

Abstract
Background
Data describing therapeutic outcomes in patients with non-small cell lung cancers (NSCLC) with BRAF mutations remains limited.
Methods
We conducted a retrospective cohort study of 31 patients with metastatic NSCLC treated at Duke University Hospital who had been identified by next-generation sequencing methods to bear a BRAF mutation in their tumor in order to evaluate clinical response to immunotherapy and chemotherapy.
Results
Sixty-five percent of patients identified in this cohort were current or former smokers. Fourteen (45.2%) of patients had a BRAF V600E mutation and 17 (54.8%) had a non-V600E mutation. Median progression-free survival (PFS) in the 23 patients who received first-line chemotherapy was 6.4 months [95% confidence interval (CI), 2.3 to 13.0]. Overall survival (OS) in patients who received first-line chemotherapy showed a median survival of 18 months (95% CI, 7.4 to 28.6). OS comparing patients who had never received immunotherapy at any point was 18.4 months (95% CI, 4.1 to NE) compared to 19.0 months (95% CI, 9.9 to 28.6) in those who had received immunotherapy. We did not find a statistically significant difference in OS in patients with BRAF V600E, BRAF amplification, or non-V600E mutations. There was also no difference in OS in patients treated with targeted BRAF inhibitors compared to those who were not treated with targeted BRAF inhibitors.
Conclusions
We describe therapeutic outcomes for patients with metastatic NSCLC with BRAF mutations treated with either cytotoxic chemotherapy or immunotherapy. Although the sample size is small, the survival curves do not suggest improved clinical activity in this population when treated with immunotherapy.
Keywords: Immunotherapy, chemotherapy, retrospective cohort study, V600E, amplification
Introduction
BRAF mutations comprise approximately 2–4% of mutations seen in non-small cell lung cancer (NSCLC) (1). The BRAF protein is a nonreceptor serine/threonine kinase that acts in the ERK/MAPK pathway and is activated downstream of the Ras protein, leading to cell growth and survival, and has been shown to play a role in the development and maintenance of tumorigenic activity (2). BRAF mutations have been identified across a diverse array of cancers, most commonly in melanoma (3), but in addition to NSCLC (4,5), are also seen in colorectal cancer (6), ovarian cancer (7), and papillary thyroid cancer (8). The most frequent BRAF genetic alteration is the V600E mutation, first discovered and described in melanoma, and has been shown to represent approximately 50–60% of BRAF mutations in NSCLC (1,9,10). There has now also been emerging interest in different classes of BRAF mutations, which include Class I (V600E mutations), Class II mutations (kinase-activating non-V600E mutations), and Class III mutations (kinase-impaired non-V600E mutations that increase ERK signaling or RAS activity), with data that suggests more aggressive behavior in non-V600E mutant NSCLC (11,12).
Molecular agents that specifically target the V600E mutation have been used in metastatic melanoma with success (13,14). Importantly, combination dabrafenib and trametinib is now approved for use in NSCLC harboring a V600E mutation and is associated with clinical benefit based on recent clinical phase two studies (15,16). In a separate non-randomized phase two clinical trial that enrolled 36 patients with previously untreated metastatic BRAF V600E mutant NSCLC to receive combination dabrafenib and trametinib, 64% of patients achieved an investigator-assessed overall clinical response (16). The efficacy of this approach is reflected in the National Comprehensive Cancer Network (NCCN) NSCLC Guidelines, which move BRAF from an emerging target to a recommended test (17). The guideline also recommended against first-line pembrolizumab in patients with BRAF V600E even if PD-L1 expression was ≥50%. Since 2015, phase three trials Checkmate 057, KEYNOTE 010, and OAK have compared the PD-1 inhibitor nivolumab, the PD-1 inhibitor pembrolizumab, and PD-L1 inhibitor atezolizumab, respectively, against docetaxel with improvements in overall survival (OS) (18-20). However, molecular genotype is known to impact clinical efficacy of immunotherapy. In a meta-analysis of two of these three key trials and one phase two trial, all patients with wildtype EGFR showed a significantly increased response to immune checkpoint inhibitors compared to docetaxel, but this benefit was not seen in the EGFR-mutant subgroup (21). Given the poor response to immune checkpoint inhibitors in this subgroup of patients, while BRAF mutations are more commonly seen in current or former smokers compared to other EGFR-mutant NSCLC (1), it does also represent an oncogenic driver mutation, and it is unclear if immune checkpoint inhibition may also be ineffective in BRAF-mutant NSCLC.
