

Abstract
Aims/Introduction
To investigate the clinical and genetic characteristics of Chinese patients with a phenotype consistent with maturity‐onset diabetes of the young type 2 and explore the pathogenic mechanism of their hyperglycemia.
Materials and Methods
We studied 12 probands and their extended families referred to our center for screening mutations in the glucokinase gene (GCK). Clinical data were collected and genetic analysis was carried out. The recombinant wild‐type and mutant glucokinase were generated in Escherichia coli. The kinetic parameters and thermal stability of the enzymes were determined in vitro.
Results
In the 12 families, 11 GCK mutations (R43C, T168A, K169N, R191W, Y215X, E221K, M235T, R250H, W257X, G261R and A379E) and one variant of uncertain significance (R275H) were identified. R191W was detected in two unrelated families. Of the 11 GCK mutations, three mutations (c.507G>C, K169N; c.645C>A, Y215X; c.771G>A, W257X; NM_000162.3, NP_000153.1) are novel. Basic kinetics analysis explained the pathogenicity of the five mutants (R43C, K169N, R191W, E221K and A379E), which showed reduced enzyme activity with relative activity indexes between ~0.001 and 0.5 compared with the wild‐type (1.0). In addition, the thermal stabilities of these five mutants were also decreased to varying degrees. However, for R250H and R275H, there was no significant difference in the enzyme activity and thermal stability between the mutants and the wild type.
Conclusions
We have identified 11 GCK mutations and one variant of uncertain significance in 12 Chinese families with hyperglycemia. For five GCK mutations (R43C, K169N, R191W, E221K and A379E), the changes in enzyme kinetics and thermostability might be the pathogenic mechanisms by which mutations cause hyperglycemia.
Keywords: Enzyme kinetics, Glucokinase, Maturity‐onset diabetes of the young
Introduction
Glucokinase is one of the four hexokinase isoforms present in humans, and is highly expressed in pancreatic β‐cells, hepatocytes and the brain. This enzyme catalyzes the first reaction of glycolysis by converting glucose into glucose‐6‐phosphate with adenosine triphosphate (ATP) as the second substrate. Although it is also known as hexokinase IV or hexokinase D, glucokinase has particular kinetics compared with the other hexokinase isoforms, including a low affinity for glucose (concentration of glucose at which the enzyme is half maximally activated, S0.5, 7–9 mmol/L), a cooperativity with glucose (Hill coefficient close to 1.7) and a lack of inhibition by its end‐product, glucose‐6‐phosphate. These particular kinetics allow pancreatic β‐cells to change the glucose phosphorylation rate over a range of physiological glucose concentrations (4–15 mmol/L)1 and make glucokinase the glucose sensor of pancreatic β‐cells. Therefore, as the glucose sensor, glucokinase plays a crucial role in the regulation of glucose‐stimulated insulin secretion and integrates glucose metabolism with insulin secretion.
Genetic studies have shown that mutations in the glucokinase (GCK) gene are responsible for various glucose regulation disorders. Heterozygous inactivating GCK mutations cause maturity‐onset diabetes of the young type 2 (MODY2), whereas heterozygous activating mutations cause persistent hyperinsulinemic hypoglycemia of infancy, and homozygous inactivating mutations cause permanent neonatal diabetes mellitus. More than 600 mutations have been reported up to now2. The most frequent mutations are heterozygous inactivating GCK mutations, which are the causes of MODY2. The alterations of the enzyme kinetic parameters are the principal pathophysiological mechanism of the disorders2.
Patients with MODY2 are characterized by mild stable fasting hyperglycemia (5.5–8.0 mmol/L)3, which is present after birth. However, because of the lack of typical symptoms of diabetes and diabetic complications4, it is not easy to make an early diagnosis. Special attention should be paid to these patients, and a correct diagnosis is important because there are implications for treatment, prognosis and the patient's family members. In the present study, we report 12 pedigrees of Chinese origin with fasting hyperglycemia, identifying 12 mutations in the GCK gene and attempting to clarify the pathogenic mechanism of their diseases.
