

Abstract
Balanced rates of mitochondrial division and fusion are required to maintain mitochondrial function, as well as cellular and organismal homeostasis. In mammals, the cellular machines that mediate these processes are dynamin-related GTPases; the cytosolic DRP1 mediates division, while the outer membrane MFN½ and inner membrane OPA1 mediate fusion. Unbalanced mitochondrial dynamics are linked to varied pathologies, including cell death and neurodegeneration, raising the possibility that small molecules that target the division and fusion machines to restore balance may have therapeutic potential. Here we describe the discovery of novel small molecules that directly and selectively inhibit assembly-stimulated GTPase activity of the division dynamin, DRP1. In addition, these small molecules restore wild type mtDNA copy number in MFN1 knockout mouse embryonic fibroblast cells, a phenotype linked to deficient mitochondrial fusion activity. Thus, these compounds are unique tools to explore the roles of mitochondrial division in cells, and to assess the potential therapeutic efficacy of rebalancing mitochondrial dynamics in pathologies associated with excessive mitochondrial division.
Keywords: DRP1, Mitochondria, Small molecule inhibitor, Fission, Dynamics
1. Introduction
Mitochondria are dynamic organelles that display morphologies ranging from elongated and interconnected tubular networks to small fragments, depending on the relative rates of mitochondrial division and fusion. Cycles of mitochondrial division and fusion are thought to maintain compartment homogeneity while also allowing for incorporation of smaller organelles via biogenesis and for removal of damaged organelles via mitophagy [1]. These processes are mediated by large multi-domain dynamin-related GTPases (DRPs) that function via self-assembly to remodel diverse membranes in cells. The DRPs that mediate mitochondrial fusion are MFN1/MFN2 and OPA1, which catalyze fusion of the mitochondrial outer and inner membranes, respectively [2]. Mitochondrial division is mediated by another DRP, DRP1, which is the catalytic component of the mitochondrial division machine that self-assembles on the surface of mitochondria at points of constriction marked by ER-mitochondrial contacts and facilitates membrane scission [3].
The DRP1 monomer is an extended structure in which the GTPase (G) domain together with the bundling signal element (BSE) is positioned on top of a helical stalk; on the opposing end is the insert B domain, which is thought to regulate assembly via various effectors [3]. DRP1 is an obligate dimer, and both dimers and tetramers are formed via interfaces located in the stalk region [4]. DRP1 tetramers further assemble into helical structures in a manner that is stimulated by GTP, and in vitro also by membrane bilayer templates containing the mitochondrial derived lipid, car-diolipin [5,6]. GTP is thought to trigger conformational changes via the BSE that mediate G-G domain interactions across helical rungs, which in turn catalyze assembly-stimulated GTP hydrolysis required for membrane scission activity and oligomer disassembly [3].
The concurrent and opposing activities of the division and fusion DRPs are required for the maintenance of physiological homeostasis. Deletion of either division or fusion DRPs unbalances mitochondrial dynamics, and in mice causes embryonic lethality [7,8]. In humans, mutations in DRP1 cause early infant mortality or childhood epileptic encephalopathy [9,10], while mutations in MFN2 and OPA1 cause the neurodegenerative diseases, Charcot Marie Tooth 2A (CMT2A) and dominant optic atrophy (DOA) [11], respectively. Unbalanced mitochondrial dynamics is also observed in response to a variety of stresses, as the DRPs that mediate mitochondrial dynamics are highly regulated and integrated into cellular signaling pathways. In cultured human cells, UV exposure, cycloheximide treatment, and starvation stimulate mitochondrial fusion, resulting in the formation of a hyperfused mitochondrial structure and improved cell survival, presumably by buffering defects and improving energy production efficiency [12]. Conversely, mitochondrial division is stimulated during cellular pathological stresses such as in neurodegeneration and in apoptosis, where activation of DRP1 recruitment to mitochondria has been shown to play a regulatory role in the permeabilization of the mitochondrial outer membrane and subsequent cell death [13].
Rebalancing mitochondrial dynamics by inhibiting mitochondrial division may therefore be therapeutic in many diseases with excessive mitochondrial fission, including kidney diseases [14], ophthalmic disorders [15], neurological diseases [16], and cancer [17]. Indeed, in mouse models with attenuated mitochondrial division, with associated severe cardiomyopathy, rebalancing mitochondrial dynamics by also decreasing fusion rescued both heart dysfunction and the associated respiratory function at the organ level [18,19].
