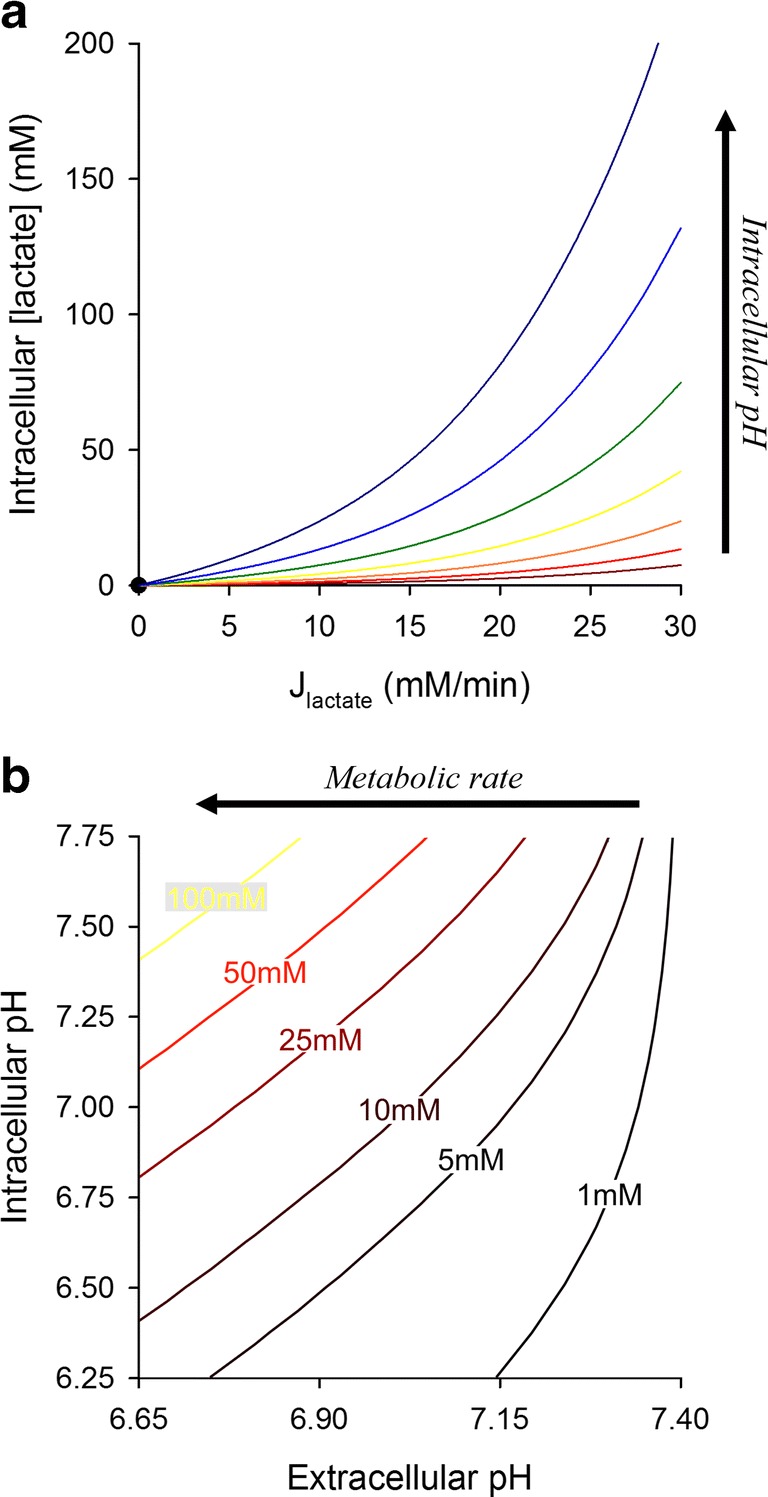
Using the mathematical model to map the relationship between pHi, pHe, and intracellular lactate. Simulations were based on the model for a glycolytic cells with high MCT and exofacial CA activity, placed 150 μm away from a capillary. pHi and lactic acid production were varied between 6.25 and 7.75, and 0 and 30 mM/min, respectively. (a) Intracellular lactate concentration as a function of metabolic lactate production rate; each line represents a different starting pHi. (b) Results replotted as a contour map. Contours show the combination of pHi and pHe that yield a particular concentration of lactate in cytoplasm. The highest degree of intracellular lactate retention is attained with high metabolic rates, when the inward pH gradient is large (i.e., pHi>pHe)
Predicting a cell’s steady state pH
The discussion of pH regulation so far has focused on how metabolic acid production influences steady-state pH in the intra- and extracellular compartments of tissue (Fig. 6a(left)). In parallel, pH feeds back on metabolic rate through the inhibitory effect of intracellular H+ ions on glycolytic enzymes (Fig. 6a(right)) [21]. For example, phosphofructokinase, the enzyme catalyzing the rate-limiting step of glycolysis, manifests a steep pH-sensitivity. The relationship between pHi and glycolysis can be modeled with a curve such as that shown in Fig. 6b. The pHi-metabolism relationship (where pHi is the independent variable) and the inverse metabolism-pHi relationship (where metabolic rate is the independent variable) can be superimposed to obtain the mathematical solution describing steady state pHi and metabolic rate. This can be visualized as the point of crossover of the two relationships. Increasing MCT activity (in the absence of active transport) allows pHi and metabolic rate to increase