Methods
Participants
A total of 10 probands with suspected MODY2 were collected from 2010 to 2014 at Peking Union Medical College Hospital, Beijing, China. The selection criteria were persistent and stable fasting hyperglycemia (5.5–10.0 mmol/L), glycosylated hemoglobin A1c <8%, negative search for the markers of type 1 diabetes islet cell antibodies, glutamic acid decarboxylase antibodies and insulin auto‐antibodies with or without insulinoma‐associated‐2 autoantibodies (tyrosine phosphatase antibodies). One proband was from our earlier study of gestational diabetes mellitus5. This participant was suspected to have MODY2 because of her fasting hyperglycemia and a small increment during a 2‐h oral glucose tolerance test. One proband was diagnosed with neonatal diabetes mellitus at 5 months. His fasting blood glucose was 29.4 mmol/L, and both of his parents have stable fasting hyperglycemia. All the participants underwent diagnostic screening for GCK mutations. Informed written consent was obtained from all the participants. For participants aged <18 years, written consent was obtained from either a parent or legal guardian. The study was approved by the ethics committee of Peking Union Medical College Hospital (IRB No. B137).
Phenotypic characterization
The medical history of probands and their extended family members were acquired in detail. The body mass index was calculated. The measurement of the plasma glucose concentration and glycosylated hemoglobin A1c were taken in the local hospital and registered. High‐sensitivity C‐reactive protein was measured by the immunoturbidimetric method on a Beckman AU5800 analyzer (Beckman Coulter Inc., Brea, CA, USA) at the Department of Clinical Laboratory of Peking Union Medical College Hospital6. The working ranges of the assays were 0.08–80.00 mg/L. The method has an intra‐assay coefficient of variation of 5.73% at 0.23 mg/L, and interassay coefficient of variation of 5.76% at 0.23 mg/L. High‐sensitivity C‐reactive protein values >10 mg/L were considered likely to represent an acute inflammatory response and were excluded from further analysis. The results of the oral glucose tolerance test using an anhydrous glucose load of 75 g were recorded.
Mutation screening and in silico functional analysis
Genomic deoxyribonucleic acid (DNA) was isolated from peripheral blood lymphocytes using the Biomed Gene kit (Biomed Ltd, Beijing, China). All the 10 exons, the intron‐exon boundaries and the promoter sequences7 of the GCK gene were amplified by polymerase chain reaction using the primers shown in Table S1, and then sequenced bi‐directionally on an ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences were compared with the reference genomic GCK sequence (NM_000162.3) using the Human BLAT Search online of University of California Santa Cruz (http://genome.ucsc.edu/cgi-bin/hgBlat). The Single Nucleotide Polymorphism Database (dbSNP, release 147) and the ClinVar Database (release 2017) were used to determine whether the identified variant was novel. All missense mutations were analyzed in silico using SIFT and PolyPhen2 (http://sift.jcvi.org/ and http://genetics.bwh.harvard.edu/pph/, respectively).
Preparation of recombinant proteins
A 1,410 bp human pancreatic β‐cell glucokinase complementary DNA clone was synthesized (Biomed Ltd), and then was ligated into BamHI and EcoRI sites of plasmid pGEX‐2T (GE Healthcare, Piscataway, NJ, USA) to generate the wild‐type recombinant vector. The recombinant vector would generate wild‐type fusion protein with GST tag when it was expressed in Escherichia coli. Mutations were introduced to the wild type by site‐directed Fast Mutagenesis System (TransGene Biotech, Beijing, China). The sequences of oligonucleotides used to modify the wild type are shown in Table S2. The coding sequences of the wild type and each mutant were confirmed by DNA sequencing.