Given that many pathologies are linked to altered mitochondrial dynamics, therapeutic interventions that rebalance dynamics hold promise. Previous work characterized a compound, mdivi-1, discovered in a yeast based screen for inhibitors of the yeast mitochondrial division dynamin, Dnm1 [20]. Although mdivi-1 has been shown to block mitochondrial fragmentation and DRP1 recruitment during apoptosis in mammalian cells, as well as to mitigate a variety of disease-related phenotypes in mammalian models, recent work has indicated that it has off-target effects [21]. Thus, to explore the therapeutic potential of selective DRP1 inhibition, we describe here a novel class of 1H-pyrrole-2-carboxamide compounds that directly inhibit assembly-stimulated DRP1 GTPase activity in vitro.
2. Materials and methods
2.1. Protein production
Human DRP1 (1—699), dynamin-1 (1—752), and OPA1 (195—960) were expressed with cleavable N-terminal His6-tags in E. coli and purified via affinity chromatography using Ni-NTA resin (Viva Biotech). The His6-tag was removed followed by a second Ni-NTA resin purification for dynamin-1 and OPA1.
2.2. Liposome preparation
Chloroform solutions of soybean polar lipid extract (Avanti) and bovine heart cardiolipin (Avanti) were mixed in an 80:20 molar ratio in a round bottom flask, and the chloroform was evaporated. Lipids were further desiccated under vacuum at room temperature for 1 h, and then hydrated in 10 mM HEPES pH 7.0/100 mM KCl at a final concentration of 2 mg/mL at room temperature for 2 h, with periodic vortexing. Lipids were extruded through a 1 μm Nuclepore membrane (Whatman) using a Mini-Extruder (Avanti) to produce liposomes.
2.3. Malachite green reagent preparation
Malachite green reagent was prepared by separately dissolving 34 mg malachite green carbinol base (Sigma) in 40 mL of 1 N HCl and 1 g of ammonium molybdate (Sigma) in 15 mL of 4 N HCl. Solutions were vortexed and placed on a rotational shaker for 1 h at room temperature. The volume of each solution was increased to 50 mL using H2O, and then combined and filtered through a 0.22 μm filter. The amount of reagent prepared was scaled as appropriate.
2.4. Liposome-dependent GTPase activity assay
For routine screening in 96-well format, the GTPase activity of DRP1, dynamin-1, or OPA1 was assessed by preincubating enzyme (0.6 μM) with liposomes (0.1 mg/mL) and vehicle (DMSO) or inhibitor as indicated for 20 min at room temperature. Reactions were initiated by the addition of GTP (1 mM) in a microtiter plate at 37 °C. The final buffer composition was 10 mM Hepes pH 7.0, 100 mM KCl, 4mM MgCl2, 1 mM DTT, 1% DMSO. Reactions were stopped with EDTA at 125 μM final concentration. Samples were then incubated with malachite green reagent for 5 min at room temperature, and the absorbance at 650 nm was measured. The change in absorbance was converted to the amount of free phosphate in the reaction, from which the rate of GTPase activity was determined. 1C50 values and max % inhibition were determined using 4-parameter fits with GraphPad Prism 6.0. For high-throughput screening in 1536-well format, 0.5 μM enzyme and 100 μM GTP were utilized, and activity was monitored via fluorescence (excitation 530 nm/emission 630 nm).
2.5. Compound synthesis
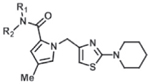
Compounds 2 and 3 were synthesized from piperidine-1-carbothioamide (a), leading to the common intermediate d after a 3-step sequence (Supplementary Scheme 1). Conventional amide coupling between carboxylic acid d and aniline produced 3. However, these coupling conditions failed to produce 2 when using 4-aminobenzoic acid, presumably due to the less reactive amino group. This problem was circumvented by a 2-step process: 4-iodoaniline was coupled with d to produce e, which then underwent a carbonylation reaction to produce 2. All compounds in Tables 1 and 2 were synthesized in analogous fashion.
Table 1.
