

Abstract
Disrupting a protein’s sequence by cleavage or insertion of a hinge domain forms the basis for protein engineering tools, including fragment complementation, circular permutation, and domain swapping. Despite the utility of these designs, their widespread implementation has been limited by the difficulty in choosing where to interrupt the protein sequence: the resulting fragments often aggregate or fail to reassemble. Here, we show that an optimal site exists within ribose binding protein (RBP) that, when disrupted, results in the most efficient formation of fragment-complemented and domain-swapped species. Cleaving RBP at this site also produces a highly stable, cooperatively folded circular permutant. This hot-spot site was identified by an experimental approach involving selection among competing folds. We find that efficiency in the case of RBP is determined by kinetic factors (survival of the first) rather than thermodynamics (survival of the fittest). Together with emerging computational tools, this limited data set defines a pathway for designing robust platforms for molecular switches and biosensors based on the aforementioned protein modifications.
Significance
The ability to manipulate a protein’s function by disrupting its sequence and controlling when and how it reforms its native structure plays a dominant role in the design of molecular switches and biosensors. A challenge in developing these tools is to find interruption sites that allow for rapid and efficient reassembly of the fragments. In this work, we show that cleaving ribose binding protein at a single, optimal site allows the two halves to reassemble via three commonly employed protein engineering methodologies—fragment complementation, circular permutation, and domain swapping—with high efficiency. We propose that analogous hot-spot disruption sites exist in other proteins and that these can be identified by recently developed experimental and computational methods.
Introduction
Protein fragment complementation, circular permutation, and domain swapping are versatile tools in the protein engineer’s repertoire. These methodologies form the basis for technologies such as protein complementation assays, induced reassembly, molecular switches, and biosensors (1, 2, 3, 4, 5). Although the three manipulations produce structures with markedly different chain topologies, they all begin with a similar modification: disruption of the polypeptide backbone at a chosen site. In fragment complementation and circular permutation, the disruption consists of breaking the chain (Fig. 1 A). The two pieces then refold and bind either in trans or in cis, respectively, with circular permutation being enabled by a peptide linker bridging the amino and carboxy termini of the original protein. In domain swapping, the chain is not broken, but the three-dimensional structure is nevertheless interrupted at the indicated position (Fig. 1 A). The disruption site serves as the hinge region at which the two halves of a monomeric protein detach, then cross over to refold with an identically split monomer to generate a swapped dimer (or oligomer).
Figure 1.

Fragment complementation, circular permutation, and domain swapping. (A) These three protein engineering methods begin by disrupting a protein’s sequence at a position denoted by the black circle (typically within a surface loop). The linker peptide in the circular permutant and the hinge region in the domain-swapped dimer are colored green and black, respectively. (B) The five N-terminal fragments of RBP (blue) and five C-terminal fragments (red) created for this study are represented by their amino-acid sequences and arranged as pairs of exactly complementary fragments (homo complexes). The gray box demarcates the region duplicated in the overlap complexes and absent in the gap complexes. Position 97 is indicated by the dashed line. (C) The complexes with the longest segments of duplicated sequence (RBP1–124 + RBP60–277) and missing sequence (RBP1–59 + RBP125–277) are shown to illustrate overlap and gap complexes, respectively. Circular permutation (CP97) and Ub insertion (RU97) at position 97 are depicted with linker peptide in green and Ub as a box, respectively, as the lower two sequences. (D) Locations of split sites in RBP (Protein Data Bank [PDB]: 2IOY) are shown as gray spheres. Colors represent the same regions as in (B). Ribose is shown as white sticks.
Despite the widespread use of these protein engineering tools, there is little guidance as to how one should choose a split point or hinge region in a given protein such that it undergoes fragment complementation, circular permutation, and domain swapping as efficiently as possible. Here, we define efficiency as forming quickly and with high yield while preserving the function and thermodynamic stability of the parent protein. On the face of it, the answer seems simple: select a flexible, solvent-exposed loop so that secondary structural elements (α-helices and β-strands) are not interrupted and that the newly generated termini or hinge region does not introduce charges or otherwise disrupt tertiary interactions in the protein’s core. In practice, however, there are multiple surface loops in typical protein, and cleaving the chain at many of these sites fails to produce complexes and permutants that fold into a stable structure in a timely manner (6, 7, 8). Disruption sites have been typically selected by trial-and-error experiment, with computational tools being largely unavailable until recently (9).
Fersht and co-workers introduced a protein engineering experiment (herein designated “overlap selection”) that has the potential for identifying the site in a given protein that generates the most stable and efficiently formed circular permutant, fragment-complemented complex, and domain-swapped species (10). The overlap selection method entails deleting a segment from the C-terminus of a target protein such that the native structure is lost or strongly destabilized. An analogous segment is deleted from the N-terminus to generate a C-terminal fragment. The only way that the stability and full structure of the native state can be reconstituted is if the N- and C-fragments bind and complement. This approach is distinct from standard protein complementation assays because the two fragments are created such that they contain a region of overlapping sequence, which enables the crossover point to be at any position within the duplicated region. These native complexes can be considered the result of fragment complementation or domain swapping, in which the crossover point is the cleavage point or the hinge region, respectively. The crossover site is best identified by NMR chemical shift mapping, which can discriminate between the native structure of complexes and partially folded structures of the fragments and allows the location to be determined with high precision (10, 11). Mass spectrometry of proteolytically digested complexes has also been employed (12).
During the overlap selection experiment, the N- and C-fragments sample all accessible conformations, including unfolded, partially folded, misfolded, and aggregated. Thus, if a single native species prevails, it has successfully avoided kinetic traps and is the most thermodynamically stable of the possible native complexes (survival of the fittest) or is a metastable structure that folds the fastest (survival of the first). In either case, one obtains the answer to the question of where to disrupt the amino-acid sequence to generate the complex that reconstitutes the native fold with the greatest efficiency in the conditions of the experiment. These conditions can be chosen to select for maximal efficiency in various environments such as high temperature or cell lysates.