The wild‐type and mutant proteins were expressed in E. coli, as described previously8, 9, 10, 11. The recombinant vectors were transformed into E. coli BL21. Cultures were grown at 37°C in Terrific Broth media containing ampicillin (100 μg/mL) to an optical density at 600 nm of approximately 2. The shaker temperature was then lowered to 22°C, and the expression of fusion proteins were induced with 200 μmol/L isopropyl‐β‐D‐thiogalactoside. After 18 h of induction at 22°C, the cells were harvested. Cell lysis and protein extraction were implemented with B‐PER® Bacterial Protein Extraction Reagent (Thermo, Rockford, IL, USA). Fusion protein was purified by affinity chromatography using a Glutathione Sepharose 4B column (GE Health). Purified protein was stored at −80°C in 50 mmol/L Tris‐HCl buffer, pH 8.0, containing 200 mmol/L KCl, 10 mmol/L glutathione and 5 mmol/L dithiothreitol8. The purity of the preparations was routinely screened with sodium dodecyl sulfate polyacrylamide gel electrophoresis. The protein concentrations were determined by Coomassie Blue binding assay with Coomassie Plus (Bradford) Assay Kit (Thermo).
Enzyme kinetic analysis
The activity of the glucokinase was measured spectrophotometrically on a Thermo Scientific Multiskan GO spectrophotometer (Thermo), using a nicotinamide adenine dinucleotide phosphate‐coupled assay with glucose‐6‐phosphate dehydrogenase. Stock enzyme was diluted to approximately 250 μg/mL with 50 mmol/L Tris‐HCl buffer, pH 8.0. The reaction medium included 100 mmol/L Tris‐HCl, pH 7.4, 6 mmol/L MgCl2, 2 mmol/L dithiothreitol, 0.1% bovine serum albumin, 150 mmol/L KCl, 0.4 mmol/L nicotinamide adenine dinucleotide phosphate, 2.5 units/mL glucose‐6‐phosphate dehydrogenase and substrate concentrations as specified8, 12. One unit of enzyme activity is defined as the amount of enzyme that phosphorylates 1 μmol of glucose per minute at 30°C. A total of 12 glucose concentrations (0.1–200 mmol/L) with 5 mmol/L ATP were used to determine the kinetic variables for glucose, and nine concentrations of ATP (0.02–5 mmol/L) with 200 mmol/L glucose were used to determine the kinetic variables for ATP. The Hill equation was utilized to calculate the V max, glucose S0.5 and Hill coefficient (h) of glucokinase towards glucose13. The Michaelis–Menten equation was applied to calculate the K m value for ATP13. The concentration of glucose at which the inflection point occurs was calculated as described by Kesavan12. The relative activity index of each mutant was estimated as previously described13 with 2.5 mmol/l intracellular ATP. Kinetic parameters were analyzed by SigmaPlot software (version 12.0; Systat Software, College Station, TX, USA).
Thermolability assays
There were two protocols to test the thermal stability of the enzyme: (i) the enzyme solutions were incubated for 30 min at different temperatures ranging from 30 to 60°C, and analyzed at 30°C with 200 mmol/L glucose and 5 mmol/L ATP; and (ii) the enzyme solutions were incubated for different periods of time from 5 to 30 min at 47.5 or 52°C (R250H and R275H) and then tested.
Statistical analysis
Data were analyzed by Stata software (version 13.0; StataCorp LP, College Station, TX, USA). Continuous variables were reported as the mean ± standard error of the mean. Discontinuous variables were reported as medians with a given range. For each independent enzyme preparation, all assays were carried out in triplicate. Comparisons of the continuous variables between the two groups were carried out by independent sample t‐tests. P‐values <0.05 were considered significant.
Results
GCK mutation screening and clinical characterization
Mutation screening identified 12 mutations in the 12 probands (Table 1). A total of 10 were missense mutations and two were nonsense mutations. Although mutation K169N and Y215X have been reported in other families with hyperglycemia, they are both novel mutations at the level of nucleotide mutation (c.507G>C, K169N; c.645C>A, Y215X; NM_000162.3, NP_000153.1). W257X is a novel mutation that has not been reported before.
Table 1.