Structure-activity relationship of amide constituent of 1H-pyrrole-2-carboxamide compounds. Compounds were characterized in the malachite green GTPase activity assay, using a 10-point dose response at 3-fold dilution starting from 10 μM, and the IC50 values and max % inhibition were determined using a 4-parameter fit of this data with GraphPad Prism 6.0. DYN1 = dynamin-1. ND = not determined.
 |
IC50 (μM) | Max % inhibition | ||||
|---|---|---|---|---|---|---|
| R1 | R2 | DRP1 | OPA1 | DYN1 | ||
| 1 | H | 6.1 | >100 | >100 | 90 | |
| 2 | H | 2.4 | >100 | >100 | 84 | |
| 3 | H | 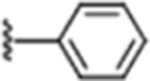 |
2.3 | >100 | >100 | 55 |
| 4 | H | 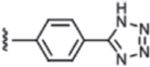 |
1.0 | >100 | >100 | 88 |
| 5 | Me | >100 | ND | ND | 12 | |
| 6 | H | 0.72 | >100 | >100 | 83 | |
| 7 | H | 1.2 | >100 | >100 | 42 | |
Table 2.
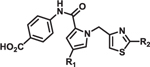
Structure-activity relationship of pyrrole and thiazole constituents of 1H-pyrrole-2-carboxamide compounds. IC50 values and max % inhibition were determined as in Table 1. DYN1 = dynamin-1.
 |
IC50 (μM) | Max % inhibition | ||||
|---|---|---|---|---|---|---|
| R1 | R2 | DRP1 | OPA1 | DYN1 | ||
| 8 | Cl | 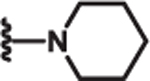 |
2.3 | >100 | >100 | 77 |
| 9 | Ph | 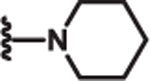 |
1.5 | >100 | >100 | l9 |
| 10 | Me | 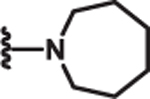 |
2.1 | >100 | >100 | 80 |
| 11 | Me | 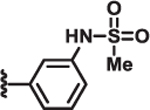 |
>100 | >100 | >100 | 6 |
2.6. Mammalian cell growth and transfection
Wildtype and MFN1 knockout (mfn1−/−) mouse embryonic fibroblast cells (MEFs) (ATCC) were grown in high-glucose Dubec-co’s Modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were seeded at ~8.0 × 104 per mL in 100 mm round bottom standard tissue culture dishes (Corning) 24 h before transient transfection. Drp1K38A-mCherry plasmid transfections were performed for 5 h in Optimem with 30 μL Lipofectamine 2000 reagent (1nvitrogen) per dish.
2.7. mtDNA quantitation
Cells were seeded as above. For experiments with compounds, MEFs were treated with compound or DMSO as indicated for 48 h. For experiments with the DRP1 K38A allele, Drp1K38A-mCherry transfected MEFs were sorted for mCherry-positive cells after 26 h of transfection using the Astrios Cytek xP5 (FACScan) 5 color cytometer (Beckman Coulter). DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen) according to manufacturer instructions. Quantitative PCR (qPCR) reactions were prepared using SSoAdvanced Universal SYBR Green Supermix (Bio-Rad), 500 nM mtDNA or nDNA primers [22], and 100 ng of isolated gDNA. Data was analyzed setting nDNA as reference and normalized to vehicle controls using the delta delta Ct method [23].
3. Results
3.1. Identification ofinhibitors ofassembly-stimulated DRP1 GTPase activity in vitro
It has been shown that liposomes containing cardiolipin enhance DRP1 GTPase activity [5,6]. Consistent with these previous findings, we observed significant enhancement of human DRP1 GTPase activity by liposomes containing 20% cardiolipin (Fig. 1a). In addition, as expected, DRP1 GTPase activity was cooperatively dependent on the concentration of DRP1 (Fig. 1b). These observations indicate that, under our conditions, DRP1 GTP hydrolysis was at least in part the result of assembly-stimulated GTPase activity. Using these parameters, the assay was utilized to screen a proprietary 5 × 105 compound library (Astellas Pharma). Primary screening used a cut-off of 30% GTPase inhibition at 20 μM compound concentration, and resulted in a hit rate of 0.17% with Z′ factor greater than 0.85. Primary hits were further evaluated in serial dilution, and compounds with IC50 values less than 10 μM for DRP1 and greater than 50 μM for dynamin-1 and OPA1, two closely related DRPs, were prioritized.
Fig. 1.