in tandem (Fig. 6b: 1 to 2). A further up-lift is attained by incorporating active transport (Fig. 6b: 2 to 3), and even more so if the transporter is adjusted to a higher set point pH (Fig. 6b: 3 to 4). This simplified analysis can be helpful in explaining the dynamic interplay between metabolism and pH.
Fig. 6.
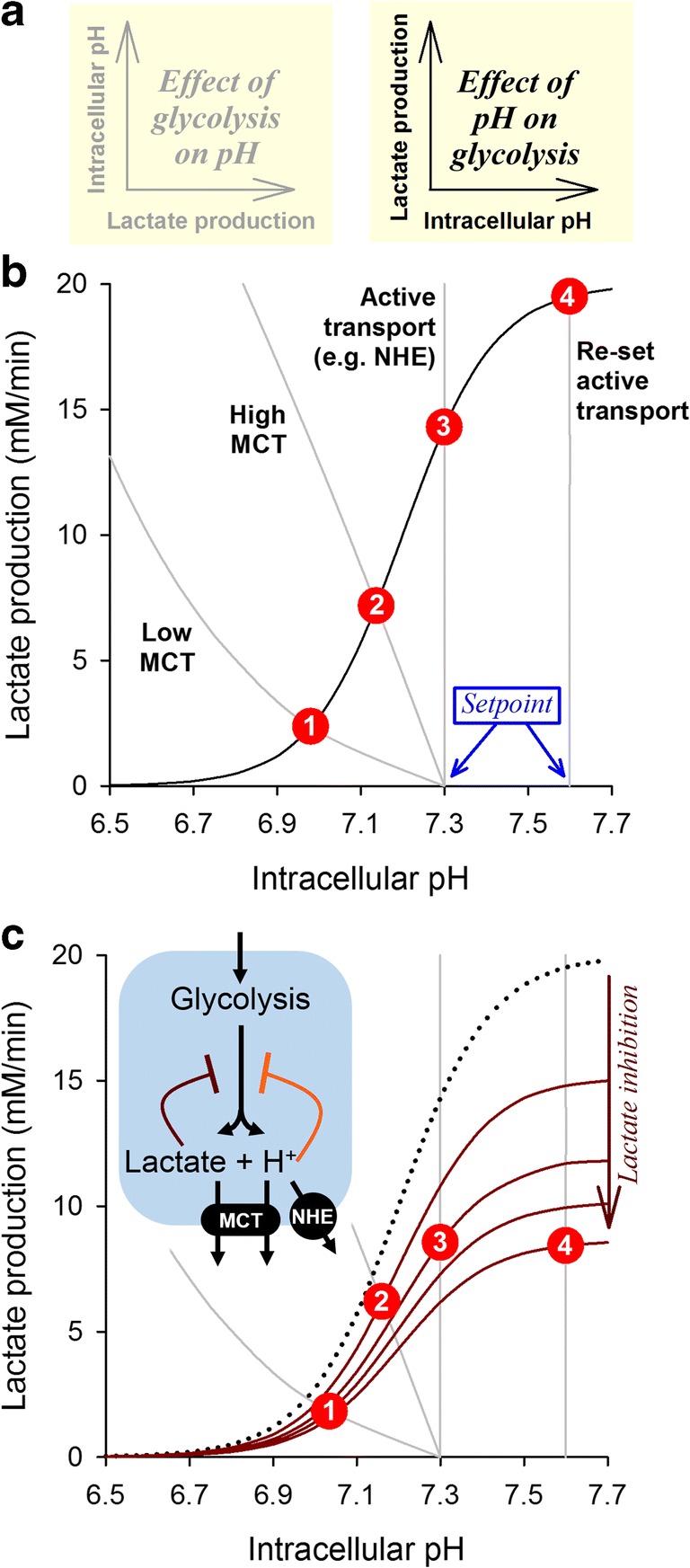
Using a graphical approach to infer steady-state pHi and metabolic rate. (a)Left: Lactate production affects intracellular pH, as described in Fig. 3 and Fig. 4. In this representation, metabolic rate is considered to be the independent variable. Right: Intracellular pH affects lactate production through the inhibition of glycolysis. In this representation, pHi is considered to be the independent variable. (b) The pHi-metabolism and metabolism-pHi relationships are akin to two equations; the mathematical solution to these can be inferred graphically from the point of crossover. Superimposing the relationship between pHi and glycolytic rate (black curve) with the relationship describing the effect of glycolytic rate on pHi (gray curve) for a cell, located 150 μm away from its nearest capillary, with high CA expression and either (1) low MCT activity, (2) high MCT activity, (3) high MCT activity and active transport with a set point of pH=7.3, or (4) high MCT activity and active transport with a set point of pH=7.6. The points of crossover (red ;circles) are the mathematical solutions of these four pairs of equations. Steady-state pHi and metabolic rate increase in tandem when MCT activity is raised and when active transport is engaged to an alkaline set point. (c) In addition to the allosteric inhibitory effect of H+ ions on glycolytic enzymes, another influence is end-product inhibition of glycolysis by the accumulation of lactate. This is expected to scale-down the