Several retrospective cohort studies have examined the clinical course of patients with NSCLC with BRAF mutations, but have had inconclusive or contradictory results regarding the prognostic implications of V600E or non-V600E mutations (1,9). In particular, although the natural clinical course of this group of patients has been studied, the clinical response of NSCLCs bearing BRAF mutations to standard chemotherapy, molecular therapy, or immunotherapy is not well-described. Knowledge of clinical outcomes can aid in clinical decisions regarding optimal sequencing of therapeutic options. By investigating the natural history of these cancers in response to the different therapies we have available, we hope to provide data that can guide clinical decision-making for subjects harboring these mutations.
Methods
Patient selection
Patients who had been noted to have BRAF mutations by various next-generation sequencing methods between 2014 and 2017 at the Duke University Medical Center were selected for this study. Specifically, patients who have been identified to have a BRAF mutation by at least 1 of 3 following methods were eligible for this study: (I) ION-TORRENT next-generation sequencing hot-spot panel; (II) tissue samples sent to FoundationOne for next generation sequencing; (III) plasma-based genotyping via Guardant360 which isolates circulating tumor DNA. Patient demographics including age, sex, race, smoking status, stage at diagnosis of NSCLC, date of biopsy-proven diagnosis, treatment history, and PD-L1 expression, if available, were collected. The stage of the NSCLC was determined according to the American Joint Committee on Cancer (AJCC) staging system, 7th edition. Patients were followed from the date of diagnosis until death or until last known encounter with the health system. From the original population of patients that was collected through both our tissue-based and plasma-based next-generation sequencing methods, eight patients were excluded from this cohort due to the presence of known primary driver mutations (i.e., EGFR T790M, L858R, exon 19 deletion), six patients were excluded as they were not diagnosed with or did not develop stage IV disease, one patient was excluded due to known small-cell lung cancer (SCLC) in addition to newly diagnosed NSCLC, which made it difficult to distinguish response to therapy, and one patient was excluded due to enrollment in an active clinical trial at the time of data collection to avoid use of unpublished data. No patients were lost to follow up. This was approved by the Duke University Health System Institutional Review Board (Protocol ID: Pro00087996) prior to data collection.
Statistical analysis
Progression-free survival (PFS) was defined as the time from the date of treatment started to the date of progression or death, whichever came first. Progression of disease was investigator-assessed, based upon overall assessment by the primary oncologist as documented in medical record or interval imaging results. RECIST measurement was not performed. OS was defined as the time from the date of diagnosis to the date of death from all causes.
Kaplan-Meier product limit method was used to estimate PFS and OS. The estimators were used to graphically describe PFS and OS. From these product limit estimates, median survival times and their 95% confidence intervals (CIs) were derived. Log-Rank p-values were provided. We performed univariate Cox proportional hazards models to estimate unadjusted hazard ratios (HRs) and their 95% CIs to compare PFS and/or OS for patients who received different types of treatments of first, second, and third line of therapies, and to compare OS for patients of different BRAF mutation types. As an exploratory analysis, we also conducted multivariate Cox proportional hazards models to estimate adjusted HRs and their 95% CIs. The adjusted factors included age at diagnosis, sex, smoking history (except for modeling of PFS for third line therapy), disease stage, and race (not included in modeling on PFS for third line therapy). All analyses were performed on SAS 9.4 for Windows, Cary, NC.
Results
Patient characteristics
A total of 31 consecutively NGS-tested BRAF-positive NSCLC patients were included in the study population. The baseline demographics of the study population are depicted in Table 1. The mean age of patients at time of diagnosis was 63 years (range, 35–83); 18 (58.1%) were female; 26 (83.9%) patients were Caucasian, and 65% were current or former smokers. Twenty-eight (90.3%) NSCLCs were adenocarcinomas based on histology, and 29 (93.5%) were stage IV at time of diagnosis, with an additional 2 who were evaluated from time of diagnosis of metastatic disease.