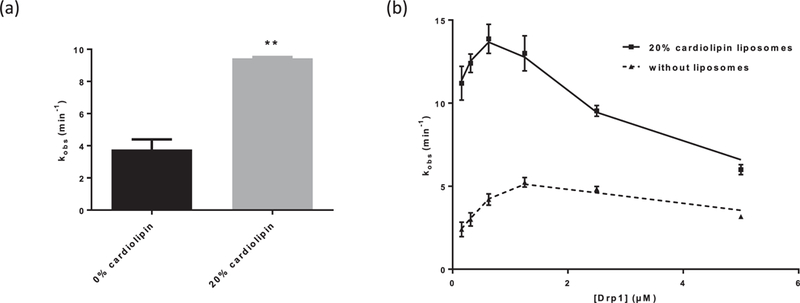
Optimization of malachite green GTPase activity assay. (a) Inclusion of 20% cardiolipin in liposomes leads to significant stimulation of DRP1 GTPase activity. **p < 0.01, unpaired t-test (n = 3). (b) DRP1 GTPase activity is cooperatively dependent on the concentration of DRP1 (n = 3).
Compound 1 was prioritized from the high-throughput screen based on our criteria (Table 1), and additional chemistry was performed to explore which features of its structure were critical for potency (IC50) and efficacy (degree of maximal inhibition) in the DRP1 GTPase activity assay. A 2-fold improvement in potency was observed when the 4-carboxylbenzyl in 1 was replaced with 4-carboxylphenyl (2) (Table 1). Removal of the carboxylic acid group led to compound 3, which showed potency similar to compound 2, but we observed a decrease in maximal inhibition (55% for 3 vs 84% for 2). Based on these initial findings, subsequent optimization efforts focused on 2 and 3.
Isosteric replacement of the carboxylic acid with a tetrazole led to a compound (4) with similar potency and efficacy of DRP1 inhibition as compound 2. Adding a methyl group to the amide N (5) eliminated DRP1 inhibitoryactivity. An additional 3-fold increase in potency was observed when the carboxylic acid was extended in space by introducing a biphenyl group (6). Compound 7, in which the carboxylic acid group was replaced with a 2-pyridyl, showed potency similar to 2, but as with compound 3, the maximum inhibition decreased (42% for 7 compared to 84% for 2).
Additional SAR investigation examined the influence of substitutions on the pyrrole and thiazole rings (Table 2). Replacing the pyrrole with a phenyl group significantly diminished potency, as did methyl substitutions on the 3-or 5-position on the pyrrole ring (IC50 > 25 μM; data not shown). However, chloro (8) and phenyl (9) substitutions on the 4-position of the pyrrole ring were tolerated. Replacing the piperidinyl group with an azepine (10) led to a compound with potency comparable to compound 2. However, removal of the piperidinyl moiety (data not shown) or replacing it with an aryl group (11) led to loss of DRP1 inhibition.
3.2. Partial DRP1 inhibitors are selective for assembly-stimulated hydrolysis
We further characterized active compounds with respect to their mechanism of DRP1 inhibition in vitro. We observed a consistent separation of active compounds into two classes based on degree of maximum inhibition (efficacy) in the DRP1 GTPase activity assay: full inhibitors with a maximum % inhibition greater than 75%, such as 4 and 6, and partial inhibitors with a maximum % inhibition of 40—60%, such as compound 7 (Table 1, Fig. 2a). Both types of inhibitors contrasted with mdivi-1, which did not inhibit oligomeric DRP1 GTPase activity under our assay conditions (Fig. 2a).
Fig. 2.
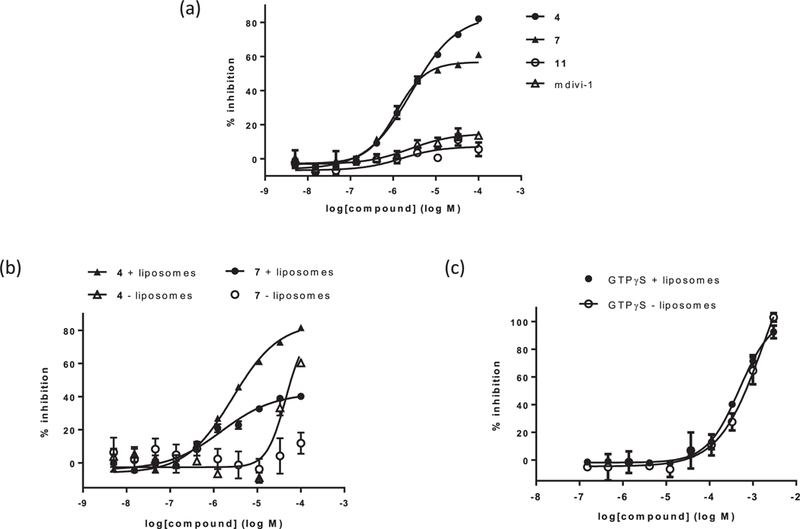
Compound characterization in malachite green GTPase activity assay. (a) Dose response curves in the presence of liposomes for three 1H-pyrrole-2-carboxamide compounds and mdivi-1. Compound 4 is a full inhibitor, with maximum inhibition of greater than 75% DRP1 activity. Compound 7 is a partial inhibitor, with maximum inhibition of 40—60% DRP1 activity. Compound 11 and mdivi-1 do not inhibit DRP1 up to 100 μM compound concentration. (b) Dose response curves in the presence and absence of liposomes for 4 and 7. The potencies of both compounds decrease in the absence of liposomes; 7 is inactive in the absence of liposomes, while 4 is still active at higher concentrations. (c) Dose response curves in the presence and absence of liposomes for GTPγS. The potency of GTPγS is unaffected by liposomes.