pHi-metabolism curve and produce a different crossover point, particularly at high pHi when lactate accumulation is expected to be substantial. In the example illustrated (assuming an inhibitory constant Ki of 10 mM for lactate), the inhibitory effect produced by intracellular lactate retention at pHi > 7.3 offsets the disinhibition of glycolysis by low [H+]; consequently, the highest possible metabolic rate is attained in the range 7.1–7.3
Given that metabolism is a limiting factor for cancer cell proliferation, it would seem desirable for tumors to express high levels of MCT and to offset pHi to an alkaline level by active transport. However, the metabolic rate plotted in Fig. 6b does not consider the effect of intracellular lactate accumulation (cf. Fig 5), which could exert end-product inhibition on glycolysis [21]. Because this thermodynamic consequence is not inherently cooperative, its effect on metabolic rate is expected to be smaller than the allosteric inhibition of enzymes by H+ ions. However, at profoundly alkaline pHi, the allosteric disinhibition of glycolytic enzymes plateaus and the inhibitory effect of lactate accumulation becomes overriding. This effect of lactate can be modeled as a down-scaling of the pHi-metabolic rate curve, as shown in Fig. 6c. A somewhat surprising consequence of the dual inhibition by lactate and H+ ions is that a profoundly alkaline cytoplasm may not necessarily be conducive for a high metabolic rate, because the inhibitory effect of lactate retention may cancel-out the benefit of enzyme disinhibition at low [H+]. This interaction may explain why most tumors have a pHi in the mildly alkaline range, around 7.2 [16, 69]: a tested compromise between a pHi that is sufficiently alkaline to disinhibit glycolysis but not at a level that would overload the cytoplasm with lactate anions.