Table 1. Patient demographics and baseline clinical characteristics.
| Characteristics | Total (N=31) |
|---|---|
| Age at diagnosis | |
| Mean ± SD | 63±11 |
| Median [range] | 65 [35–83] |
| Sex, n (%) | |
| Female | 18 (58.1) |
| Male | 13 (41.9) |
| Race, n (%) | |
| African American | 2 (6.5) |
| Asian/Pacific Islander | 2 (6.5) |
| Caucasian | 26 (83.9) |
| Native American | 1 (3.2) |
| Smoking history, n (%) | |
| No | 11 (35.5) |
| Former | 16 (51.6) |
| Current | 4 (12.9) |
| Pack years | |
| Mean ± SD | 39±29 |
| Median [range] | 40 [2–120] |
| Histology, n (%) | |
| Adenocarcinoma | 27 (87.1) |
| Adenocarcinoma (h/o squamous cell carcinoma) | 1 (3.2) |
| Carcinoma | 1 (3.2) |
| Poorly differentiated carcinoma | 1 (3.2) |
| Sarcomatoid carcinoma | 1 (3.2) |
BRAF mutation profile
Fourteen of the 31 patients in this cohort had V600E mutations. Of the 17 non-V600E mutations, four patients had Class II mutations, which possess intermediate to high kinase activity, and two patients had Class III mutations, which possess low or lack kinase activity (12), five had BRAF amplifications, and the remaining six had silent mutations or mutations of unknown significance (Figure 1). Specifically, these six remaining mutations were identified as R354Q (n=2), I463T (n=1), and 3 silent mutations (also noted in Table 2). Four mutations were found to reside in the p-loop, which stabilizes ATP binding and maintains an inactive state (12). A schematic of the BRAF protein with its relevant domains, and the relative frequencies of mutations noted in this cohort has been provided in Figure 2.
Figure 1.

BRAF mutation genotypes.
Table 2. BRAF mutations and next-generation sequencing method.
| Type of BRAF mutation | Total (N=31), n (%) | Hotspot panel (N) | FoundationOne (N) | Guardant360 (N) |
|---|---|---|---|---|
| V600E | 14 (45.2) | 7 | 5 | 2 |
| BRAF amplification | 5 (16.1) | 0 | 0 | 5 |
| G469V | 2 (6.5) | 0 | 1 | 1 |
| R354Q | 2 (6.5) | 0 | 0 | 2 |
| G469R | 1 (3.2) | 1 | 0 | 0 |
| G469A | 1 (3.2) | 1 | 0 | 0 |
| D594G | 1 (3.2) | 1 | 0 | 0 |
| N581S | 1 (3.2) | 0 | 0 | 1 |
| I463T | 1 (3.2) | 0 | 0 | 1 |
| Silent mutations | 3 (9.7) | 0 | 0 | 3 |
Figure 2.

BRAF gene and mutations observed in this cohort.
BRAF mutations were identified by an institutional lung hotspot panel, FoundationOne tissue sampling, and Guardant360 serum sampling. Using these methods, the lung hotspot panel identified 7 of the 16 BRAF V600E-mutant NSCLCs, FoundationOne identified five, and Guardant360 identified two (Table 2). The methods by which the non-V600E mutations were also identified are also visible in Table 2. All five of the BRAF amplifications were identified using the plasma-based genotyping with Guardant360. On average, the BRAF amplifications were noted to have 2.32 copy numbers, with a range from 2.25 to 2.37.
Eight patients with EGFR mutations accepted to be oncogenic drivers or evidence of the presence of an EGFR driver (T790M, L858R, or exon 19 deletion) were detected to also have BRAF mutations. None of these patients were included in our analyses as it was assumed that these BRAF mutations had emerged as acquired resistance mutations, and thus their EGFR mutations would independently affect their responses to therapy. Given previously described data that had noted reduced responses in patients with EGFR-mutant NSCLC, these patients were therefore excluded to avoid confounding from their concomitant EGFR mutation leading to the expected reduced responses to immune therapy. The most common EGFR driver mutations were an exon 19 deletion, present in four patients, and T790M mutation, present also in four patients. An L858R mutation was present in three patients. With regard to their BRAF mutational genotype, six had BRAF amplification, one had a BRAF D287V mutation, and one had a splice BRAF mutation. There was a single patient who was found to have concomitant NSCLC and SCLC, who did also have two separate BRAF mutations, one of which was V600E and another which was a silent mutation.