We previously applied the overlap selection methodology to Thermoanaerobacter tengcongensis ribose binding protein (RBP; 277 amino acids) (11). Two constructs were made in which residues 125–277 and 1–59 were deleted, generating the N-terminal fragment RBP1–124 and C-terminal fragment RBP60–277, respectively (Fig. 1 C, top sequence). Most of the NMR cross peaks in the 15N heteronuclear single-quantum correlation (HSQC) NMR spectrum of RBP60–277 were well-dispersed (but distinct from those in the wild-type (WT) spectrum), indicating that RBP60–277 retained significant but non-native structure. RBP1–125 showed a large cluster of broad, overlapped peaks near the center of the spectrum, suggesting that tertiary structure was mostly lost. The sequence overlapped by the two fragments consists of 65 amino acids (gray box in Fig. 1 B and gray structure in Fig. 1 D). The fragments chose to domain swap at a single site mapped between residues 94 and 101, which comprises the fourth of five surface loops in the overlapping region (centered at positions 60, 70, 85, 97, and 125; Fig. 1 D). This result was unexpected because we had found that RBP readily formed domain-swapped dimers and oligomers when forced to swap at positions 60 or 125 by insertion of Ub (11). That the protein instead opted to swap at an uncharacterized surface loop when released from this constraint suggested that loop 97 is a hot spot for natural domain swapping and fragment complementation.
Here, we determine the molecular basis for why position 97 is a hot spot for RBP swapping and complementation, and we also test the hypothesis that this site is optimal for circular permutation. The results raise the possibility that a site exists within a given protein sequence that, when cleaved or otherwise disrupted, allows domain-swapped, fragment-complemented, and circularly permuted species to form with maximal efficiency. Our findings further suggest that the overlap selection experiment can be used to locate this site.
Materials and Methods
Gene construction and protein purification
Genes were constructed using standard methods and were fully sequenced. All constructs were expressed in Escherichia coli BL21(DE3) as described (12) and purified on Ni2+-NTA agarose (Bio-Rad, Hercules, CA) following the manufacturer’s protocol. RBP fragments lacking fluorescent protein labels were coexpressed in E. coli (as homo pairs) and purified as the complex. The fragments were then separated by dissolving the lyophilized complex in 7.5 M GdnHCl (pH 3.0, 1 h), adjusting pH to 8.0 with Tris base, then passing the solution through an Ni2+-NTA column equilibrated in 7.5 M GdnHCl (pH 8.0). The N-fragment was collected from the unbound fractions, and the bound C-fragment was eluted with 0.3 M imidazole. All proteins were judged to be >95% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
CD experiments
Samples consisted of 2 μM of each fragment in 10 mM sodium phosphate (pH 7.0), 0.15 M NaCl, 0.1 mM EDTA, and 0.005% TWEEN-20. Complexes were incubated for 16 h before scans. Data were collected at 22°C using a 1 cm pathlength cuvette and an Aviv model 420 spectropolarimeter (Lakewood, NJ).
FRET experiments
Samples for binding affinity measurements were prepared by serially diluting individual CyPet- and YPet-labeled proteins (200 nM initial concentration), then mixing the two fragments at a constant 1:1 molar ratio. Complexes were allowed to equilibrate for 3–5 days at 22°C. FRET efficiency was assessed by exciting CyPet at 414 nm (1–3 nm bandpass) and measuring the ratio of acceptor/donor emission at 525 and 475 nm, respectively (4–6 nm bandpass). The acceptor/donor ratio for the uncomplexed fragments was obtained by scanning the fragments individually, then summing their spectra. This value (0.34) was fixed in the fits of the data to the one-site quadratic binding equation.
Association rates were measured by mixing CyPet-labeled N-fragment (50 nM) with an excess of YPet-labeled C-fragment (0.5–8 μM). Fluorescence was recorded as above, and the time-dependent increase in FRET ratio was fitted to a single exponential function to obtain kobs. The acceptor/donor ratio at time zero was fixed at 0.34. Dissociation kinetics were characterized by mixing CyPet- and YPet-labeled fragments (1 μM each, 6 h incubation), then diluting the complex to 4 nM in the presence of 80 nM free, unlabeled C-fragment. Aliquots were withdrawn every 6–24 h for 5 days, and their FRET ratios determined as above. All fluorescence data were collected at 22°C on a Horiba Fluoromax-4 fluorometer (Kyoto, Japan), with the exception of kon data for complexes containing RBP1–96, which were acquired on a Bio-Logic SFM4000 stop-flow fluorometer (Seyssinet-Pariset, France).
NMR experiments
Uniformly 15N-labeled proteins (99 atom %) were purified as above from 15N cultures prepared as described (11). Proteins were dialyzed against ddH2O, lyophilized, and resuspended in 20 mM sodium phosphate (pH 7.0), 0.1 M NaCl. Protein concentrations were 0.6 mM (WT RBP and RBP1–96 + RBP97–277) and 1 mM (RBP1–96 and RBP97–277). 1 mM ribose was added to the WT RBP and RBP1–96 + RBP97–277 samples. 15N and 1H resonance assignments of WT RBP are from (11). Data were recorded on a Bruker AVANCE III HD 800 MHz NMR spectrometer (Billerica, MA) equipped with a 5 mm TCI cryoprobe.
Domain-swapping experiments
E. coli cultures expressing domain-swapped constructs were grown and induced under identical conditions to facilitate comparable expression levels. Fractions of pure protein from the Ni2+-NTA column were diluted to 20 μM, and 10 mM ribose was added to stabilize the RBP domains. The samples were incubated in 0.2% SDS loading dye for either 5 m at room temperature or 30 m at 95°C (fully denatured loading controls), then run on Mini Protean TGX 4–15% gradient gels (Bio-Rad) or manually poured 4–20% SDS-PAGE gradient gels.
Results
Our approach was to break the RBP chain at all five surface loops between residues 60–125, then characterize the structures, stabilities, folding kinetics, and complementation binding affinities of the resulting split complexes, circular permutants, and domain-swapped species. For the complementation studies, we created the 10 N- and C-terminal fragments shown in Fig. 1 B, arranged as five exactly complementary pairs (homo complexes). We also evaluated all possible hetero pairs (10 “overlap” complexes and 10 “gap” complexes). Examples of overlap and gap complexes are depicted in Fig. 1 C. Circular permutants were generated by cleaving RBP at the same positions as in Fig. 1 B and joining the original N- and C-termini with a 30-residue flexible linker; these are designated CP60, CP70, CP85, CP97, and CP125. To evaluate domain swapping, we inserted ubiquitin (Ub) into the five loops in Fig. 1 D. The long N-to-C distance (38 Å) of Ub destabilizes monomeric RBP and stabilizes its folding as domain-swapped dimers and oligomers, with the hinge region consisting of the Ub domain at the indicated position (11, 13). The RBP-Ub constructs are named RU60, RU70, RU85, RU97, and RU125. Representative CP and RU constructs are illustrated in Fig. 1 C, and amino-acid sequences of the constructs are included in Fig. S1.