Conclusions
Since the milestone discoveries of cellular pH regulation by Roger Thomas, Walter Boron, Richard Vaughan-Jones, Andrew Halestrap, and many others, our understanding of acid-base homeostasis has developed to a fine level of molecular detail thanks to breakthroughs in physiology, molecular biology, and genetics. Complex systems, like pH regulation, are not intuitive to understand, and can be misinterpreted if our analytical framework is not adequately integrative, i.e., when it considers a subset of components of the system in isolation. Although therapeutic interventions aimed at disturbing pH regulation are typically targeted to meet the criteria for clinical translation, their effects on pHi and pHe will be highly context-sensitive, and depend on factors such as metabolic rate, diffusion distances, and the repertoire of other pH-regulating molecules. This problem highlights the need to characterize pH regulation in as much detail as possible, and use calibrated mathematical models to identify a suitable Achilles heel for targeted disruption. To make such models accurate yet accessible, they must be simple to understand and supply with parameters, but not any simpler (Albert Einstein, 1950).
The analyses shown in this review are based on representative parameters obtained from the literature and must not be generalized to all cases of tumors; rather, the graphical illustrations should be used a didactic guides for explaining the scope of various elements of pH regulation in influencing pH and lactate concentration. The modeling scenarios discussed herein assume that cells behave as independent units in terms of pHi regulation. Most cells in the body are, however, diffusively coupled by means of channels, such gap junctions formed by connexins. Such coupling would result in syncytial behaviors of clusters of cells, but the relevance of this to cancer is likely to be limited to special cases, because gap junctional coupling tends to be low or absent in tumors [70], possibly due to the tumor-suppressing effect that has been attributed to connexins [71, 72]. Nonetheless, there are cases of well-coupled cancer cells, and in such instances, pH regulation would operate in a syncytial mode [73, 74].
Some key points borne from the analyses presented herein are paraphrased below:
CO2 permeation across membranes is fast and unlikely to be a substantial barrier to CO2 movement. Consequently, no significant gradients in CO2 partial pressure are expected between cells and their immediate microenvironment.
Interstitial diffusion distances in poorly-perfused tissues can impose a meaningful resistance to CO2 movement. CO2 diffusion can be facilitated by a parallel flux of HCO3− and H+ ions, but only in the presence of extracellular carbonic anhydrase (CA) activity. This CA-catalyzed CO2 clearance will alkalinize cytoplasm and acidify extracellular spaces.
In contrast to CO2, lactic acid crosses lipid bilayers very slowly and therefore its permeation must be assisted by H+-monocarboxylate co-transporters (MCT); otherwise, lactic acid and lactate will accumulate intracellularly to untenable levels, even in well-perfused cells.
Lactic acid diffusion across the interstitium is a resistance in series with membrane permeation, and therefore cannot be augmented by MCT expression at the cell surface. Since lactic acid almost fully ionizes, a rate-limiting step to its venting is likely to be the diffusion of H+ ions, which is greatly restricted in biological fluids. This limiting step can be assisted by CO2/HCO3− buffer, which acts as a mobile H+ shuttle, if there is adequate extracellular CA activity.
Overall, exofacial CA isoforms improve acid venting from cells by facilitating diffusion. However, this beneficial effect will only be meaningful in the context of long diffusion distances. Thus, it is not possible to demonstrate a meaningful CA-related effect on pHi regulation in isolated cells or well-stirred monolayers, where extracellular diffusion distances are negligible.
Cells that express extracellular-facing CA isoforms and MCT at their membrane improve their bandwidth for venting acidic end-products, but their pHi will become subservient to metabolic rate and diffusion distance in a manner that does not meet the strict criteria for true pHi homeostasis. These criteria are met by the inclusion of active transporters that generate uphill movement of H+ ions (or their chemical equivalents; e.g., HCO3−) across membranes. Active transport can thus uncouple pHi and pHe from the constraints of passive equilibration.