For first-line chemotherapy, 23 patients were analyzed for OS. Twenty-three patients received first-line chemotherapy and 3 patients received first-line immunotherapy. All patients in the first-line chemotherapy group received platinum-doublet chemotherapy (most commonly, carboplatin-pemetrexed with or without bevacizumab, but several patients received cisplatin instead of carboplatin, and docetaxel or etoposide rather than pemetrexed). Four patients received non-pemetrexed-based regimens. Of the three patients who received immunotherapy first-line, all received anti-PD1 monotherapy. All three patients had stage IV disease, two of which were adenocarcinomas and one of which was sarcomatoid. Briefly, 16 patients received one line of therapy, 10 patients received two to three lines of therapy, and five patients received four or more lines of therapy.
PD-L1 expression and tumor mutational burden (TMB)
PD-L1 expression was only available for 11 of 31 patients. PD-L1 expression levels ranged from <1% (calculated as zero) up to 90%, with 6 patients with PD-L1 expression levels greater than 50%. TMB was only available on five patients with the following mutation rates: 3, 4, 15, 17, 18 mutations/Mb.
PFS and OS of first-line chemotherapy and immunotherapy
The median PFS in patients who received first-line chemotherapy was 6.4 months (95% CI, 2.3 to 13.0) as depicted in Figure 3. The PFS of each of the three patients who received first-line immunotherapy was 0.17, 1.4, and 4.4 months. The median OS in patients who received first-line chemotherapy was 18.4 months (95% CI, 7.4 to 28.6) as depicted in Figure 4. The OS of each of the three patients who received first-line immunotherapy was 0.17, 6.8, and 7.5 months.
Figure 3.
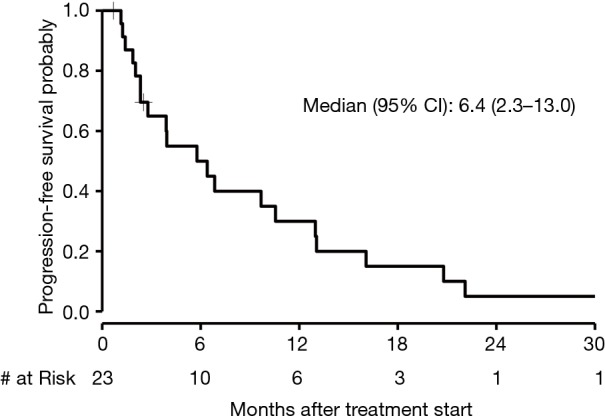
Kaplan-Meier curve of PFS of first-line chemotherapy. PFS, progression-free survival.
Figure 4.
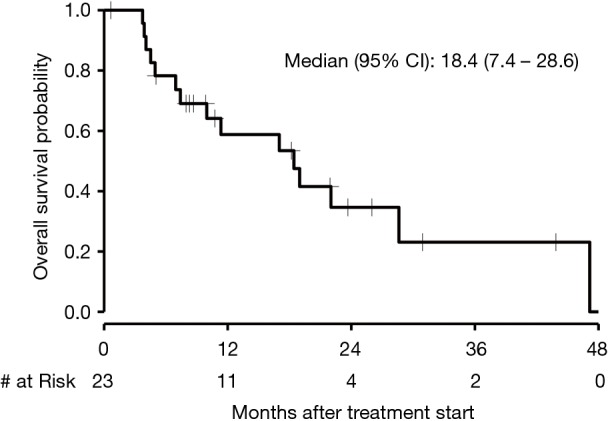
Kaplan-Meier curve of OS of first-line chemotherapy. OS, overall survival.
PFS of second-line immunotherapy
Eight patients received second-line immunotherapy and their median PFS was 2.5 months (95% CI, 1.4 to 5.9).
OS of patients who did or did not receive immunotherapy
In our cohort, 15 patients were identified to have received non-immunotherapy systemic treatment (chemotherapy or targeted therapy) without ever receiving immunotherapy. Sixteen patients were identified to have received immunotherapy at least once during their course of treatment. As depicted in Figure 5, median OS for patients who never received immunotherapy was 18.4 months (95% CI, 4.1 to NE) compared to 19.0 months (95% CI, 9.9 to 28.6) for those who received immunotherapy at least once (P=0.929).