GCK mutations identified in the probands with hyperglycemia
| Family | Exon | Nucleotide change | Amino acid change | Reported functional studies | References | Prediction of SIFT (score) | Prediction of PolyPhen2 (score) |
|---|---|---|---|---|---|---|---|
| M2 | 2 | c.127C>T | R43C | No | 31 | Damaging (0.01) | Probably damaging (1.000) |
| M26 | 5 | c.502A>G | T168A | Yes | 32, 33 | Damaging (0) | Probably damaging (0.999) |
| M16 | 5 | c.507G>C† | K169N | No | 34, 35 | Damaging (0) | Probably damaging (1.000) |
| M13, N8 | 5 | c.571C>T | R191W | No | 36, 37, 38 | Damaging (0) | Probably damaging (1.000) |
| N22 | 6 | c.645C>A† | Y215X | No | 38, 39 | – | – |
| M6 | 6 | c.661G>A | E221K | No | 40 | Tolerated (0.13) | Probably damaging (1.000) |
| M38 | 7 | c.704T>C | M235T | Yes | 2 | Damaging (0) | Possibly damaging (0.754) |
| M1 | 7 | c.749G>A | R250H | No | 20 | Tolerated (0.36) | Probably damaging (0.999) |
| M7 | 7 | c.771G>A† | W257X‡ | No | – | – | – |
| M3 | 7 | c.781G>C | G261R | Yes | 13, 41 | Damaging (0) | Probably damaging (1.000) |
| G64 | 7 | c.824G>A | R275H | No | – | Damaging (0.03) | Benign (0.011) |
| N8 | 9 | c.1136C>A | A379E | No | – | Damaging (0) | Probably damaging (0.979) |
†The nucleotide mutation is novel. ‡The amino acid mutation is novel.
All the pedigrees are shown in Figure 1. The R191W mutation was identified in two unrelated families, M13 and N8. The proband of N8 was diagnosed with neonatal diabetes mellitus, with a new compound heterozygous mutation (A379E and R191W) in the GCK gene. He inherited the A379E mutation from his father and the R191W mutation from his mother. GCK mutation carriers might present as diabetes mellitus, impaired fasting glucose or gestational diabetes mellitus. In the same family, patients with other types of diabetes and patients with MODY2 can exist simultaneously.
Figure 1.
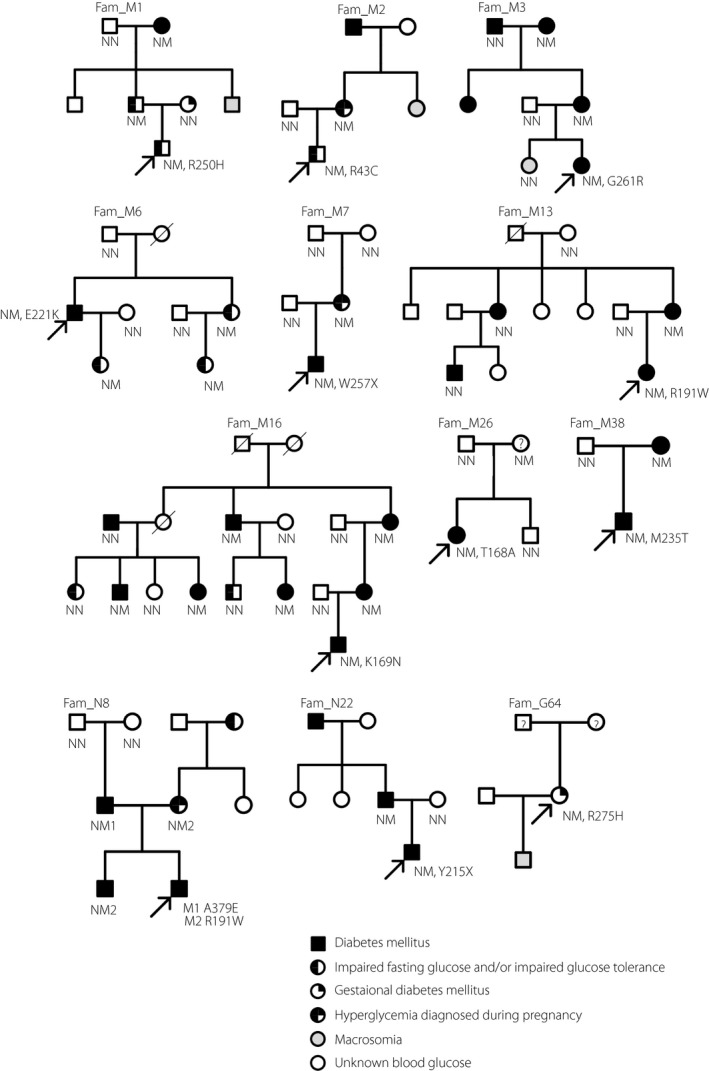
Pedigrees of Chinese families with hyperglycemia. Square symbols indicate males, and round symbols indicate females. A diagonal slash mark through the symbol indicates deceased. The black arrow indicates probands. Filled quadrant indicates the diagnosis of the specific indicated trait. Four quadrants filling indicates diabetes mellitus. Left half quadrant filling indicates impaired fasting glucose and/or impaired glucose tolerance. Right upper quadrant filling indicates history of gestational diabetes mellitus. Right lower quadrant blank indicates hyperglycemia diagnosed during pregnancy. Grey filling indicates macrosomia at birth. Question mark indicates that blood glucose is unknown.