Structural characterization of fragments and homo complexes
We first employed circular dichroism (CD) to assess the residual structure present in the N- and C-terminal fragments. The two longest N-terminal fragments, RBP1–124 and RBP1–96, retain 90% of the α-helical content observed for WT RBP (on a per-residue basis) as judged by minima at 222 and 208 nm (Fig. 2 A). RBP1–84, RBP1–69, and RBP1–59 progressively lose helicity to the point where RBP1–59 is predominantly unfolded. A distinct dropoff occurs when residues 70–84 are truncated. This stretch adopts an α-helix in native RBP that docks against the helix formed by residues 41–55 (Fig. 1 D). This interaction appears to be important for maintaining residual structure in the N-terminal fragments. Turning to the C-terminal fragments, RBP60–277, RBP70–277, RBP85–277, and RBP97–277 exhibit 80% of the per-residue helical content of WT RBP (Fig. 2 B). Only the shortest fragment, RBP125–277, shows pronounced loss of helicity. Residues 97–125 adopt a short β-strand and a long α-helix in native RBP (Fig. 1 D). One or both of these structures are critical to the folding of the C-terminal fragments.
Figure 2.

Characterization of RBP fragments and complexes thereof. Residual structure of isolated N-terminal (A) and C-terminal (B) fragments detected by CD are shown. (C) Cleaving RBP at position 97 maximizes the combined helical structure in the resulting fragments. Units of y axes are 103 deg cm2 dmol−1. (D) Equilibrium binding of homo fragments monitored by FRET acceptor/donor emission ratio is shown. Lines are best fits to the one-site binding equation. Error bars are SDs (n = 3). (E) and (F) depict the kinetics of homo fragment binding, with symbols representing the same proteins as in (D). Lines are best fits to the linear portions of the data. Error bars are SDs (n = 3).
When the molar ellipticities of the N- and C-terminal fragments are plotted as a function of cleavage position, it becomes apparent that position 97 is a “tipping point” beyond which truncating the sequence to either side causes the remaining fragment to lose helical structure (Fig. 2 C). Cleaving RBP at positions 60, 70, 85, and 97 yields identically well-folded C-terminal fragments, but the first three N-terminal fragments are significantly less structured than RBP1–96. Cleaving RBP at position 125 does not increase the helical content of the N-terminal fragment beyond that of RBP1–96, but it causes the C-terminal fragment RBP124–277 to unfold. Thus, splitting RBP at loop 97 results in the most balanced outcome, in which the resulting N- and C-terminal fragments retain as much combined secondary structure as possible.
We next tested whether the homo pairs can regenerate native RBP by comparing the CD spectra of the mixture to the sum of the individual spectra shown in Fig. 2, A and B. Mixing the fragments increases the CD signal intensity in all cases, suggesting that they complement and that additional folding occurs upon binding (Fig. S2). Relatively little additional folding is observed for the complexes of RBP1–96 + RBP97–277 and RBP1–84 + RBP85–277 compared to the other three complexes, consistent with the finding that their N- and C-fragments are already highly helical in isolation. To establish whether the complexes are functional, we recorded thermal denaturation curves in the absence and presence of ribose. All complexes exhibit high thermal stability, with apparent Tm values exceeding 75°C (Fig. S3 A). Tm values shift by ≥10°C in the presence of 1 mM ribose (Fig. S3 B), indicating that the native RBP fold has been restored in all cases.
We recorded 15N HSQC NMR spectra to further characterize the structures of RBP1–96, RBP97–277, and RBP1–96 + RBP97–277. The HSQC cross peaks of the complex are well-dispersed and sharp, symptomatic of a folded protein (Fig. 3). The majority of the resonances overlay with those of WT RBP, indicating that the two structures are similar. The exceptions are residues 100–103, which are located in the loop flanking position 97, and two stretches of amino acids from 73–82 to 137–138. Residues 73–82 and 137–138 comprise an α-helix and a loop, respectively, both of which are in direct contact with the loop surrounding the cleavage site. The RBP97–277 fragment likewise appears to be well-structured (Fig. S4 A). 178 backbone resonances out of a possible 192 are resolved and sharp, although few align with those of the complex or WT RBP. The crosspeaks of RBP1–96 are dispersed but exhibit varying degrees of broadening (Fig. S4 B). At lower contours, a large, poorly resolved mass of resonances is apparent in the center of the spectrum. The structure of RBP1–96 thus appears to be folded but dynamic, with motions occurring over a range of timescales.
Figure 3.

15N-HSQC NMR spectra of WT RBP (red) and RBP1–96 + RBP97–277 (blue). Assigned WT RBP resonances that do not overlay with a corresponding RBP1–96 + RBP97–277 crosspeak are indicated by residue number. Samples contain 1 mM ribose, and temperature is 50°C.
Homo fragment binding affinities and kinetics
Because all pairs of homo fragments were able to regenerate stable and active molecules, we investigated whether any one complex might be favored because of thermodynamic or kinetic considerations. To do so, we fused cyan fluorescent protein (CyPet) to the N-termini of the N-fragments and yellow fluorescent protein (YPet) to the C-termini of the C-fragments and monitored binding by Förster resonance energy transfer (FRET) (14). The five homo complexes form with high but variable affinity (Fig. 2 D). Four of these (RBP1–59 + RBP60–277, RBP1–69 + RBP70–277, RBP1–84 + RBP85–277, and RBP1–124 + RBP125–277) exhibit Kd values of 0.45–104 nM (upper values on the diagonal of Table 1). Nanomolar affinity can therefore be achieved with either the N-terminal fragment (RBP1–59 and RBP1–69) or the C-terminal fragment (RBP125–277) being largely unfolded. The tightest binding is observed when N- and C-fragments are both highly helical, with Kd of the RBP1–96 + RBP97–277 interaction being too low to measure (<0.1 nM).
Table 1.