Figure 5.
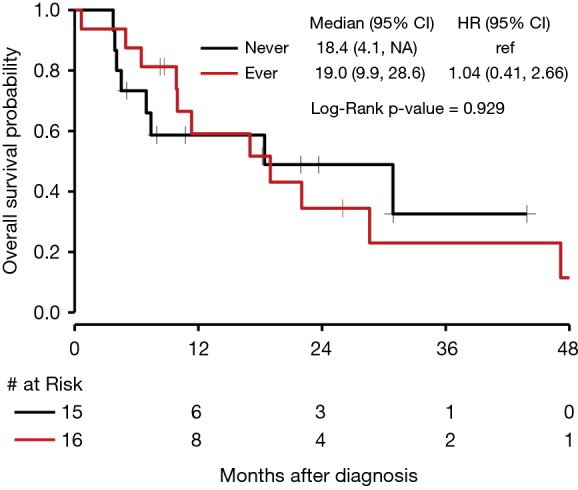
Kaplan-Meier curves of OS of patients who received or never received immunotherapy. OS, overall survival.
Concomitant chemotherapy and immunotherapy
There were two patients who received frontline combination chemotherapy and immunotherapy. Both were diagnosed with stage IV adenocarcinoma, with PFS of 1.5 and 2.1 months, and OS of 6.6 and 5.6 months.
Clinical outcomes of targeted BRAF inhibitors and outcomes in different BRAF mutations including BRAF amplification
There was no significant difference in OS for those who received targeted BRAF inhibitors compared to those who did not with a P value of 0.44 (Figure 6). The median OS for NSCLC with BRAF V600E mutations was 28.6 months (95% CI, 7.4 to NE). The median OS for BRAF amplification was 19.0 months (95% CI, 4.5 to 47.1) with a HR of 1.6 (95% CI, 0.6 to 4.7) compared to BRAF V600E. The median OS for BRAF mutations that were neither V600E nor BRAF amplification (as seen in Figure 1, 6 of the 12 patients who fit this category had Class II or III mutations) was 9.9 months (95% CI, 3.9 to 63.0), with a HR of 1.6 (95% CI, 0.5 to 5.3) compared to BRAF V600E. Seven patients received either dabrafenib, vemurafenib, or combination dabrafenib/trametinib at some point during their course of treatment for their NSCLC, and all patients who received these therapies had V600E mutations. Of these seven patients, five had partial response, 1 had stable disease or mixed response, and 1 had disease progression. There was no significant difference in OS for V600E and non-V600E mutations (Figure 7). As there were five patients with a BRAF amplification as their main BRAF mutation, these were separated from the non-V600E mutations and independently compared to V600E mutations, but again without a significant difference in OS.
Figure 6.
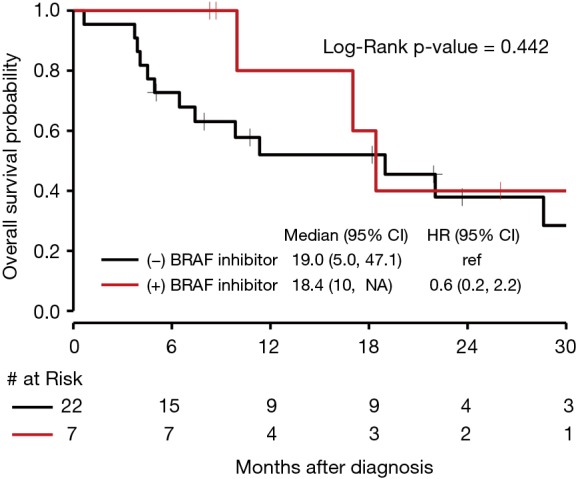
Kaplan-Meier curve of OS for patients treated with targeted BRAF inhibitors vs. patients not treated with targeted BRAF inhibitors. OS, overall survival.
Figure 7.