The clinical and biochemical characteristics of the GCK mutation carriers are presented in Table 2. Their median age of diagnosis of hyperglycemia was 24 years. The blood glucose concentrations and glycosylated hemoglobin A1c values are similar to those in previous studies of MODY214.
Table 2.
Clinical and biochemical parameters of heterozygous GCK mutation carriers
| Family | GCK mutation | Age at diagnosis (years) | Patient sex | BMI (kg/m2)† | FPG (mmol/L) | 2 h‐OGTT (mmol/L) | HbA1c (%) | hsCRP (mg/L)‡ |
|---|---|---|---|---|---|---|---|---|
| M2 | R43C | 18 | Male | 16.6 | 6.2 | 8.5 | 6.5 | 0.19 |
| M26 | T168A | 24 | Female | 20.1 | 7.0 | 9.3 | 6.3 | 0.15 |
| M16 | K169N | 4 | Male | 14.4 (P20) | 5.7 | 8.3 | 6.6 | 0.08 |
| M16‐m | K169N | 25 | Female | 20.6 | 6.5 | – | 6.6 | 0.50 |
| M13 | R191W | 21 | Female | 16.8 | 6.6 | 11.6 | 6.2 | 0.19 |
| M13‐m | R191W | 44 | Female | 20.1 | 8.5 | 12.9 | 6.6 | 0.10 |
| N8‐m | R191W | 30 | Female | 15.2 | 6.7 | – | 6.5 | 0.73 |
| N22 | Y215X | 10 | Male | 13.8 (P4) | 6.6 | 12.3 | 7.2 | 0.31 |
| M6 | E221K | 32 | Male | 22.8 | 7.7 | 11.0 | 6.7 | – |
| M38 | M235T | 6 | Female | 16.1 (P75) | 7.4 | 6.5 | 6.5 | – |
| M1 | R250H | 5 years 3 months | Male | 13.7 (P9) | 5.2 | 10.5§ | 5.0 | 0.10 |
| M1‐f | R250H | 32 | Male | 23.9 | 5.5 | 7.1 | 6.7 | – |
| M7 | W257X | 5 days | Male | – | 7.5 | – | 7.0 | – |
| M3 | G261R | 8 years 9 months | Female | 16.2 (P55) | 6.9 | 9.1 | 5.9 | 0.25 |
| M3‐m | G261R | 31 | Female | 19.3 | 6.7 | 7.1 | 6.5 | 0.36 |
| G64 | R275H | 30 | Female | 20.1 | 5.9 | 7.9 | – | – |
| N8‐f | A379E | 32 | Male | 24.2 | 6.6 | 11.3 | 6.6 | 0.35 |
†For participants aged <18 years, body mass index (BMI) percentile for age and sex‐matched control is given in parentheses. ‡The reference range was 0.00–3.00 mg/L. §Plasma glucose at 2 h after a steamed bread meal test. f, father; FPG, fasting plasma glucose; HbA1c, glycosylated hemoglobin A1c; hsCRP, high‐sensitivity C‐reactive protein; m, mother; OGTT, plasma glucose at 2 h after a standard oral glucose tolerance test (1.75 g per kg, maximum 75 g).