Active transporters will produce a meaningful correction to pHi if the H+/H+-equivalent flux they generate is adequately high. The magnitude of this flux depends on maximal turnover and allosteric modulation by H+ ions. For typical metabolic rates, fluxes greater than several mM/min are necessary for the pHi regulatory system to achieve adequate homeostatic power.
Given that acid-loading by metabolism is the primary threat to pH housekeeping, it may seem counterproductive for cells to express acid-loading transporters at the membrane. However, these regulated acid-loading fluxes are mandated for balancing acid-extrusion and stabilizing pHi at a particular level.
The energy consumed by acid-extruding active transporters relates to the magnitude of the regulatory acid-loading fluxes that must work against them. The ATP-cost of this balancing act places a limit on how responsive a cell’s pHi regulatory system can become. Typical ATP consumption rates are in the high μM/min to low mM/min range.
Various enzyme-catalyzed processes can be ascribed specific pHi-optima; glycolytic rate is, overall, faster at higher pHi. However, underperfused glycolytic tissues may not necessarily find it beneficial to maintain an alkaline pHi because this leads to a greater retention of lactate in cytoplasm, which itself may exert end-product inhibition on glycolysis. This reasoning may explain why the cell’s set point pHi tends to decrease at low pHe: a pre-emptive action to limit the degree of lactate accumulation.
Funding information
The work was supported by the European Research Council, SURVIVE #723997.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sorensen SPL. Enzymstudien. II. Mitteilung. Über die Messung und die Bedeutung der Wasserstoffionenkoncentration bei enzymatischen Prozessen. Biochemische Zeitschrift. 1909;21:131–394. [Google Scholar]
- 2.Srivastava J, Barber DL, Jacobson MP. Intracellular pH sensors: design principles and functional significance. Physiology (Bethesda) 2007;22:30–39. doi: 10.1152/physiol.00035.2006. [DOI] [PubMed] [Google Scholar]
- 3.Schonichen A, Webb BA, Jacobson MP, Barber DL. Considering protonation as a posttranslational modification regulating protein structure and function. Annual Review of Biophysics. 2013;42:289–314. doi: 10.1146/annurev-biophys-050511-102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White KA, Ruiz DG, Szpiech ZA, Strauli NB, Hernandez RD, Jacobson MP, Barber DL. Cancer-associated arginine-to-histidine mutations confer a gain in pH sensing to mutant proteins. Science Signaling. 2017;10(495):eaam9931. doi: 10.1126/scisignal.aam9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Persi E, Duran-Frigola M, Damaghi M, Roush WR, Aloy P, Cleveland JL, Gillies RJ, Ruppin E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nature Communications. 2018;9(1):2997. doi: 10.1038/s41467-018-05261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature Reviews. Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 7.Hochachka PW, Mommsen TP. Protons and anaerobiosis. Science. 1983;219(4591):1391–1397. doi: 10.1126/science.6298937. [DOI] [PubMed] [Google Scholar]
- 8.Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nature Reviews. Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 9.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Research. 1989;49(23):6449–6465. [PubMed] [Google Scholar]
- 12.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature Reviews. Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 13.Kallinowski F, Schlenger KH, Runkel S, Kloes M, Stohrer M, Okunieff P, et al. Blood flow, metabolism, cellular microenvironment, and growth rate of human tumor xenografts. Cancer Research. 1989;49(14):3759–3764. [PubMed] [Google Scholar]
- 14.Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiotherapy and Oncology. 1984;2(4):343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 15.Gillies RJ, Liu Z, Bhujwalla Z. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. The American Journal of Physiology. 1994;267(1 Pt 1):C195–C203. doi: 10.1152/ajpcell.1994.267.1.C195. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths JR, Stevens AN, Iles RA, Gordon RE, Shaw D. 31P-NMR investigation of solid tumours in the living rat. Bioscience Reports. 1981;1(4):319–325. doi: 10.1007/BF01114871. [DOI] [PubMed] [Google Scholar]