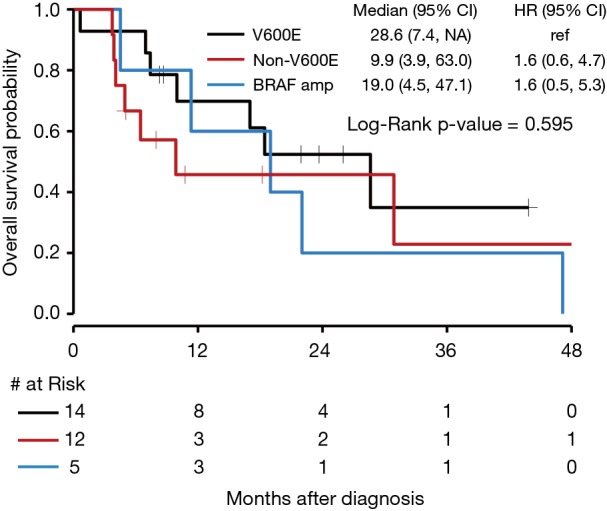
Kaplan-Meier curve of OS by BRAF mutation genotype. OS, overall survival.
Discussion
This single-center retrospective cohort study of patients with metastatic NSCLC bearing BRAF mutations notes clinical responses that are comparable in immunotherapy compared to chemotherapy, and at least in the few patients administered first-line immunotherapy, numerically inferior to the mean PFS and OS in first-line chemotherapy. Because of the small cohort size and the relatively recent approval of immunotherapy as first-line therapy, PFS and OS curves were documented only for patients who received chemotherapy first-line, and the measured survivals rather than curves were recorded for the three patients who received immunotherapy as first-line treatment. We further compared OS in patients who had at any point received immunotherapy, regardless of line of treatment, and patients who never received immunotherapy but instead received other systemic therapy, and while the study is underpowered, this showed no clear benefit to immunotherapy over chemotherapy. This is a finding that likely merits further research to determine the optimal regimen for patients in this cohort.
Checkmate 057, KEYNOTE 010, and OAK are three seminal randomized-controlled trials comparing a monoclonal antibody targeting PD-L1 or PD-1 against docetaxel in patients with previously-treated NSCLC (18-20). Each of these studies showed a superiority in OS when treated with the immune checkpoint inhibitor compared to docetaxel, but not in PFS. This apparently contradictory improvement in OS but similar rates of progression between these two therapies have been attributed to a number of things, including the concept of pseudoprogression, a delay in the antitumor activity, prolonged immune activation which provides benefit in spite of tumor progression (22), or small subset of patients experiencing the majority of the benefit. While our study was underpowered to compare long-term survival benefit in seen in non-BRAF mutant NSCLC with immunotherapy, the survival curves comparing those who received chemotherapy or targeted therapy alone compared to those who had at some point received immunotherapy demonstrate numerically similar median OS, without the survival benefit that has been seen in many populations in NSCLC in response to immunotherapy.
Recently, additional data has been published in which Dudnik et al. examined patients with BRAF mutations, noting an association with high PD-L1 expression, low/intermediate TMB and microsatellite-stable status (23). Although the sample size was small and baseline characteristics tended to favor patients treated with immunotherapy, the authors suggest that PFS was comparable to the outcomes seen in Checkmate 057, KEYNOTE 010 and OAK (23).
Importantly, multiple studies have demonstrated reduced response rates to immune therapies in patients with oncogenic EGFR mutation or ALK alterations (19,20). The mechanism behind the reduced response of NSCLC with oncogenic EGFR mutations to immunotherapy is not well-understood, although it has been suggested that these cancers may have a lower TMB, perhaps because a single EGFR driver mutation may suffice for tumor development, and a lower TMB may decrease response to immune checkpoint inhibitors (24). Interestingly, in vitro studies have shown that EML4-ALK fusion and mutant EGFR may actually induce PD-L1 expression in NSCLC cell lines, suggesting that these known oncogenic drivers may mediate oncogenesis in part through escape from the immune system (25). Regardless, further research into the interplay between the development of driver gene alterations and the tumor immune environment will be needed. As the TMB was not available on the vast majority of the patients in this cohort, we were unable to evaluate whether TMB may play a role in the response of these tumors to therapy.
