

Abstract
A vaccine that prevents transmission of infection is urgently needed in the fight against tuberculosis (TB). Results of clinical trials have been disappointing. Major problems include lack of biomarkers and understanding of the mechanisms of disease and protection. A more fundamental problem is that the scientific community seldom recognizes that primary and post-primary TB are distinct disease entities. Nearly all vaccine candidates have been designed and tested in models of primary TB, while transmission of infection is mediated by post-primary TB. Post-primary TB is seldom studied because no animal develops the disease as it exists in humans. Nevertheless, mice, guinea pigs and rabbits all develop infections that at certain points appear to be models of human post-primary TB. Slowly progressive pulmonary TB in immunocompetent mice is an example. It is characterized by an alveolitis with infected foamy macrophages that have multiple characteristics of the human disease. We demonstrated that inclusion of an immune modulating agent, lactoferrin, with a BCG vaccine in this model induced a sustained reduction in lung pathology, but not numbers of organisms in tissue. Since the animals die of expanding pathology, this demonstrates the feasibility of using selected animal models for studies of vaccines against post-primary TB.
Keywords: Tuberculosis, vaccine, post-primary, lactoferrin, pathology
In 2009, the National Academy published a monograph “A New Biology for the 21st Century” stating that molecular biology of the 20th century was too narrowly focused to successfully address problems in areas such as evolution, morphogenesis and infectious disease (1). “Advancing from identifying parts to defining complex systems is well beyond its present capabilities.” The New Biology requires integration of the knowledge from many disciplines to permit deeper understanding of biological systems.
Carl Woese, a thought leader in this area put it differently: “Science is impelled by two main factors, technological advance and a guiding vision (overview). A properly balanced relationship between the two is key to the successful development of a science. Without the proper technological advances the road ahead is blocked. Without a guiding vision there is no road ahead” (2). Biology hits the ‘wall of biocomplexity’ meaning that simply defining molecular components is insufficient to understand higher level biologic functions. The discipline of molecular biology has produced an astounding harvest in areas such as the gene and the nature of the cell. However, it chose to ignore other problems such as evolution and the nature of biological form that are fundamentally not understandable as collections of parts typically dismissing them as inconsequential (2). This becomes a problem as we try to address issues such as the pathogenesis of tuberculosis that exceed capabilities of molecular biology.
Tuberculosis is an excellent example.
Prior to the rise of molecular biology, investigators had a ‘guiding vision’ of TB, but lacked ‘technological advances’. They understood the pathology, radiology and clinical course of each stage of TB, but could not understand the biologic processes. Today, we have many ‘technological advances’, but have forgotten the ‘guiding vision’ and replaced it with a convenient but erroneous notion that granulomas are the only key lesion of TB (3). The result is that research on the pathogenesis of TB has hit the ‘wall of biocomplexity’. Modern publications on the pathogenesis of TB describe increasing numbers of mechanisms and contributing parts, but with an inaccurate guiding vision these pieces cannot be assembled to form a coherent explanation (4).
Most contemporary research on TB follows the guiding vision (overview or paradigm) that granulomas are the important lesion of all TB (3). This is a late 20th century concept that has no support among those who actually studied tissues of people with untreated pulmonary TB (5–7). As documented in a recent review, M. tuberculosis (MTB) produces, not just one, but two distinct disease entities known as primary and post-primary TB (8). Both are necessary for the continued survival of MTB. Primary TB mediates protective immunity to disseminated infection while post-primary TB causes tissue damage that results in formation of cavities. Primary TB has been extensively studied in humans and animals. Post-primary TB is seldom recognized or studied. It begins as an asymptomatic early infiltrate that may resolve or progress by bronchogenic spread and necrosis to become caseous pneumonia that fragments to produce cavities or is retained to produce post-primary granulomas and fibrocaseous disease (9). Primary and post-primary TB differ in histopathology, x-ray appearance, genetic predisposition and immune status of the host, age of onset, organ distribution, clinical course and susceptibility to protection induced by BCG (8). MTB is a highly successful human parasite because it produces both primary and post-primary TB as distinct disease entities in humans.