- 17.De Brabander M, Geuens G, Nuydens R, Willebrords R, De Mey J. Microtubule stability and assembly in living cells: the influence of metabolic inhibitors, taxol and pH. Cold Spring Harbor Symposia on Quantitative Biology. 1982;46(Pt 1):227–240. doi: 10.1101/sqb.1982.046.01.026. [DOI] [PubMed] [Google Scholar]
- 18.Isfort RJ, Cody DB, Asquith TN, Ridder GM, Stuard SB, LeBoeuf RA. Induction of protein phosphorylation, protein synthesis, immediate-early-gene expression and cellular proliferation by intracellular pH modulation. Implications for the role of hydrogen ions in signal transduction. European Journal of Biochemistry. 1993;213(1):349–357. doi: 10.1111/j.1432-1033.1993.tb17768.x. [DOI] [PubMed] [Google Scholar]
- 19.Morita T, Nagaki T, Fukuda I, Okumura K. Clastogenicity of low pH to various cultured mammalian cells. Mutation Research. 1992;268(2):297–305. doi: 10.1016/0027-5107(92)90235-t. [DOI] [PubMed] [Google Scholar]
- 20.McConkey DJ, Orrenius S. Signal transduction pathways in apoptosis. Stem Cells. 1996;14(6):619–631. doi: 10.1002/stem.140619. [DOI] [PubMed] [Google Scholar]
- 21.Hu X, Chao M, Wu H. Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation. Signal Transduction and Targeted Therapy. 2017;2:16047. doi: 10.1038/sigtrans.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature Reviews. Cancer. 2012;12(7):487–493. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatenby RA, Gillies RJ, Brown JS. Evolutionary dynamics of cancer prevention. Nature Reviews. Cancer. 2010;10(8):526–527. doi: 10.1038/nrc2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szpiech ZA, Strauli NB, White KA, Ruiz DG, Jacobson MP, Barber DL, Hernandez RD. Prominent features of the amino acid mutation landscape in cancer. PLoS One. 2017;12(8):e0183273. doi: 10.1371/journal.pone.0183273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webb BA, Forouhar F, Szu FE, Seetharaman J, Tong L, Barber DL. Structures of human phosphofructokinase-1 and atomic basis of cancer-associated mutations. Nature. 2015;523(7558):111–114. doi: 10.1038/nature14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 27.White KA, Grillo-Hill BK, Esquivel M, Peralta J, Bui VN, Chire I, Barber DL. Beta-catenin is a pH sensor with decreased stability at higher intracellular pH. The Journal of Cell Biology. 2018;217(11):3965–3976. doi: 10.1083/jcb.201712041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vercoulen, Y., Kondo, Y., Iwig, J. S., Janssen, A. B., White, K. A., Amini, M., Barber D. L., Kuriyan J., Roose J. P. (2017). A Histidine pH sensor regulates activation of the Ras-specific guanine nucleotide exchange factor RasGRP1. Elife. 10.7554/eLife.29002. [DOI] [PMC free article] [PubMed]
- 29.Choi CH, Webb BA, Chimenti MS, Jacobson MP, Barber DL. pH sensing by FAK-His58 regulates focal adhesion remodeling. The Journal of Cell Biology. 2013;202(6):849–859. doi: 10.1083/jcb.201302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boedtkjer E, Bunch L, Pedersen SF. Physiology, pharmacology and pathophysiology of the pH regulatory transport proteins NHE1 and NBCn1: similarities, differences, and implications for cancer therapy. Current Pharmaceutical Design. 2012;18(10):1345–1371. doi: 10.2174/138161212799504830. [DOI] [PubMed] [Google Scholar]