Binding Affinities and Association Rates of Homo Complexes, Overlap Complexes, and Gap Complexes
| RBP1–59 | RBP1–69 | RBP1–84 | RBP1–96 | RBP1–124 | |
|---|---|---|---|---|---|
| RBP60–277 | 104 ± 4∗ | 1.62 ± 0.05 | 2.4 ± 0.3 | <0.1 | 4.0 ± 0.1 |
| 119 ± 16∗ | 780 ± 40 | 1480 ± 60 | (2.90 ± 0.05) × 105 | 530 ± 20 | |
| RBP70–277 | ND | 4.0 ± 0.1∗ | 7.2 ± 0.8 | <0.1 | 2.7 ± 0.1 |
| 340 ± 30∗ | 900 ± 60 | (2.54 ± 0.04) × 105 | 620 ± 20 | ||
| RBP85–277 | ND | 5.3 ± 0.2 | 9.3 ± 0.4∗ | <0.1 | 4.5 ± 0.5 |
| 540 ± 30 | 1490 ± 70∗ | (2.91 ± 0.04) × 105 | 620 ± 30 | ||
| RBP97–277 | ND | ND | 79 ± 30 | <0.1∗ | 3.0 ± 0.5 |
| 590 ± 40 | (2.13 ± 0.04) × 105∗ | 570 ± 10 | |||
| RBP125–277 | ND | ND | ND | 2.98 ± 0.47 | 0.45 ± 0.05∗ |
| (4.26 ± 0.08) × 105 | 1260 ± 30∗ |
Kd (units of nM) and kon (italics; units of M−1 s−1) are listed at the top and bottom of each cell, respectively. Kd errors are SDs of three independent measurements. kon errors are those calculated from the linear regression fits (three independent data sets). ND, binding not detected. ∗ indicates homo complexes (diagonal). Overlap complexes are above the diagonal. Gap complexes are below the diagonal.
Although all homo pairs complement with high affinity, comparison of association rates reveals pronounced differences. Plots of the pseudo-first-order association rate (kobs) versus [C-fragment] are hyperbolic-shaped for all homo complexes except for RBP1–96 + RBP97–277 (Fig. 2 E), which remains linear over the concentrations tested (Fig. 2 F). The hyperbolic curves suggest that for these proteins, the rate-limiting step is fragment binding at low [C-fragment] but shifts to a concentration-independent process at high concentration. This latter step is likely folding of one or both free fragments because conformational changes that occur after complex formation would not result in a significant FRET change. Second-order rate constants (kon), calculated from the slopes of the linear portions of the plots, are listed as the lower values (in italics) on the diagonal of Table 1. The four homo pairs excluding RBP1–96 + RBP97–277 exhibit kon values (120–1510 M−1 s−1) much lower than that expected for a diffusion-limited encounter between two folded proteins, which can be as high as 109 M−1 s−1 for uniformly reactive spheres (15) but is in the range of 105–106 M−1 s−1 for more realistic protein models (16, 17). By contrast, RBP1–96 + RBP97–277 bind with a rate (2 × 105 M−1 s−1) that is closer to the theoretical diffusion-limited value, suggesting that these fragments may be pre-ordered for rapid binding by virtue of their more native-like structures. The CD data of the isolated fragments support this interpretation (Fig. 2, A and B).
Overlap and gap complexes
We next measured Kd values for all possible combinations of N- and C-terminal fragments. The overlap complexes occupy the region in Table 1 above the diagonal. Within a given row, the complexes to the right of the diagonal consist of the same C-fragment mixed with progressively longer and more overlapping N-fragments. The observed trend is exemplified in the first row. Increasing the length of the redundant sequence enhances binding relative to the RBP1–59 + RBP60–277 homo pair, with the maximal effect (Kd < 0.1 nM) seen for RBP1–96. This finding is consistent with the above hypothesis that a well-folded N-terminal fragment is required for the highest affinity complementation. Surprisingly, extending RBP1–96 by 27 amino acids to generate RBP1–124 weakens the interaction with RBP60–277 (as well as with the other C-fragments). Residues 97–124 form a short β-strand and a 17-residue α-helix in the native RBP structure, and CD spectra suggest that this helix is folded in RBP1–124 as well as in RBP60–277 (Fig. 2, A and B). A simple explanation is that the duplicated structures clash with each other and sterically interfere with binding. One of these sequences may need to unfold for association to occur. In this scenario, RBP1–96 represents the optimal case in which the overlapping sequence is long enough to establish native-like structure in the N-terminal fragment but not too long to allow formation of duplicate structures that inhibit fragment binding.
Turning to the columns in Table 1, the complexes above the diagonal consist of the same N-fragment mixed with progressively more overlapping C-fragments. In the first four columns, only a modest reduction in Kd is apparent, consistent with the idea that complementation affinity is largely driven by the structure of the N-fragment. The last column shows the opposite result: the overlap complexes of RBP1–124 all form with reduced affinity compared to the homo complex. This result can be explained by the above hypothesis in which residues 97–124 adopt native-like structure and interfere with binding if present on both fragments.
The gap complexes occupy the region of Table 1 below the diagonal. As expected, they associate more weakly than the corresponding homo complex. Only the gap species with the shortest deletions (those closest to the diagonal) are able to complement at all; the remainder show little or no interaction at the highest protein concentration tested (10 μM). The single exception is that the RBP1–69 + RBP70–277 homo complex and RBP1–69 + RBP85–277 gap complex form with equal affinity. Residues 70–84 appear to play a lesser role in complex formation.
The kinetic binding data provide further evidence of the importance of the N-fragment to complementation. Two trends are apparent. First, kon values within each column of Table 1 tend to be similar, suggesting that the identity of the N-terminal fragment largely dictates the rates at which the complexes form. For example, RBP1–96 binds RBP97–277, the three overlapping C-terminal fragments, and even the gap fragment RBP125–277 with approximately equal rates. RBP125–277 is predominantly unfolded, suggesting that folding of the C-fragments is rapid and does not limit the rate of complex formation. The second trend is that kon of the overlap complexes correlates with the foldedness of the N-fragment. Within each row of Table 1, starting from the homo complex on the diagonal, kon increases as the same C-fragment is paired with N-fragments of increasing length and residual structure up to RBP1–96. kon slows dramatically with RBP1–124, consistent with the idea that residues 97–124 are folded in both fragments and sterically inhibit binding.
We attempted to measure dissociation rates by mixing the CyPet- and YPet-labeled fragment complexes with a 20-fold excess of unlabeled fragment and monitoring loss of FRET efficiency. Dissociation half-times of all homo and overlap complexes were slow (1–5 days), as expected for affinities in the nanomolar range.