It has long been known that the most important risk factor for the type of TB disease is the existence of prior TB infection (5, 10). Post-primary TB is defined as the type of disease that develops after primary TB. It remains confined to the lungs, spreads via the airways as bronchogenic TB and causes far more necrosis than primary TB (6, 11). The fundamental pathologic and radiologic features of post-primary TB that were reported by many investigators in the preantibiotic era were ignored in the early days of molecular biology and have now been largely forgotten.
Failure to recognize the differences between primary and post-primary TB impedes vaccine research. For example, global leaders in TB vaccine development from six countries recently wrote that an improved vaccine is ‘an inevitability’ because the Bacille Calmette Guerin (BCG) vaccine can “provide decades of protection against tuberculosis disease” (12). The paper did not mention the fact that while BCG is effective against primary TB, it has little or probably no effect on post-primary TB. They reported that we now have greater ability than ever to manipulate vaccines to induce desired immune responses including choice of vaccine technology, choice of antigen, choice of adjuvant and route of administration. The big question is what type of immune response should be induced? There are many options for this question, but no way other than clinical trials to decide among them since we have no biomarkers of protective immunity for adult pulmonary TB. With little knowledge of postprimary TB, research on vaccines has ‘hit the wall of biocomplexity’. While new knowledge is being generated at an unprecedented rate, little progress is being made on the fundamental questions of the nature of host immunity to post-primary TB or to the goal of producing a vaccine that inhibits transmission of infection (4, 13–16).
Since transmission of infection occurs during late stage TB, investigators have logically sought to develop vaccines with late stage antigens (17). Transcriptomics has been used to identify genes expressed during each of the disease stages (18–20). Great care has been exercised to insure that the antigens are recognized by human T cells as well as those generated by experimental animals (21). This led to identification of vaccine antigen candidates that represent ‘late-stage’ molecular targets. Many of these antigens, alone or in combination with early stage antigens, have proven successful as vaccines demonstrating improved survival, bacterial load and/or reduced extra pulmonary dissemination in animal models (21–24). While some results are impressive, their relevance for human disease is doubtful. Prevention of extra pulmonary TB and bacterial burden a few weeks after infection are functions of primary TB, not post-primary TB. Furthermore, tissue damage in TB is caused largely by immune responses (25). It is important to develop vaccines that do not augment tissue damage (26, 27). Finally, animal models for TB vaccine development have not been designed to mimic postprimary TB (26–28).
Animal models of post-primary TB:
The most prevalent conception today is that TB is a ‘war of attrition’ between MTB and the host. Can MTB divide faster than they are killed by activated macrophages or do they evade, overwhelm and eventually kill the macrophages so that they are free to divide extracellularly (14). This conception misapprehends the human post-primary disease. There is no consistent correlation between the numbers of MTB in tissue and the severity of disease in people (5). Many people with post-primary TB die with paucibacillary infections in which very few viable mycobacteria are demonstrable by either culture or AFB staining. Subclinical pulmonary lesions, the early infiltrates, frequently develop asymptomatically for months before onset of clinical TB (8, 29). Most early infiltrates regress and resolve completely while others progress to caseous pneumonia and clinical post-primary TB.
Several animal models of TB show patterns of organisms in tissue and pathology that resemble developing post-primary TB. Progressive pulmonary TB in rabbits, mice and guinea pigs is not due to increasing numbers of viable bacilli but is due to a continuous host response to mycobacterial products (30). After containing the initial infection, animals develop a low load of MTB in their lungs. This number of organisms remains constant for months until the animals die of progressive pathology. In recognizing this, North wrote “A central problem in tuberculosis research is to explain why immunity to infection does not enable mice, guinea pigs, rabbits or susceptible humans to resolve lung infection and thereby stop development of the disease” (31). This is characteristic of post-primary, not primary TB.