The development of immune checkpoint inhibitors has been based upon the observation that the immune system plays a role in regulating tumor progression. Multiple solid tumors have been shown to overexpress PD-L1, which is found on T-cells and helps create an immunosuppressive environment that presumably protects the tumor against the host’s immune system (26-28). In general, patients with stronger levels of pre-treatment PD-L1 expression tend to have stronger responses to immunotherapy (29-31), but cancers with low or no PD-L1 expression have been shown to also respond to immunotherapy (29,30). In the OAK study mentioned above, the benefit seen with atezolizumab in treating NSCLC was seen independent of PD-L1 expression, with a benefit in OS seen in low or negative PD-L1 expression patients treated with atezolizumab over docetaxel, although higher PD-L1 expression cohorts derived greater survival benefit (18). PD-L1 expression in our population was available in only a limited number of patients, and thus difficult to interpret whether it may have influenced clinical outcomes one way or the other.
Our population did not show a significant difference in mortality between patients with NSCLC harboring BRAF V600E and non-V600E mutations, including BRAF amplification. The genetic profile of our cohort had slightly more non-V600E BRAF mutations than V600E mutations, with 53% of our patients bearing non-V600E mutations, which is consistent with previous reports (32). Indeed, BRAF amplifications, which have not been well-studied, composed 16.1% of the mutations seen in our patient population although no significant difference in survival was seen between different BRAF mutations. A number of BRAF amplifications were also present in patients who already had an identified EGFR driver mutation, and are thus not a part of our survival analyses as their clinical behavior is likely contingent upon their EGFR mutation. Given that a BRAF mutation’s oncogenic properties are driven by constitutive activation of its pathway, a BRAF amplification could give rise to increased activity of this pathway as well. Thus far, there have been no studies into the clinical significance of BRAF amplification in tumor development and progression. Finally, there were a number of mutations of unknown significance, which evenly received immunotherapy at some point, or chemotherapy only. Due to increased sensitivity of next-generation sequencing, the authors anticipate that more mutations of unknown significance will now be identified, although these mutations are unlikely to be as clinically sensitizing as Class I, II, or III mutations. It remains to be seen if BRAF amplifications or these other mutations will provide any prognostic or therapeutic significance, and future research will be needed.
There now exist several generations of BRAF V600E inhibitors. As previously mentioned, phase II trials of combination dabrafenib, a BRAF V600E inhibitor, and trametinib, a MEK inhibitor, in patients with V600E and previously untreated NSCLC are currently underway and showing promising results with most recently reported overall responses of 64% in the patients recruited (16). Finally, a second generation of BRAF inhibitors aimed at bypassing resistance to first-generation BRAF inhibitors is under study. These have been tested in xenograft models, and demonstrate activity against splice variants and BRAF amplification as well as V600E mutations (33-35). There was a trend towards improved survival in patients treated with BRAF inhibitors in the first 18 months of therapy compared to those who did not receive BRAF inhibitors in our study, which given that there was no statistical difference in survival between those who had V600E mutations and those who did not, is suggestive of an improved therapeutic response. These targeted agents that are currently undergoing clinical and preclinical testing represent a new and promising avenue for treating this population of patients.
Multiple limitations are applicable to our study given the retrospective design and possibilities of underlying selection bias and the presence of cohort imbalances. These limitations that must be considered when comparing pooled data of OS curve comparing patients who either did or did not receive immunotherapy at some point during their treatment. In addition, disease progression was assessed by the investigator in our analysis. A larger number of subjects would certainly generate a more reliable statistical comparison, however this remains challenging given the relative rarity of this mutation across the NSCLC patient population. In conclusion, although this data is underpowered and is based upon a small cohort in a single-center analysis and larger prospective trials are needed, patients with NSCLC harboring BRAF mutations do not clearly demonstrate improved clinical activity with immunotherapy.
Acknowledgments
Funding for publication costs of this manuscript was provided by the Duke Stead Resident Research Grant.
Ethical Statement: This study was approved by the Duke University Health System Institutional Review Board (Protocol ID: Pro00087996) prior to data collection. A waiver of informed consent was approved as part of this Institutional Review Board protocol.
Footnotes
Conflicts of Interest: TE Stinchcombe has consulted for Novartis. NE Ready is on ad board honoraria for BMS, Abbvie, Merck, Genentech, Celgene, and Novartis. J Crawford serves as a consultant/independent contractor for Amgen, Enzychem, Merck and Pfizer; he receives grant support from Amgen, AstraZeneca, and Bayer; he is a chair/DSMB member for Beyond Spring, G1 Therapeutics, Janssen, Merrimack, Mylan, and Roche. RJ Nagy and RB Lanman are employees and stockholders of Guardant Health. JM Clarke has received research funding from Bristol-Meyers Squibb, Medpacto, and is the site principal investigator with funding received from Genentech, Spectrum, Adaptimmune, Bayer and Abbvie; he has served as an advisory for Astra Zeneca, Guardant, and Merck and he has been a speaker for Merck and Guardant. The other authors have no conflicts of interest to declare.