- 31.Swietach P, Vaughan-Jones RD, Harris AL, Hulikova A. The chemistry, physiology and pathology of pH in cancer. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2014;369(1638):20130099. doi: 10.1098/rstb.2013.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Reviews. Drug Discovery. 2008;7(2):168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 33.Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nature Reviews. Cancer. 2013;13(9):611–623. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- 34.Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Reviews. 2007;26(2):299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- 35.Andersen AP, Moreira JM, Pedersen SF. Interactions of ion transporters and channels with cancer cell metabolism and the tumour microenvironment. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 2014;369(1638):20130098. doi: 10.1098/rstb.2013.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee AH, Tannock IF. Heterogeneity of intracellular pH and of mechanisms that regulate intracellular pH in populations of cultured cells. Cancer Research. 1998;58(9):1901–1908. [PubMed] [Google Scholar]
- 37.Hulikova A, Vaughan-Jones RD, Swietach P. Dual role of CO2/HCO3(−) buffer in the regulation of intracellular pH of three-dimensional tumor growths. The Journal of Biological Chemistry. 2011;286(16):13815–13826. doi: 10.1074/jbc.M111.219899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swietach P, Hulikova A, Vaughan-Jones RD, Harris AL. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene. 2010;29(50):6509–6521. doi: 10.1038/onc.2010.455. [DOI] [PubMed] [Google Scholar]
- 39.Hulikova A, Harris AL, Vaughan-Jones RD, Swietach P. Regulation of intracellular pH in cancer cell lines under normoxia and hypoxia. Journal of Cellular Physiology. 2013;228(4):743–752. doi: 10.1002/jcp.24221. [DOI] [PubMed] [Google Scholar]
- 40.Hulikova A, Swietach P. Rapid CO2 permeation across biological membranes: implications for CO2 venting from tissue. The FASEB Journal. 2014;28(7):2762–2774. doi: 10.1096/fj.13-241752. [DOI] [PubMed] [Google Scholar]
- 41.Swietach P, Wigfield S, Cobden P, Supuran CT, Harris AL, Vaughan-Jones RD. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. The Journal of Biological Chemistry. 2008;283(29):20473–20483. doi: 10.1074/jbc.M801330200. [DOI] [PubMed] [Google Scholar]
- 42.Wagner BA, Venkataraman S, Buettner GR. The rate of oxygen utilization by cells. Free Radical Biology & Medicine. 2011;51(3):700–712. doi: 10.1016/j.freeradbiomed.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu H, Ying M, Hu X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget. 2016;7(26):40621–40629. doi: 10.18632/oncotarget.9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yizhak K, Le Devedec SE, Rogkoti VM, Baenke F, de Boer VC, Frezza C, et al. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Molecular Systems Biology. 2014;10:744. doi: 10.15252/msb.20134993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey KM, Wojtkowiak JW, Cornnell HH, Ribeiro MC, Balagurunathan Y, Hashim AI, et al. Mechanisms of buffer therapy resistance. Neoplasia. 2014;16(4):354–364. doi: 10.1016/j.neo.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. American Journal of Physiology. Cell Physiology. 2007;292(1):C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 47.Giang AH, Raymond T, Brookes P, de Mesy Bentley K, Schwarz E, O'Keefe R, Eliseev R. Mitochondrial dysfunction and permeability transition in osteosarcoma cells showing the Warburg effect. The Journal of Biological Chemistry. 2013;288(46):33303–33311. doi: 10.1074/jbc.M113.507129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng G, Zielonka J, McAllister D, Tsai S, Dwinell MB, Kalyanaraman B. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. British Journal of Cancer. 2014;111(1):85–93. doi: 10.1038/bjc.2014.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boron WF. Sharpey-Schafer lecture: gas channels. Experimental Physiology. 2010;95(12):1107–1130. doi: 10.1113/expphysiol.2010.055244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Missner A, Pohl P. 110 years of the Meyer-Overton rule: predicting membrane permeability of gases and other small compounds. Chemphyschem. 2009;10(9–10):1405–1414. doi: 10.1002/cphc.200900270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Opavsky R, Pastorekova S, Zelnik V, Gibadulinova A, Stanbridge EJ, Zavada J, et al. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics. 1996;33(3):480–487. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 52.Pastorek J, Pastorekova S, Callebaut I, Mornon JP, Zelnik V, Opavsky R, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene. 1994;9(10):2877–2888. [PubMed] [Google Scholar]