Lung infections in the mouse, guinea pig, and rabbit do not resolve but persist at stationary levels from approximately days 40-60 of infection, and onward. A stationary level of MTB is also characteristic of developing human post-primary TB. The infection is maintained by the continuous expression of TH1 immunity as evidenced by the demonstration that depleting mice with stationary lung infection of CD4+ T cells results in a resumption of MTB growth, as does treatment with a NOS2 inhibitor (32). Stationary lung infection has been shown to be associated with the presence in the lungs of replicating CD4+ T cells capable of making IFN-γ in response to MTB antigens. (33, 34). The lesions in these animals are models of the asymptomatic early infiltrate of developing human post-primary TB (8). TH1 immunity restricts systemic pathology, but cannot prevent development of pulmonary disease.
It is generally assumed that primary MTB infection fails to resolve because of the generation of an inadequate level of TH1 immunity. Accordingly, the purpose of vaccination is to induce a sufficient level of MTB-specific TH1 cells. Most attempts to design a vaccine that is more protective than BCG are based on the assumption that BCG is of insufficient immunogenicity (35). Such vaccines typically enable vaccinated mice to maintain an MTB challenge infection at about one log lower level than in unvaccinated mice. However, the lower level of lung infection eventually causes progressive pathology (34). In addition, using chemotherapy to reduce the MTB load in the lungs by 2 logs does not enable immunity to cause the much lower level of infection to resolve (34).
The data from humans also suggests that simply increasing the magnitude of TH1 immunity is the wrong approach, that MTB actually needs and uses our strongest immune responses for its benefit. Recovery from TB does not cause protection, but leaves a person more susceptible to new infection (36, 37) . Humans cured of tuberculosis by chemotherapy can become reinfected with a different strain of MTB within weeks (37). Finally, young immune-competent adults with the strongest tuberculin skin tests are far more susceptible to clinical TB than those with small skin test reactions demonstrating that a strong immune response can be detrimental (38–40). MTB has evolved to avoid destruction by innate and adaptive immune mechanisms of immune-competent humans and to use our immune responses to induce lesions that facilitate transmission (31)
The foregoing implies that development of a vaccine that prevents post-primary TB, and thereby blocks development of disease and transmission of infection, will be difficult. However, there is hope. At least 95% of early infiltrations of post-primary TB resolve spontaneously in immune-competent people (5, 41). If we knew why, it might be possible to design a vaccine or other therapy to make them all regress and thereby have a tool to eradicate MTB completely. North identified a persistent low-level infection (a signature of developing post-primary TB) in most of the commonly used animal models of TB even though they have different pathologies (31). The common features of these models could be valuable in identifying the key components.
Studies with experimental TB vaccines typically measure CFUs, weight loss and survival (21). However, in human TB, there is no consistent relationship between disease pathology and numbers of organisms (5). Similarly, death from TB in many immune-competent animal models is due to increasing immunopathology, not increasing numbers of organisms (30). We reported that slowly progressive TB in C57/BL6 mice is a model of the preclinical stage of human post-primary TB (9). In addition, we reported that a vaccine can be modified to inhibit development of the characteristic pathology of post-primary TB in that model (42).
This was accomplished by inclusion of lactoferrin with BCG in vaccination of mice. Lactoferrin is an iron binding protein present in mucosal secretions and secondary granules of neutrophils (43, 44). It possess a wide variety of immune regulatory activities (45), including increasing lymphocyte and natural killer cell activity (46), and increasing surface expression of antigen presentation and co-stimulatory molecules in both MTB-infected macrophages (47, 48) and dendritic cells (47, 49, 50).