Kinetic inhibition analysis revealed both classes of compounds to be uncompetitive relative to GTP, indicating that they preferentially bind to DRP1 in the presence of GTP, consistent with an ability to inhibit assembly-stimulated hydrolysis (Fig. 3). Also consistent with this, both types of inhibitors were significantly less potent in the absence of cardiolipin-containing liposomes (Fig. 2b). This mode of inhibition is in contrast to GTPγS, a nonhydrolyzable GTP analog, which was unaffected by liposomes (Fig. 2c). However, unlike partial inhibitors, full inhibitors, such as compound 4, retained GTPase inhibitory activity at higher concentrations even in the absence of liposomes (Fig. 2b). In addition, with the partial inhibitors, we observed that the degree of maximum inhibition correlated with the degree of DRP1 GTPase rate enhancement dependent on the cardiolipin-containing liposomes, which was typically 2—3 fold (Fig. 1a). These observations suggest that the partial inhibitors exclusively inhibit DRPl’s assembly-stimulated GTP hydrolysis, while the full inhibitors are additionally capable of inhibiting the non-oligomeric DRP1 dimers and tetramers.
Fig. 3.
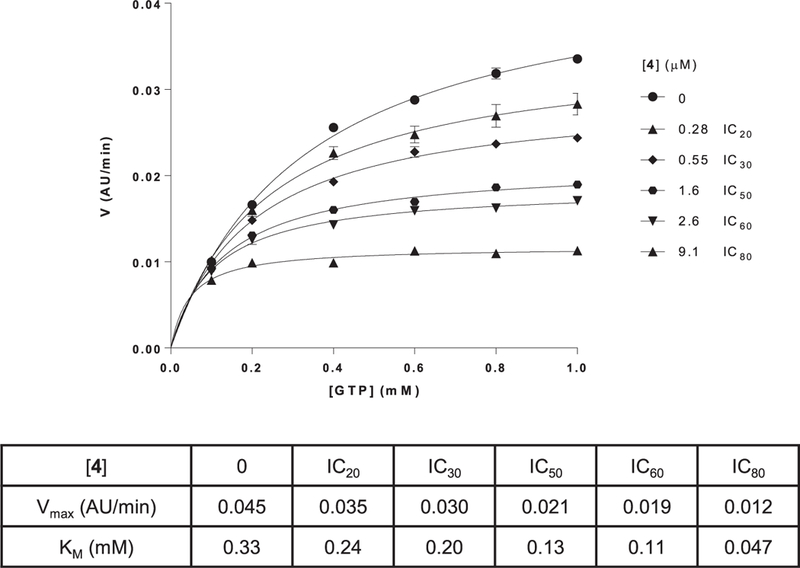
Mechanism of inhibition study on compound 4 relative to GTP shows an effect on both Vmax and KM, indicating an uncompetitive mode of binding. Analogous results were obtained with other active 1H-pyrrole-2-carboxamide compounds. Best-fit values were generated with GraphPad Prism 6.0.
3.3. Inhibitors are selective for DRP1 over other DRP family members
DRPs are a family of proteins with related structure and mechanism. Thus, we determined the specificity of our 1H-pyrrole-2-carboxamide DRP1 inhibitors by examining their effects on the GTPase activities of two closely related DRPs, the human endocytic dynamin, dynamin-1, and the mitochondrial inner membrane fusion dynamin, OPA1. Our analysis indicated that all active compounds were at least 2 orders of magnitude more potent inhibitors of DRPl relative to dynamin-1 and OPA1 (Table 1, Table 2). This is in contrast to GTPγS, which inhibits all 3 enzymes with comparable weak potency of 300—500 μM, consistent with previous characterizations (data not shown). Thus, these data indicate that our compounds are highly selective DRP1 inhibitors.