In silico analysis of all missense mutations
Analysis of SIFT and PolyPhen2 showed that for the majority of missense mutations there was consensus between the two different bioinformatic tools, but the three mutations, E221K, R250H and R275H, have different protein function predictions (Table 1). Because the functional characterization of the T168A, M235T and G261R mutations have been reported in the literature2, the remaining missense mutations were selected for the kinetic and thermolability analysis. The Y215X and W257X mutations are predicted to cause loss of normal protein function either through protein truncation or nonsense‐mediated messenger ribonucleic acid decay. We consider the two mutations to be pathogenic and no longer suitable for functional analysis.
Kinetic properties of recombinant glucokinases
Each of the purified recombinant proteins was homogeneous and had an apparent molecular mass of ~75 kDa. The protein yield and kinetic profiles are shown in Table 3. Most of the mutant proteins have a lower yield, except for K169N and R275H. Four mutants (K169N, R191W, E221K, A379E) were shown to be kinetically inactivating, with a decreased rate of catalysis (K cat) and decreased affinity for glucose (increased S0.5). Mutants R191W, E221K and A379E also showed decreased affinity for ATP (increased K m). Mutant R43C had a similar affinity of substrates to the wild type, but its catalytic activity (K cat) was decreased significantly. Mutant K169N produced the strongest effect on enzyme activity because of its extremely low affinity for glucose (S0.5 of ~500 mmol/L). Data analysis for this mutant was limited, and therefore the kinetic results are approximations. K169N might have lost the cooperativity for glucose, as shown by an approximate Hill coefficient close to 1. Although the mutants R250H and R275H have a similar or marginally increased affinity for glucose and slightly decreased K cat, their overall activity was not seriously affected compared with the wild type.
Table 3.
Kinetic constants of human recombinant wild‐type and mutant β‐cell glutathione S‐transferase–glucokinase fusion proteins
| Preparation | Protein yield (mg/L) | S0.5 for glucose (mmol/L) | Hill coefficient (h) | Inflection point (mmol/L) | K m for ATP (mmol/L) | K cat (/s) | Relative activity index |
|---|---|---|---|---|---|---|---|
| WT | 85.8 ± 8.7 | 7.63 ± 0.21 | 1.42 ± 0.06 | 2.22 ± 0.25 | 0.30 ± 0.01 | 20.9 ± 2.1 | 1.000 |
| R43C | 65.2 ± 3.9* | 7.47 ± 0.12 | 1.44 ± 0.03 | 2.26 ± 0.18 | 0.29 ± 0.02 | 8.9 ± 0.9*** | 0.429 ± 0.073*** |
| K169N† | 84.6 ± 5.7 | 516.90 ± 29.46*** | 0.95 ± 0.07** | NO | NO | 0.4 ± 0.1*** | NO |
| R191W | 47.7 ± 8.1** | 35.27 ± 2.20*** | 1.40 ± 0.11 | 9.66 ± 1.90 | 0.42 ± 0.01*** | 6.5 ± 1.0*** | 0.037 ± 0.006*** |
| E221K | 42.3 ± 7.6** | 11.07 ± 0.60*** | 1.30 ± 0.05 | 2.55 ± 0.33 | 0.39 ± 0.02** | 14.1 ± 1.8* | 0.477 ± 0.057*** |
| R250H | 68.2 ± 3.3* | 7.30 ± 0.50 | 1.33 ± 0.05 | 1.72 ± 0.23 | 0.33 ± 0.03 | 14.1 ± 2.4* | 0.854 ± 0.232 |
| R275H | 79.6 ± 12.8 | 6.80 ± 0.36* | 1.50 ± 0.21 | 2.26 ± 0.73 | 0.26 ± 0.04 | 14.6 ± 2.3* | 0.725 ± 0.174 |
| A379E | 54.5 ± 5.3** | 13.30 ± 0.44*** | 1.38 ± 0.04 | 3.52 ± 0.35 | 0.54 ± 0.04*** | 10.5 ± 1.1** | 0.233 ± 0.025*** |
Data presented as the mean ± standard error of the mean from three separate enzyme expressions. K cat values refer to the turnover at 30°C. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the wild type. †Due to the severity of the kinetic inactivation, data measurement was difficult. The inflection point is a mathematical derivative of glucose S0.5 and therefore was not submitted for statistical analysis. ATP, adenosine triphosphate; NO, data not obtainable; WT, wild type.