As expected, vaccination with BCG produced about one log reduction of CFU counts in the lungs of mice subsequently challenged with virulent MTB and a sustained IFN-γ recall responses to mycobacterial antigens (51). Surprisingly, the inclusion of lactoferrin with the BCG vaccine had no significant effect on the numbers of organisms in tissue, but dramatically reduced the alveolitis in tissue surrounding the resolved granulomas, (Figure 1). This is significant because, like the disease in many humans, it is the spreading alveolitis, and not the numbers of organisms that eventually kills the animals. The alveolitis is composed primarily of interstitial lymphocytes and foamy alveolar macrophages that are sparsely infected with MTB demonstrated by AFB staining. The lactoferrin adjuvanted BCG vaccine was able to induce long-lasting pathological protection (greater than 6 months) and limited proinflammatory mediators in lung tissue without reducing CFU’s more than the BCG alone (49). Since the onset of clinical disease in human post-primary TB are due to expansion of this type of pathology, not to increasing numbers of MTB (30), then these animals may represent a model where mice continue indefinitely with subclinical or latent disease. If this could be transferred to humans, it would be the basis for an anti-transmission vaccine.
Figure 1. C57BL/6 mice immunized with BCG and lactoferrin demonstrate long lasting diminished inflammation and destructive pulmonary histopathology upon aerosol challenge with virulent MTB.
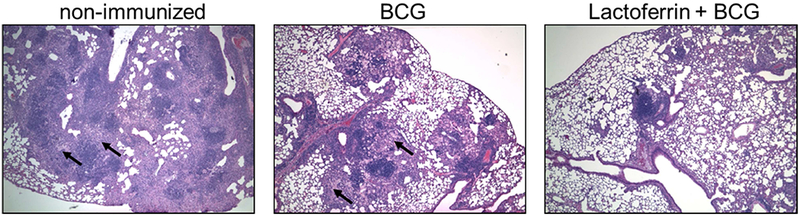
Mice demonstrated reduced histological manifestation of disease in the BCG and recombinant human lactoferrin immunized group at day 150 post challenge with <100 CFU Erdman strain (methods detailed in (49, 51)). Activated monocytes are apparent in the BCG and non-immunized groups (arrows). Lactoferrin adjuvant immunized mice revealed reduction in alveolitis with evidence of lymphocytic clusters and limited focal pockets of inflamed monocytes. H&E staining (20×; N≥5 mice per group).
MTB does not follow the usual rules of infection and immunity. It has evolved with humans to produce primary and post-primary disease as separate disease entities both of which are required for continued survival of the organism (8). Primary TB protects the host from disseminated infection. Post-primary TB, in contrast, manipulates our strongest immune response in parts of the lung to produce cavities that support massive numbers of organisms in proximity position to be exhaled to infect new people. Vaccines that simply increase the natural response have not shown efficacy against post-primary TB. Fortunately, most nascent post-primary lesions regress spontaneously so that progression to clinical disease is a rare event. The challenge is to learn why these lesions regress and to find ways to make all lesions regress in a similar manner.
In conclusion, progress on understanding the pathogenesis of post-primary TB has been inhibited by an excessively narrow focus on molecular biology. We must abandon the concept that granulomas are the important lesion of all TB, and reactivate knowledge of the actual human disease learned over nearly 200 years of study of the pathology, radiology and clinical presentation of untreated TB. We must use this knowledge with our best modern tools to advance understanding of long standing questions of immunity, susceptibility and pathogenesis of TB. As an example, we have shown that use of an adjuvant molecule (lactoferrin) with BCG can inhibit the characteristic pathology of developing postprimary TB in a mouse model. Other models of parts of post-primary TB have been described (27, 28). We suggest that further studies in such models will lead to a better understanding of immunity that protects most adults from clinical TB, and help establish the basis for a vaccine that prevents transmission of infection.
Acknowledgements
Research was supported by the National Institute of Allergy and Infectious Diseases (R42AI051050-02 and R42-AI117990-02).
This work was presented in part at the Texas Tuberculosis Research Symposium (TTRS) 2018, El Paso, Texas, USA, sponsored by the Texas Tech University Health Sciences Center El Paso
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
None declared.
• Each article needs to have a financial disclosure line, usually within the author bio section. Publication of this supplement was supported by The University of Texas Health Science Center at Houston.