3.4. DRP1 inhibitors reverse defects in mtDNA copy number in cells deficient in mitochondrial fusion
To investigate the efficacy of the 1 H-pyrrole-2-carboxamide DRP1 inhibitors in a more physiological context, we sought a cellular readout of DRP1 inhibition. We reasoned that rebalancing dynamics by inhibiting DRPl-mediated mitochondrial division would rescue a cellular phenotype linked to decreased mitochondrial fusion. MFN1 is a key mediator of mitochondrial outer membrane fusion, which acts in opposition of DRP1 in mammalian cells to promote mitochondrial distribution and content mixing [2]. Loss of MFN1 in MEFs leads to mitochondrial fragmentation and cellular loss of the mitochondrial genome (mtDNA) [24]. Consistent with this, qPCR analysis showed that mfn1−/− MEFs were deficient in mtDNA copy number as compared to wildtype MEFs (Fig. 4a).
Fig. 4.
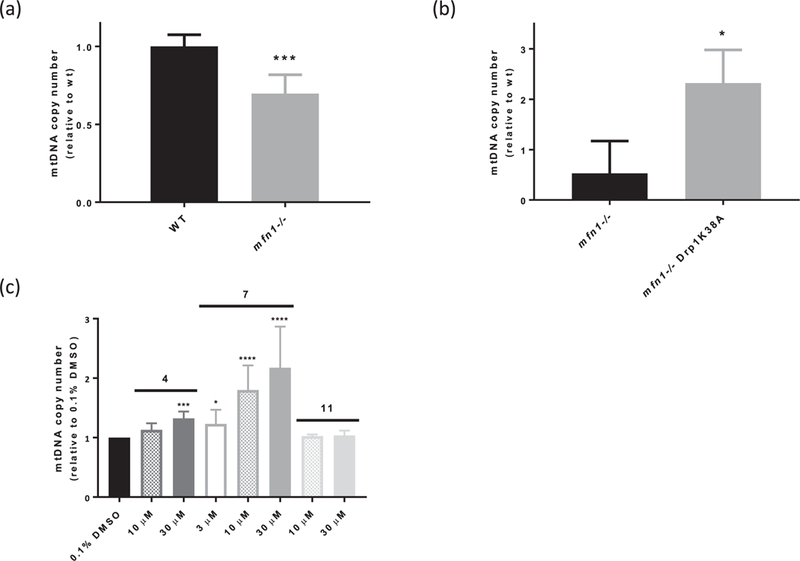
Inhibition of DRP1 rescues mtDNA copy number defect in mfn1−/− MEFs. (a) mfn1−/− MEFs have decreased mtDNA copy number relative to wildtype (WT) cells. ***p < 0.001, unpaired t-test (n = 6). (b) Transiently expressing Drp1K38A-mCherry rescues mtDNA copy number defect in mfn1−/− MEFs. *p < 0.05, unpaired t-test (n = 3). (c) Two active 1H-pyrrole-2-carboxamide compounds, 4 and 7, rescue mtDNA copy number defect in mfn1−/− MEFs in dose-dependent fashion, while an inactive compound from the same scaffold, 11, does not. ****p < 0.0001, ***p < 0.001, *p < 0.05, one-way ANOVA followed by Dunnett’s test vs. 0.1% DMSO (n = 4).
We examined the effect of DRP1 inhibition by qPCR in mfn1−/− MEFs expressing a dominant negative DRP1 K38A allele. We observed an increase in mtDNA copy number in mfn1−/− MEFs relative to both control mfn1−/− and wildtype MEFs, indicating that loss of mtDNA in cells with attenuated mitochondrial fusion can be mitigated by inhibiting division (Fig. 4b). Therefore, we examined the effects of several of our active inhibitors in cells using qPCR, and observed a dose-dependent increase in mtDNA copy number in mfn1−/− cells up to a similar degree as expression of the K38A allele (Fig. 4c). In contrast, treatment with the structurally similar but inactive compound 11 had no effect on mtDNA copy number in mfn1−/− cells (Fig. 4c). Taken together, these data suggest that our inhibitors are effective at rebalancing mitochondrial dynamics in mfn1−/− MEFs at concentrations consistent with their inhibitory effects on DRP1 GTPase activity.