Circular permutants
The extremely slow unfolding rates of RBP circular permutants preclude the measurement of their free energies of unfolding (ΔGunf) by GdnHCl denaturation (12). Accordingly, we measured ΔGunf in the background of the destabilizing F217A + D218S mutations (Fig. 4 A) (18). WT RBP is very stable even with these mutations (ΔGunf = 18.7 kcal/mol; Table 2). This is due to its unusually steep unfolding transition (m-value), which has been previously noted (19). CP97 exhibits the greatest m-value among the CPs and consequently has the highest ΔGunf. Relative stability can also be assessed by comparing the midpoints of GdnHCl denaturation (Cm), of which CP60 displays the largest. CP125 is the least stable by both ΔGunf and Cm criteria.
Figure 4.

Thermodynamic and kinetic characterization of circular permutants. (A) Stabilities of CPs determined by GdnHCl denaturation. All proteins shown contain the F217A + D218S mutations. Lines are best fits of the CD signal (222 nm) to the linear extrapolation model (26). (B) Folding and (C) unfolding rates after dilution from 6 M GdnHCl into buffer and from buffer into GdnHCl, respectively, with the final GdnHCl concentrations indicated on the x axes. Rates were obtained by fitting the decay curves to a single exponential function. Error bars are standard deviations for three independent measurements.
Table 2.
Stabilities of Circular Permutants
| Variant | ΔGunf (kcal mol-1) | m (kcal mol-1 M-1) | Cm (M) |
|---|---|---|---|
| WT | 18.7 ± 1.2 | 9.0 ± 0.7 | 2.09 ± 0.03 |
| CP60 | 3.2 ± 0.4 | 1.9 ± 0.1 | 1.69 ± 0.09 |
| CP70 | 3.75 ± 0.05 | 2.7 ± 0.1 | 1.37 ± 0.03 |
| CP85 | 2.9 ± 0.2 | 1.9 ± 0.1 | 1.50 ± 0.09 |
| CP97 | 5.4 ± 0.3 | 4.0 ± 0.2 | 1.39 ± 0.01 |
| CP125 | 2.7 ± 0.1 | 2.0 ± 0.1 | 1.30 ± 0.01 |
All variants contain the F217A + D218S mutations. Errors are SDs of three independent measurements.
We measured folding (kfold) and unfolding (kunf) rates of the CPs by change in CD signal at 222 nm. Folding and unfolding curves fit adequately to single exponential functions and the logarithms of kfold and kunf decrease and increase linearly with [GdnHCl] as expected (Fig. 4, B and C). Extrapolated to zero denaturant, the folding rates of the five CPs are nearly identical (ranging from 0.44 to 0.66 s−1), as are their unfolding rates (1.1 × 10−5 − 3.7 × 10−5 s−1). CP folding can be modeled as fragment complementation, with the two homo fragments held at a fixed local concentration by the 30-AA flexible linker. The docking rate is then approximated by the second-order rate constant of the fragments (Table 1) multiplied by their local concentration (∼1 mM (20)). The observed kfold values are significantly slower than these calculated rates, implying that CP folding is rate-limited by folding of one or both fragments before docking or additional folding of the complex once docked. The latter scenario is more likely because kfold values are constant among CPs and show no correlation with the degree to which the isolated fragments are folded or unfolded. Thus, the mechanism of CP folding and fragment complementation appears to be rapid folding and docking of N- and C-fragments, the rate of which varies dramatically with split site, followed by a slow final folding step that does not depend on split site.
Domain swapping
RBP does not naturally domain swap. To induce RBP to form domain-swapped complexes with the hinge regions at the specified split points, we employed the “lever-assembler” design that we previously developed (21, 22). Freshly purified RU proteins were diluted to 20 μM, incubated in 0.2% SDS loading buffer for 5 m, and run on a 4–15% SDS-PAGE. Essentially all of the protein molecules remain folded in SDS, either as native monomers (39.3 kDa) or domain-swapped complexes that manifest as a ladder above the monomers (Fig. 5 A). Closer analysis of the monomer region by 4–20% SDS-PAGE confirms that only a faint band from each RU variant runs at the same position as the boiled and fully denatured controls (Fig. 5 B).
Figure 5.
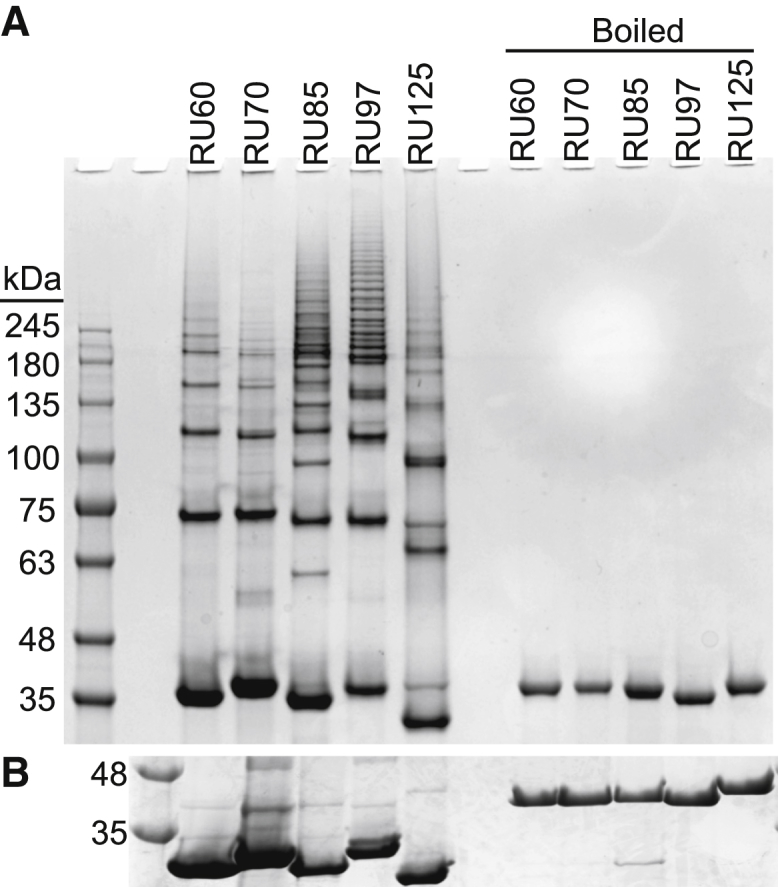
Domain swapping of RU variants assayed by SDS-PAGE. (A) 4–15% SDS-PAGE gel illustrating domain-swapped and monomeric (39.3 kDa) RU proteins is shown. (B) The monomeric region of 4–20% polyacrylamide gel is shown, indicating that the protein monomers, like the domain-swapped complexes, remain folded in SDS.