References
- 1.National_Research_Council. 2009. A New Biology for the 21st Century: Ensuring the United States Leads the Coming Biology Revolution. The National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- 2.Woese CR 2004. A new biology for a new century. Microbiology and molecular biology reviews : MMBR 68: 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toossi Z, and Ellner J. 2000. Pathogenesis of tuberculosis In Tuberculosis: Current Concepts and Treatment, Second Edition Friedman L, ed. CRC Press, New York: 19–48. [Google Scholar]
- 4.Bhatt K, Verma S, Ellner JJ, and Salgame P. 2015. Quest for correlates of protection against tuberculosis. Clinical and vaccine immunology : CVI 22: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rich A 1951. The Pathogenesis of Tuberculosis, Second Edition Charles C Thomas, Springfield, Illinois. [Google Scholar]
- 6.Medlar EM 1955. The behavior of pulmonary tuberculous lesions; a pathological study. Am Rev Tuberc 71: 1–244. [PubMed] [Google Scholar]
- 7.Canetti G 1968. Biology of the mycobacterioses. Pathogenesis of tuberculosis in man. Ann N Y Acad Sci 154: 13–18. [DOI] [PubMed] [Google Scholar]
- 8.Hunter R 2018. The Pathogenesis of Tuberculosis: The early infiltrate of post-primary (adult pulmonary) tuberculosis - A distinct disease entity. Frontiers in immunology 9: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter RL, Jagannath C, and Actor JK. 2007. Pathology of postprimary tuberculosis in humans and mice: contradiction of long-held beliefs. Tuberculosis (Edinb) 87: 267–278. [DOI] [PubMed] [Google Scholar]
- 10.Pottenger FM 1927. The Interpretation of Clinical Pulmonary Tuberculosis in Terms of Allergy. Trans Am Climatol Clin Assoc 43: 145–161. [PMC free article] [PubMed] [Google Scholar]
- 11.Dannenberg AM Jr. 2006. Pathogenisis of human pulmonary tuberculosis Insights from the rabbit model. ASM press, Washington, DC. [Google Scholar]
- 12.Voss G, Casimiro D, Neyrolles O, Williams A, Kaufmann SHE, McShane H, Hatherill M, and Fletcher HA. 2018. Progress and challenges in TB vaccine development. F1000Res 7: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geadas C, Stoszek SK, Sherman D, Andrade BB, Srinivasan S, Hamilton CD, and Ellner J. 2017. Advances in basic and translational tuberculosis research: Proceedings of the first meeting of RePORT international. Tuberculosis (Edinb) 102: 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, Menzies D, and Raviglione M. 2016. Tuberculosis. Nat Rev Dis Primers 2: 16076. [DOI] [PubMed] [Google Scholar]
- 15.Schrager LK, Chandrasekaran P, Fritzell BH, Hatherill M, Lambert PH, McShane H, Tornieporth N, and Vekemans J. 2018. WHO preferred product characteristics for new vaccines against tuberculosis. The Lancet. Infectious diseases 18: 828–829. [DOI] [PubMed] [Google Scholar]
- 16.Schrager L 2015. Developing vaccines to prevent sustained infection with Mycobacterium tuberculosis: Conference proceedings: November 7, 2014, National Institute of Allergy and Infectious Diseases, Rockville, Maryland USA: Vaccine. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalo-Asensio J, Aguilo N, Marinova D, and Martin C. 2017. Breaking Transmission with Vaccines: The Case of Tuberculosis. Microbiol Spectr 5. [DOI] [PubMed] [Google Scholar]
- 18.Deffur A, Wilkinson RJ, and Coussens AK. 2015. Tricks to translating TB transcriptomics. Annals of translational medicine 3: S43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehra S, Pahar B, Dutta NK, Conerly CN, Philippi-Falkenstein K, Alvarez X, and Kaushal D. 2010. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PloS one 5: e12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen Thi le T, Sarmiento ME, Calero R, Camacho F, Reyes F, Hossain MM, Gonzalez GS, Norazmi MN, and Acosta A. 2014. Immunoinformatics study on highly expressed Mycobacterium tuberculosis genes during infection. Tuberculosis (Edinb) 94: 475–481. [DOI] [PubMed] [Google Scholar]