Although treatment with both full and partial inhibitors increased mtDNA copy number in mfn1−/− cells, we found that the partial inhibitors were more potent than full inhibitors with identical biochemical IC50 values (such as 7 vs. 4) (Fig. 4c). Full inhibitors, including compound 4, had reasonable kinetic solubility, in vitro rat liver microsome stability and Caco-2 permeability, indicating that a lack of cellular permeability or stability is unlikely to account for this observed difference in cellular efficacy (Table 3). These observations suggest that, in a cellular context, it may be advantageous to selectively target oligomeric DRP1, consistent with the idea that assembled DRPs are responsible for their associated membrane remodeling activities.
Table 3.
In vitro ADME results for selected inhibitors.
| Kinetic solubility at pH l.4 (μM) | RLM Clint (μL/min/mg protein) | Caco-2 permeability (Papp A>B/B>A 10−6cm-s−1) | |
|---|---|---|---|
| 2 | 186 | 6.8 | 1l/13 |
| 4 | 19l | 21 | 18/46 |
4. Discussion
The role of mitochondrial dysfunction in the pathology of a wide variety of disease states is increasingly appreciated [25]. Mitochondrial dynamics are important in maintaining overall mitochondrial health, with healthy mitochondria having a tubular morphology spanning a range of lengths from more elongated to shorter tubes. Under diverse in vitro and in vivo stress conditions, the shift of mitochondria to severely fragmented spheres has been observed [11,25]. Compounds that selectively modulate mitochondrial dynamics with a defined mechanism of action are useful to elucidate the role of this shift in morphology in the propagation of the stress response, as well as in other normal and pathological biological processes.
Of the proteins involved in modulating mitochondrial dynamics, DRP1 is the key enzymatic component mediating mitochondrial fission. DRP1 is a cytosolic enzyme recruited to the mitochondrial membrane in response to numerous cellular signals, where it forms oligomers around the mitochondrial membrane that mediate mitochondrial membrane scission [3]. We designed a biochemical screen to identify small molecule inhibitors of oligomeric DRP1, which yielded a family of lH-pyrrole-2-carboxamide compounds following SAR exploration of a prioritized hit (Table 1, Table 2). Among these compounds, we identified both full and partial inhibitors, and we speculate that the partial inhibitors selectively inhibit the oligomeric form of DRP1, while the full inhibitors can also inhibit the non-assembled form of the enzyme (Fig. 2b). These two distinct classes of DRP1 inhibitors may be useful to elucidate the relative contributions of different structural forms of DRP1 to its mechanistic action and cellular function. Indeed, partial inhibitors with similar biochemical IC50 values to full inhibitors were more potent in rescuing the mtDNA defect in mfn1−/− MEFs than the full inhibitors (Fig. 4c), suggesting that it may be therapeutically beneficial to selectively target the oligomeric, membrane-bound form of DRP1.
It has been demonstrated that inhibiting mitochondrial fragmentation via DRP1 inhibition using a dominant negative allele can ameliorate cellular stress responses, including delaying apoptosis, supporting a role for DRP1 early in the apoptotic response pathway and suggesting that inhibition of DRP1 could be acutely protective against stress-activated apoptosis [26]. In addition, another potential therapeutic application of DRP1 inhibition could be in cases where there is an imbalance in mitochondrial dynamics resulting from defects in mitochondrial fusion. In several mouse models, it has been shown that rebalancing of mitochondrial dynamics can reverse defects at the whole organism level [18,19]. Our observations are consistent with these results: the mtDNA copy number deficit in cells with attenuated mitochondrial fusion (Fig. 4a, [24]) was robustly reversed by expression of a dominant negative DRP1 or by treating cells with our DRP1 inhibitors (Fig. 4bc). It is of note that we did not see a robust effect of our inhibitors on the mitochondrial morphology of either wildtype or mfnl−/− MEFs (data not shown), indicating that the effect of our compounds on DRP1 function does not completely overlap with that of the dominant negative construct. Although additional work is required to understand the mechanism by which these compounds engage DRP1 in cells, they are unique tools to further explore the roles of mitochondrial division in cells, and to assess the potential therapeutic efficacy of rebalancing mitochondrial dynamics in pathologies associated with excessive mitochondrial division.