RU97 swaps to the greatest degree, as judged by the extensive laddering observed above ∼250 kDa and the relatively faint native monomer band. RU125 and RU85 follow close behind, whereas RU60 and RU70 fold mostly as monomers. Because the RBP domains of all RU constructs appear to be stable and resistant to denaturation (and subsequent rearrangement), it is likely that the distribution of swapped and monomeric species in Fig. 5 was established as the proteins were folding in the cell. The amount of laddering was not influenced by differential expression of RU variants because yields of purified proteins were similar (84–108 mg per liter of starting culture). Thus, position 97 appears to represent the most efficient site for domain swapping in vivo.
Discussion
The goal of this study is to test the hypothesis that position 97 represents the optimal location for fragment complementation, domain swapping, and circular permutation among the stretch of 65 amino acids evaluated by the overlap selection experiment. We find that bisecting RBP at position 97 yields fragments that complement with the lowest Kd value of homo pairs tested and one of the lowest of any complementation system yet reported. However, the data suggest that the outcome of the overlap selection experiment was not dictated by thermodynamic stability but by kinetic factors. The RBP1–96 + RBP97–277 complex is not the most thermally stable of the homo complexes (RBP1–59 + RBP60–277 has a higher apparent Tm). It appears to dominate over the others because it forms 100-fold faster than its closest competitor. kon is established by the N-terminal fragment, and RBP1–96 is the most efficient in this regard: it complements rapidly (kon >105 M−1 s−1) and with high affinity (Kd < 1 nM) to all of the C-terminal fragments, regardless of the latter’s length or foldedness. Given the CD results in Fig. 2, A and C, it would be tempting to conclude that greater amounts of helical structure in the N-terminal fragment equate with faster complementation. However, too much structure can be unfavorable: RBP1–124 contains 28 more residues than RBP1–96, and CD finds these residues adopt native-like helicity, but kon of the RBP1–124 + RBP125–277 homo pair is >100-fold slower than that of the RBP1–96 + RBP97–277 homo pair. Thus, RBP1–96 appears to define a “sweet spot” for rapid and high-affinity complementation.
Position 97 also emerges as the most preferred site for domain swapping in vivo. This result is unexpected, given that position 97 represents the longest loop (11 residues) in RBP. For the RU proteins to swap, the Ub domain must stretch apart the two segments of RBP on either side. Loop amino acids decouple this conformational strain by acting as flexible linkers to the point where 20 residues abolished Ub-induced domain swapping when inserted into barnase (22). The four residues comprising loop 85 and loop 125, the second-best split sites for swapping, allow for greater stretching forces and thus greater monomer destabilization. The original selection experiment found that the overlapping fragments swapped at position 97 without Ub being inserted at that site, so loop 97 appears to be an inherent hot spot for natural as well as induced swapping.
The large variations in Kd and kon that we observe for fragment binding do not translate to corresponding trends in ΔGunf and kfold of the CPs. Rather, all CPs are extremely stable and exhibit nearly identical folding and unfolding rates. This limited data set argues that a cleavage site that produces efficient complementation is predictive of efficient permutation, but a site that results in efficient permutation is not necessarily the best position for fragment complementation.
All domain-swapped and circularly permuted species exhibit extreme stability toward SDS and thermal denaturation, respectively. In addition, fragment complexes associate rapidly and dissociate over days. These findings suggest that in the overlap selection experiment, once one of the possible native structures is formed, it is likely to be kinetically trapped and slow to equilibrate with the competing native folds, even if they are of lower free energy. The presence of deep energy minima with high kinetic barriers is consistent with the exceptional stability of T. tengcongensis RBP (23).
We now address a pressing question raised by this study. Was the overlap selection experiment necessary to identify position 97 as a hot-spot site for complementation and swapping or could it have been predicted by visual inspection or computational methods? For proteins with distinct domains such as RBP, disruption sites are frequently chosen at domain boundaries. RBP consists of N- and C-terminal lobes. The major crossover point is at position 106, which is separated from position 97 by a β strand (Fig. 1 D). Cleavage at position 106 would seem to be a better choice than at position 97 because it would maintain the integrity of the N-domain β-sheet. That position 97 emerged in the overlap selection experiment indicates that domain boundaries do not necessarily make the best split points.
Hahn, Dokholyan, and colleagues recently developed a computational tool (SPELL) to predict split sites in proteins (9). This algorithm calculates the total energies of the fragments generated by scission at any given residue, then subtracts this from the energy of the intact protein to generate the “split energy.” The split energy, together with solvent-accessible area and sequence conservation data, are used to rank potential split sites. SPELL predicted the optimal site in RBP to be position 97, followed by 40, 60, and 70. Our results concur that 97 is the top choice, but we find that splitting at positions 125 and 85 yield complexes that form faster and with greater affinity than scission at position 60. Nevertheless, it is encouraging that the computational and experimental approaches converge in identifying position 97 as the hot spot for complementation. Our data suggest that kinetics play the dominant role in establishing the most efficient split site in RBP. The SPELL algorithm does not contain kinetic elements—these are significantly harder to compute than thermodynamic quantities—so it is possible that the two methods arrived at the same answer for different reasons. This idea can be tested by performing the overlap selection experiment on E. coli RBP, which is structurally similar to T. tengcongensis RBP but is much less stable and does not exhibit extreme kinetic traps (24). SPELL predicts that position 217 is the optimal split site for E. coli RBP. Our study cannot exclude the possibility of a global optimum split site occurring after residue 125 or before residue 60.
Dokholyan and co-workers also introduced a theoretical tool (H-Predictor) to predict the propensity of each amino acid in a protein to act as a hinge region for domain swapping (25). The method is based on the idea that swapping is facilitated by local unfolding of the protein around the hinge residue. H-Predictor computes the temperature at which the protein locally unfolds into two native-like subdomains around each residue, with the lowest temperature corresponding to the highest hinge propensity. Residues in loop 125 are predicted to have the greatest overall hinge propensity, followed closely by those in loop 97. The agreement with our finding that RU97 and RU125 are the most efficient swappers is remarkable, especially considering RU constructs incorporate the Ub domain as part of the hinge.