- 21.Stylianou E, Harrington-Kandt R, Beglov J, Bull N, Pinpathomrat N, Swarbrick GM, Lewinsohn DA, Lewinsohn DM, and McShane H. 2018. Identification and Evaluation of Novel Protective Antigens for the Development of a Candidate TB Subunit Vaccine. Infect Immun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aagaard C, Hoang T, Dietrich J, Cardona PJ, Izzo A, Dolganov G, Schoolnik GK, Cassidy JP, Billeskov R, and Andersen P. 2011. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat Med 17: 189–194. [DOI] [PubMed] [Google Scholar]
- 23.Lin PL, Dietrich J, Tan E, Abalos RM, Burgos J, Bigbee C, Bigbee M, Milk L, Gideon HP, Rodgers M, Cochran C, Guinn KM, Sherman DR, Klein E, Janssen C, Flynn JL, and Andersen P. 2012. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J Clin Invest 122: 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Zhang J, Liang J, Zhang Y, Teng X, Yuan X, and Fan X. 2015. Protection against Mycobacterium tuberculosis infection offered by a new multistage subunit vaccine correlates with increased number of IFN-gamma+ IL-2+ CD4+ and IFN-gamma+ CD8+ T cells. PloS one 10: e0122560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dannenberg AM Jr., and Rook GA. 1994. Pathogenesis of pulmonary tuberculosis: an interplay of tissue-damaging and macrophage-activating immmune responses. Dual mechanisms that control bacillary multiplication In Tuberculosis 2004. Bloom B, ed. ASM Press, Washington, DC: 459–483. [Google Scholar]
- 26.Russell DG 2013. The evolutionary pressures that have molded Mycobacterium tuberculosis into an infectious adjuvant. Curr Opin Microbiol 16: 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter RL, Actor JK, Hwang SA, Khan A, Urbanowski ME, Kaushal D, and Jagannath C. 2018. Pathogenesis and Animal Models of Post-Primary (Bronchogenic) Tuberculosis, A Review. Pathogens 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basaraba RJ, and Hunter RL. 2017. Pathology of Tuberculosis: How the Pathology of Human Tuberculosis Informs and Directs Animal Models. Microbiol Spectr 5. [DOI] [PubMed] [Google Scholar]
- 29.Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG, Ma S, Meermeier E, Lewinsohn DM, and Sherman DR. 2018. Incipient and Subclinical Tuberculosis: a Clinical Review of Early Stages and Progression of Infection. Clin Microbiol Rev 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dannenberg AM Jr., and Collins FM. 2001. Progressive pulmonary tuberculosis is not due to increasing numbers of viable bacilli in rabbits, mice and guinea pigs, but is due to a continuous host response to mycobacterial products. Tuberculosis (Edinb) 81: 229–242. [DOI] [PubMed] [Google Scholar]
- 31.North RJ, and Jung YJ. 2004. Immunity to tuberculosis. Annu Rev Immunol 22: 599–623. [DOI] [PubMed] [Google Scholar]
- 32.Flynn JL, Scanga CA, Tanaka KE, and Chan J. 1998. Effects of aminoguanidine on latent murine tuberculosis. J Immunol 160: 1796–1803. [PubMed] [Google Scholar]
- 33.Winslow GM, Roberts AD, Blackman MA, and Woodland DL. 2003. Persistence and turnover of antigen-specific CD4 T cells during chronic tuberculosis infection in the mouse. J Immunol 170: 2046–2052. [DOI] [PubMed] [Google Scholar]