- 53.Tureci O, Sahin U, Vollmar E, Siemer S, Gottert E, Seitz G, Parkkila AK, Shah GN, Grubb JH, Pfreundschuh M, Sly WS. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7608–7613. doi: 10.1073/pnas.95.13.7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. The Journal of Biological Chemistry. 2009;284(30):20299–20310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee SH, McIntyre D, Honess D, Hulikova A, Pacheco-Torres J, Cerdan S, et al. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. British Journal of Cancer. 2018;119(5):622–630. doi: 10.1038/s41416-018-0216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 57.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflügers Archiv. 2004;447(5):619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 58.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. The Journal of Biological Chemistry. 2006;281(14):9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 59.Swietach P, Zaniboni M, Stewart AK, Rossini A, Spitzer KW, Vaughan-Jones RD. Modelling intracellular H(+) ion diffusion. Progress in Biophysics and Molecular Biology. 2003;83(2):69–100. doi: 10.1016/s0079-6107(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 60.Irving M, Maylie J, Sizto NL, Chandler WK. Intracellular diffusion in the presence of mobile buffers. Application to proton movement in muscle. Biophysical Journal. 1990;57(4):717–721. doi: 10.1016/S0006-3495(90)82592-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Junge W, McLaughlin S. The role of fixed and mobile buffers in the kinetics of proton movement. Biochimica et Biophysica Acta. 1987;890(1):1–5. doi: 10.1016/0005-2728(87)90061-2. [DOI] [PubMed] [Google Scholar]
- 62.Cukierman S. Et tu, Grotthuss! And other unfinished stories. Biochimica et Biophysica Acta. 2006;1757(8):876–885. doi: 10.1016/j.bbabio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Wakabayashi S, Shigekawa M, Pouyssegur J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiological Reviews. 1997;77(1):51–74. doi: 10.1152/physrev.1997.77.1.51. [DOI] [PubMed] [Google Scholar]
- 64.Parker MD, Boron WF. The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiological Reviews. 2013;93(2):803–959. doi: 10.1152/physrev.00023.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas RC. Cell growth factors. Bicarbonate and pHi response. Nature. 1989;337(6208):601. doi: 10.1038/337601a0. [DOI] [PubMed] [Google Scholar]
- 66.Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Molecular Aspects of Medicine. 2013;34(2–3):494–515. doi: 10.1016/j.mam.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. The Journal of Experimental Biology. 2009;212(Pt 11):1672–1683. doi: 10.1242/jeb.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology (Bethesda) 2008;23:104–114. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 69.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. Journal of Magnetic Resonance Imaging. 2002;16(4):430–450. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 70.Loewenstein WR, Kanno Y. Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature. 1966;209(5029):1248–1249. doi: 10.1038/2091248a0. [DOI] [PubMed] [Google Scholar]
- 71.Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nature Reviews. Cancer. 2010;10(6):435–441. doi: 10.1038/nrc2841. [DOI] [PubMed] [Google Scholar]
- 72.McLachlan E, Shao Q, Wang HL, Langlois S, Laird DW. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Research. 2006;66(20):9886–9894. doi: 10.1158/0008-5472.CAN-05-4302. [DOI] [PubMed] [Google Scholar]
- 73.Dovmark TH, Hulikova A, Niederer SA, Vaughan-Jones RD, Swietach P. Normoxic cells remotely regulate the acid-base balance of cells at the hypoxic core of connexin-coupled tumor growths. The FASEB Journal. 2018;32(1):83–96. doi: 10.1096/fj.201700480R. [DOI] [PubMed] [Google Scholar]
- 74.Dovmark TH, Saccomano M, Hulikova A, Alves F, Swietach P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene. 2017;36(32):4538–4550. doi: 10.1038/onc.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]