Supplementary Material
Acknowledgements
We thank the high-throughput screening group at Astellas Pharma for their compound library screening efforts, and Taisuke Takahashi for hit evaluation. We also thank our colleagues at Syngene International, in particular Atul Tiwari and Jeyaprakash N. Seenisamy, for their work in compound synthesis and characterization. We are grateful to our colleagues at Mitobridge for valuable discussions and experimental efforts. This work was supported in part by NIH Ruth L. Kirschstein Postdoctoral Fellowship F32GM113388 and a Burroughs Wellcome Postdoctoral Enrichment Award (both to S.C.L.), and a UC Davis Molecular and Cellular Biology Segal Laboratory Undergraduate Research Award (to L.F.U.).
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.bbrc.2018.03.189.
Transparency document
Transparency document related to this article can be found online at https://doi.org/10.1016/j.bbrc.2018.03.189.
References
- [1].Gottlieb RA, Bernstein D, Mitochondrial remodeling: rearranging, recycling, and reprogramming, Cell Calcium 60 (2016) 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pernas L, Scorrano L, Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function, Annu. Rev. Physiol. 78 (2016) 505–531. [DOI] [PubMed] [Google Scholar]
- [3].Ramachandran R, Mitochondrial dynamics: the dynamin superfamily and execution by collusion, Semin. Cell Dev. Biol. (2017). http://dx.dox.org/10.1016/j.semcdb.2017.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fröhlich C, Grabiger S, Schwefel D, et al. , Structural insights into oligomerization and mitochondrial remodeling of dynamin 1-like protein, EMBO J. 32 (2013) 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bustillo-Zabalbeitia I, Montessuit S, Raemy E, et al. , Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1, PLoS One 9 (2014) e102738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Macdonald PJ, Stepanyants N, Mehrotra N, et al. , A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission, Mol. Biol. Cell 25 (2014) 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wakabayashi J, Zhang Z, Wakabayashi N, et al. , The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice, J. Cell Biol. 186 (2009) 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen H, Detmer SA, Ewald AJ, et al. , Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development, J. Cell Biol. 160 (2003) 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Waterham HR, Koster J, van Roermund CWT, et al. , A lethal defect of mitochondrial and peroxisomal fission, N. Engl. J. Med. 356 (2007) 1736–1741. [DOI] [PubMed] [Google Scholar]
- [10].Chao Y-H, Robak LA, Xia F, et al. , Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila, Hum. Mol. Genet. 25 (2016) 1846–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Archer SL, Mitochondrial dynamics — mitochondrial fission and fusion in human diseases, N. Engl. J. Med. 369 (2013) 2236–2251. [DOI] [PubMed] [Google Scholar]
- [12].Tondera D, Grandemange S, Jourdain A, et al. , SLP-2 is required for stress-induced mitochondrial hyperfusion, EMBOJ. 28 (2009) 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oettinghaus B, Licci M, Scorrano L, et al. , Less than perfect divorces: dysregulated mitochondrial fission and neurodegeneration, Acta Neuropathol. 123 (2012) 189–203. [DOI] [PubMed] [Google Scholar]
- [14].Ayanga BA, Badal SS, Wang Y, et al. , Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy, J. Am. Soc. Nephrol. 27 (2016) 2733–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim KY, Perkins GA, Shim MS, et al. , DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma, Cell Death Dis. 6 (2015) e1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guo X, Disatnik MH, Monbureau M, et al. , Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration, J. Clin. Invest. 123 (2013) 5371–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xie Q, Wu Q, Horbinski CM, et al. , Mitochondrial control by DRP1 in brain tumor initiating cells, Nat. Neurosci. 18 (2015) 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen H, Ren S, Clish C, et al. , Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy, J. Cell Biol. 211 (2015) 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song M, Franco A, Fleischer JA, et al. , Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence, Cell Metabol. 26 (2017) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cassidy-Stone A, Chipuk JE, Ingerman E, et al. , Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization, Dev. Cell 14 (2008) 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bordt EA, Clerc P, Roelofs BA, et al. , The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species, Dev. Cell 40 (2017) 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen H, Vermulst M, Wang YE, et al. , Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations, Cell 141 (2010) 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method, Methods 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [24].Chen H, McCaffery JM, Chan DC, Mitochondrial fusion protects against neurodegeneration in the cerebellum, Cell 130 (2007) 548–562. [DOI] [PubMed] [Google Scholar]
- [25].Nunnari J, Suomalainen A, Mitocondria: in sickness and in health, Cell 148 (2012) 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Frank S, Gaume B, Bergmann-Leitner ES, et al. , The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis, Dev. Cell 1 (2001) 515–525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.