- 34.Mogues T, Goodrich ME, Ryan L, LaCourse R, and North RJ. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med 193: 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufmann SH, Evans TG, and Hanekom WA. 2015. Tuberculosis vaccines: time for a global strategy. Science translational medicine 7: 276fs278. [DOI] [PubMed] [Google Scholar]
- 36.Caminero JA, Pena MJ, Campos-Herrero MI, Rodriguez JC, Afonso O, Martin C, Pavon JM, Torres MJ, Burgos M, Cabrera P, Small PM, and Enarson DA. 2001. Exogenous reinfection with tuberculosis on a European island with a moderate incidence of disease. Am J Respir Crit Care Med 163: 717–720. [DOI] [PubMed] [Google Scholar]
- 37.van Rie A, Warren R, Richardson M, Victor TC, Gie RP, Enarson DA, Beyers N, and van Helden PD. 1999. Exogenous reinfection as a cause of recurrent tuberculosis after curative treatment. N Engl J Med 341: 1174–1179. [DOI] [PubMed] [Google Scholar]
- 38.Comstock GW 1982. Epidemiology of Tuberculosis in Koch Centennial Memorial. . American Review of Respiratory Diseases 125: 8–15. [DOI] [PubMed] [Google Scholar]
- 39.Corbiere V, Pottier G, Bonkain F, Schepers K, Verscheure V, Lecher S, Doherty TM, Locht C, and Mascart F. 2012. Risk stratification of latent tuberculosis defined by combined interferon gamma release assays. PloS one 7: e43285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youmans GP 1979. Chapter 13. Relationship between delayed hypersensitivity and immunity in tuberculosis In Tuberculosis. Youmans GP, ed. W. B. Saunders Co., Philadelphia, PA: 302–326. [Google Scholar]
- 41.Pinner M 1945. Pulomonary Tuberculosis in the Adult Its Fundamental Aspects. Charles C Thomas, Springfield, Ill. [Google Scholar]
- 42.Hwang SA, Wilk KM, Budnicka M, Olsen M, Bangale YA, Hunter RL, Kruzel ML, and Actor JK. 2007. Lactoferrin enhanced efficacy of the BCG vaccine to generate host protective responses against challenge with virulent Mycobacterium tuberculosis. Vaccine 25: 6730–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Actor JK, Hwang SA, and Kruzel ML. 2009. Lactoferrin as a natural immune modulator. Curr Pharm Des 15: 1956–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kruzel ML, Actor JK, Boldogh I, and Zimecki M. 2007. Lactoferrin in health and disease. Postepy higieny i medycyny doswiadczalnej 61: 261–267. [PubMed] [Google Scholar]
- 45.Kruzel ML, Zimecki M, and Actor JK. 2017. Lactoferrin in a Context of Inflammation-Induced Pathology. Frontiers in immunology 8: 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shau H, Kim A, and Golub SH. 1992. Modulation of natural killer and lymphokine-activated killer cell cytotoxicity by lactoferrin. J Leukoc Biol 51: 343–349. [PubMed] [Google Scholar]
- 47.Hwang SA, and Actor JK. 2009. Lactoferrin modulation of BCG-infected dendritic cell functions. Int Immunol 21: 1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang SA, Kruzel ML, and Actor JK. 2009. Influence of bovine lactoferrin on expression of presentation molecules on BCG-infected bone marrow derived macrophages. Biochimie 91: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hwang SA, Kruzel ML, and Actor JK. 2015. CHO expressed recombinant human lactoferrin as an adjuvant for BCG. International journal of immunopathology and pharmacology 28: 452–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Shea KM, Hwang SA, and Actor JK. 2015. Immune Activity of BCG Infected Mouse Macrophages Treated with a Novel Recombinant Mouse Lactoferrin. Ann Clin Lab Sci 45: 487–494. [PubMed] [Google Scholar]
- 51.Hwang SA, Wilk K, Kruzel ML, and Actor JK. 2009. A novel recombinant human lactoferrin augments the BCG vaccine and protects alveolar integrity upon infection with Mycobacterium tuberculosis in mice. Vaccine 27: 3026–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]