Our results and those of Fersht with chymotrypsin inhibitor 2 (CI2) (10) indicate that overlap selection can be an effective method for generating complementing protein fragments without extensive trial-and-error experimentation. The overlapped fragments RBP1–124 + RBP60–277 bind as well or better than most of the homo pairs. Mixing overlapping peptides of CI2 (64 amino acids) revealed a single crossover point at residue 40, with the redundant amino acids being unstructured (10). This proved to be the only viable cleavage site in CI2. From these two examples, there appears to be a significant advantage and no drawback to incorporating an overlapped sequence when designing a protein complementation system. In the case of RBP, identifying the crossover point (position 97) and trimming the duplicated sequence from at least one of the fragments (residues 97–125 from the C-fragment) result in further improvements in binding affinity and association kinetics.
Conclusions
Overlap selection provides a means for identifying the site in a protein that, when disrupted, produces fragments that form complemented, circularly permuted, and domain-swapped species with high efficiency and yield. Potentially powerful aspects of the method are that it may be agnostic with regard to how efficiency is attained—whether by optimization of thermodynamic and/or kinetic factors—and experimental conditions can be chosen to reflect real-world environments. The combination of overlap selection and emerging computational tools will allow researchers to generate robust platforms for engineered protein switches and biosensors.
Author Contributions
J.-H.H. and S.N.L. designed and performed experiments. M.F.P. performed experiments. S.N.L. wrote the manuscript with critical review and comments from J.-H.H. and M.F.P.
Acknowledgments
This work was supported by National Institutes of Health grant GM115762 to S.N.L.
Editor: Doug Barrick.
Footnotes
Supporting Material can be found online at https://doi.org/10.1016/j.bpj.2019.06.002.
Supporting Material
References
- 1.Romei M.G., Boxer S.G. Split green fluorescent proteins: scope, limitations, and outlook. Annu. Rev. Biophys. 2019;48:19–44. doi: 10.1146/annurev-biophys-051013-022846. [DOI] [PMC free article] [PubMed] [Google Scholar]; Romei, M. G., and S. G. Boxer. 2019. Split green fluorescent proteins: scope, limitations, and outlook. Annu. Rev. Biophys. 48:19-44. [DOI] [PMC free article] [PubMed]
- 2.Dagliyan O., Hahn K.M. Controlling protein conformation with light. Curr. Opin. Struct. Biol. 2019;57:17–22. doi: 10.1016/j.sbi.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dagliyan, O., and K. M. Hahn. 2019. Controlling protein conformation with light. Curr. Opin. Struct. Biol. 57:17-22. [DOI] [PMC free article] [PubMed]
- 3.Ribeiro L.F., Warren T.D., Ostermeier M. Construction of protein switches by domain insertion and directed evolution. Methods Mol. Biol. 2017;1596:43–55. doi: 10.1007/978-1-4939-6940-1_3. [DOI] [PubMed] [Google Scholar]; Ribeiro, L. F., T. D. Warren, and M. Ostermeier. 2017. Construction of protein switches by domain insertion and directed evolution. Methods Mol. Biol. 1596:43-55. [DOI] [PubMed]
- 4.Ha J.H., Loh S.N. Protein conformational switches: from nature to design. Chemistry. 2012;18:7984–7999. doi: 10.1002/chem.201200348. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ha, J. H., and S. N. Loh. 2012. Protein conformational switches: from nature to design. Chemistry. 18:7984-7999. [DOI] [PMC free article] [PubMed]
- 5.Michnick S.W., Landry C.R., Tchekanda E. Protein-fragment complementation assays for large-scale analysis, functional dissection, and spatiotemporal dynamic studies of protein-protein interactions in living cells. Cold Spring Harb. Protoc. 2016;2016 doi: 10.1101/pdb.top083543. [DOI] [PubMed] [Google Scholar]; Michnick, S. W., C. R. Landry, …, E. Tchekanda. 2016. Protein-fragment complementation assays for large-scale analysis, functional dissection, and spatiotemporal dynamic studies of protein-protein interactions in living cells. Cold Spring Harb. Protoc. 2016;2016. doi: 10.1101/pdb.top083543. [DOI] [PubMed]
- 6.Topell S., Hennecke J., Glockshuber R. Circularly permuted variants of the green fluorescent protein. FEBS Lett. 1999;457:283–289. doi: 10.1016/s0014-5793(99)01044-3. [DOI] [PubMed] [Google Scholar]; Topell, S., J. Hennecke, and R. Glockshuber. 1999. Circularly permuted variants of the green fluorescent protein. FEBS Lett. 457:283-289. [DOI] [PubMed]
- 7.Yu Y., Lutz S. Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol. 2011;29:18–25. doi: 10.1016/j.tibtech.2010.10.004. [DOI] [PubMed] [Google Scholar]; Yu, Y., and S. Lutz. 2011. Circular permutation: a different way to engineer enzyme structure and function. Trends Biotechnol. 29:18-25. [DOI] [PubMed]
- 8.Guntas G., Kanwar M., Ostermeier M. Circular permutation in the Ω-loop of TEM-1 β-lactamase results in improved activity and altered substrate specificity. PLoS One. 2012;7:e35998. doi: 10.1371/journal.pone.0035998. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guntas, G., M. Kanwar, and M. Ostermeier. 2012. Circular permutation in the Ω-loop of TEM-1 β-lactamase results in improved activity and altered substrate specificity. PLoS One. 7:e35998. [DOI] [PMC free article] [PubMed]
- 9.Dagliyan O., Krokhotin A., Dokholyan N.V. Computational design of chemogenetic and optogenetic split proteins. Nat. Commun. 2018;9:4042. doi: 10.1038/s41467-018-06531-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dagliyan, O., A. Krokhotin, …, N. V. Dokholyan. 2018. Computational design of chemogenetic and optogenetic split proteins. Nat. Commun. 9:4042. [DOI] [PMC free article] [PubMed]
- 10.Ladurner A.G., Itzhaki L.S., Fersht A.R. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J. Mol. Biol. 1997;273:317–329. doi: 10.1006/jmbi.1997.1303. [DOI] [PubMed] [Google Scholar]; Ladurner, A. G., L. S. Itzhaki, …, A. R. Fersht. 1997. Complementation of peptide fragments of the single domain protein chymotrypsin inhibitor 2. J. Mol. Biol. 273:317-329. [DOI] [PubMed]