References
- 1.Marchetti A, Felicioni L, Malatesta S, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 2011;29:3574-9. 10.1200/JCO.2011.35.9638 [DOI] [PubMed] [Google Scholar]
- 2.Ji H, Wang Z, Perera SA, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res 2007;67:4933-9. 10.1158/0008-5472.CAN-06-4592 [DOI] [PubMed] [Google Scholar]
- 3.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135-47. 10.1056/NEJMoa050092 [DOI] [PubMed] [Google Scholar]
- 4.Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002;62:6997-7000. [PubMed] [Google Scholar]
- 5.Naoki K, Chen TH, Richards WG, et al. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res 2002;62:7001-3. [PubMed] [Google Scholar]
- 6.Samowitz WS, Sweeney C, Herrick J, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 2005;65:6063-9. 10.1158/0008-5472.CAN-05-0404 [DOI] [PubMed] [Google Scholar]
- 7.Singer G, Oldt R, III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 2003;95:484-6. 10.1093/jnci/95.6.484 [DOI] [PubMed] [Google Scholar]
- 8.Kimura ET, Nikiforova MN, Zhu Z, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003;63:1454-7. [PubMed] [Google Scholar]
- 9.Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669-74. 10.1097/JTO.0000000000000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardarella S, Ogino A, Nishino M, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res 2013;19:4532-40. 10.1158/1078-0432.CCR-13-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dagogo-Jack I, Martinez P, Yeap BY, et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-mutant Lung Cancer. Clin Cancer Res 2019;25:158-65. 10.1158/1078-0432.CCR-18-2062 [DOI] [PubMed] [Google Scholar]
- 12.Dankner M, Rose AA, Rajkumar S, et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 2018;37:3183-99. 10.1038/s41388-018-0171-x [DOI] [PubMed] [Google Scholar]
- 13.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507-16. 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012;367:1694-703. 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planchard D, Besse B, Groen HJ, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984-93. 10.1016/S1470-2045(16)30146-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planchard D, Smit EF, Groen HJ, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 2017;18:1307-16. 10.1016/S1470-2045(17)30679-4 [DOI] [PubMed] [Google Scholar]
- 17.Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2017;15:504-35. 10.6004/jnccn.2017.0050 [DOI] [PubMed] [Google Scholar]
- 18.Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017;389:255-65. 10.1016/S0140-6736(16)32517-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015;373:1627-39. 10.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540-50. 10.1016/S0140-6736(15)01281-7 [DOI] [PubMed] [Google Scholar]
- 21.Lee CK, Man J, Lord S, et al. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J Thorac Oncol 2017;12:403-7. 10.1016/j.jtho.2016.10.007 [DOI] [PubMed] [Google Scholar]
- 22.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009;15:7412-20. 10.1158/1078-0432.CCR-09-1624 [DOI] [PubMed] [Google Scholar]
- 23.Dudnik E, Peled N, Nechushtan H, et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J Thorac Oncol 2018;13:1128-37. 10.1016/j.jtho.2018.04.024 [DOI] [PubMed] [Google Scholar]
- 24.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ota K, Azuma K, Kawahara A, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res 2015;21:4014-21. 10.1158/1078-0432.CCR-15-0016 [DOI] [PubMed] [Google Scholar]
- 26.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother 2005;54:307-14. 10.1007/s00262-004-0593-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002;99:12293-7. 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007;56:739-45. 10.1007/s00262-006-0272-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taube JM, Klein AP, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064-74. 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847-56. 10.1158/1535-7163.MCT-14-0983 [DOI] [PubMed] [Google Scholar]
- 31.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noeparast A, Teugels E, Giron P, et al. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2016;8:60094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girotti MR, Lopes F, Preece N, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015;27:85-96. 10.1016/j.ccell.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell 2015;28:370-83. 10.1016/j.ccell.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Spevak W, Zhang Y, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015;526:583. 10.1038/nature14982 [DOI] [PubMed] [Google Scholar]