- 11.Ha J.H., Karchin J.M., Loh S.N. Engineered domain swapping as an on/off switch for protein function. Chem. Biol. 2015;22:1384–1393. doi: 10.1016/j.chembiol.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ha, J. H., J. M. Karchin, …, S. N. Loh. 2015. Engineered domain swapping as an on/off switch for protein function. Chem. Biol. 22:1384-1393. [DOI] [PMC free article] [PubMed]
- 12.Ha J.H., Shinsky S.A., Loh S.N. Stepwise conversion of a binding protein to a fluorescent switch: application to Thermoanaerobacter tengcongensis ribose binding protein. Biochemistry. 2013;52:600–612. doi: 10.1021/bi301105u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ha, J. H., S. A. Shinsky, and S. N. Loh. 2013. Stepwise conversion of a binding protein to a fluorescent switch: application to Thermoanaerobacter tengcongensis ribose binding protein. Biochemistry. 52:600-612. [DOI] [PMC free article] [PubMed]
- 13.Karchin J.M., Ha J.-H., Loh S.N. Small molecule-induced domain swapping as a mechanism for controlling protein function and assembly. Sci. Rep. 2017;7:44388. doi: 10.1038/srep44388. [DOI] [PMC free article] [PubMed] [Google Scholar]; Karchin, J. M., J.-H. Ha, …, S. N. Loh. 2017. Small molecule-induced domain swapping as a mechanism for controlling protein function and assembly. Sci. Rep. 7:44388. [DOI] [PMC free article] [PubMed]
- 14.Nguyen A.W., Daugherty P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]; Nguyen, A. W., and P. S. Daugherty. 2005. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 23:355-360. [DOI] [PubMed]
- 15.Smoluchowski M.V. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Lösungen. Z. Phys. Chem. 1917;92:129–168. [Google Scholar]; Smoluchowski, M. V. 1917. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider Losungen. Z. Phys. Chem. 92:129-168.
- 16.Schlosshauer M., Baker D. Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci. 2004;13:1660–1669. doi: 10.1110/ps.03517304. [DOI] [PMC free article] [PubMed] [Google Scholar]; Schlosshauer, M., and D. Baker. 2004. Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci. 13:1660-1669. [DOI] [PMC free article] [PubMed]
- 17.Qin S., Pang X., Zhou H.X. Automated prediction of protein association rate constants. Structure. 2011;19:1744–1751. doi: 10.1016/j.str.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; Qin, S., X. Pang, and H. X. Zhou. 2011. Automated prediction of protein association rate constants. Structure. 19:1744-1751. [DOI] [PMC free article] [PubMed]
- 18.Vercillo N.C., Herald K.J., Dattelbaum J.D. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein Sci. 2007;16:362–368. doi: 10.1110/ps.062595707. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vercillo, N. C., K. J. Herald, …, J. D. Dattelbaum. 2007. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein Sci. 16:362-368. [DOI] [PMC free article] [PubMed]
- 19.Song T., Park C. Effect of folding on the export of ribose-binding protein studied with the genetically isolated suppressors for the signal sequence mutation. J. Mol. Biol. 1995;253:304–312. doi: 10.1006/jmbi.1995.0554. [DOI] [PubMed] [Google Scholar]; Song, T., and C. Park. 1995. Effect of folding on the export of ribose-binding protein studied with the genetically isolated suppressors for the signal sequence mutation. J. Mol. Biol. 253:304-312. [DOI] [PubMed]
- 20.Krishnamurthy V.M., Semetey V., Whitesides G.M. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J. Am. Chem. Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]; Krishnamurthy, V. M., V. Semetey, …, G. M. Whitesides. 2007. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J. Am. Chem. Soc. 129:1312-1320. [DOI] [PMC free article] [PubMed]
- 21.Cutler T.A., Loh S.N. Thermodynamic analysis of an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 2007;371:308–316. doi: 10.1016/j.jmb.2007.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cutler, T. A., and S. N. Loh. 2007. Thermodynamic analysis of an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 371:308-316. [DOI] [PMC free article] [PubMed]
- 22.Cutler T.A., Mills B.M., Loh S.N. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 2009;386:854–868. doi: 10.1016/j.jmb.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cutler, T. A., B. M. Mills, …, S. N. Loh. 2009. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 386:854-868. [DOI] [PMC free article] [PubMed]
- 23.Cuneo M.J., Tian Y., Hellinga H.W. The backbone structure of the thermophilic Thermoanaerobacter tengcongensis ribose binding protein is essentially identical to its mesophilic E. coli homolog. BMC Struct. Biol. 2008;8:20. doi: 10.1186/1472-6807-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cuneo, M. J., Y. Tian, …, H. W. Hellinga. 2008. The backbone structure of the thermophilic Thermoanaerobacter tengcongensis ribose binding protein is essentially identical to its mesophilic E. coli homolog. BMC Struct. Biol. 8:20. [DOI] [PMC free article] [PubMed]
- 24.Lee H., Chi S.W., Kim H. Stability and folding of precursor and mature tryptophan-substituted ribose binding protein of Escherichia coli. Arch. Biochem. Biophys. 1996;328:78–84. doi: 10.1006/abbi.1996.0145. [DOI] [PubMed] [Google Scholar]; Lee, H., S. W. Chi, …, H. Kim. 1996. Stability and folding of precursor and mature tryptophan-substituted ribose binding protein of Escherichia coli. Arch. Biochem. Biophys. 328:78-84. [DOI] [PubMed]
- 25.Ding F., Prutzman K.C., Dokholyan N.V. Topological determinants of protein domain swapping. Structure. 2006;14:5–14. doi: 10.1016/j.str.2005.09.008. [DOI] [PubMed] [Google Scholar]; Ding, F., K. C. Prutzman, …, N. V. Dokholyan. 2006. Topological determinants of protein domain swapping. Structure. 14:5-14. [DOI] [PubMed]
- 26.Santoro M.M., Bolen D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]; Santoro, M. M., and D. W. Bolen. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 27:8063-8068. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.