Thermal stability analysis
The thermal stability of wild‐type and mutant recombinant enzymes were tested at different temperatures (Figure 2a). When the incubation temperature increased to 45°C, the enzyme activity of the wild type remained constant (the relative activity >90%), whereas that of the four mutants (R43C, R191W, E221K and A379E) rapidly fell to 50% or even less. Although the data of K169N was not shown in the figure, it was the most unstable enzyme. After a 30‐min incubation at 30°C, its activity was reduced to ~10% of that observed without this treatment. After incubation at 35°C, its activity was undetectable. Mutants R250H and R275H showed thermal stability similar to that of the wild type (Figure S1). The time‐course analysis of thermal decay showed a clear difference in the lability of the five enzymes (Figure 2b). Within the first 15 min of incubation at 47.5°C, the wild type was relatively stable (the relative activity >90%), whereas the four mutants rapidly lost their activity. After a 30‐min incubation at 47.5°C, the remaining activity of the wild type was approximately 40%, whereas that of the mutants was 10–30%.
Figure 2.
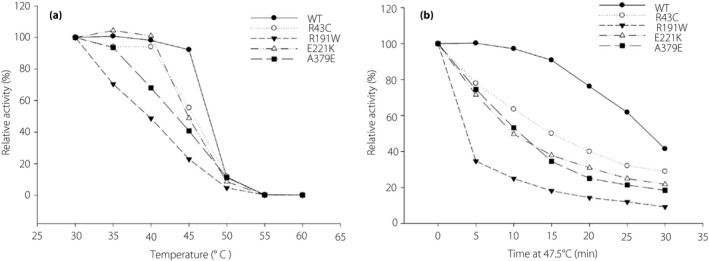
Effect of temperature on the stability of the glutathione S‐transferase–glucokinase fusion protein. Stock enzyme solutions were diluted to 220 μg/mL by 50 mmol/L Tris/HCl, pH 8.0. (a) The enzyme solutions were incubated for 30 min at different temperatures ranging from 30 to 60°C and assayed at 30°C. (b) The enzyme solutions were incubated for different periods of time from 5 to 30 min at 47.5°C. Mean ± standard error of the mean for three independent enzyme preparations are shown for each case. WT, wild type.
Discussion
In the present study, we described 12 Chinese pedigrees suspected of having MODY2. Although the GCK mutation carriers had fasting hyperglycemia and a small increment in the 2‐h blood glucose in the oral glucose tolerance test, they usually lacked typical symptoms of diabetes, such as polyuria and polydipsia. Routine or accidental blood glucose testing is the main form of discovery of hyperglycemia. The GCK mutation carriers might be misdiagnosed as type 1 diabetes, type 2 diabetes or gestational diabetes, depending on their age and physical state. These factors add to the difficulty of the correct diagnosis of MODY2.
In addition, diabetes mellitus is a common disease. In a country with a high incidence of diabetes, in China it is very common for patients with diabetes caused by GCK mutations and patients with other types of diabetes to coexist in the same family, such as family M3 and M13. Therefore, it is inappropriate to determine whether GCK mutation is pathogenic only by genotype–phenotype analysis in a large pedigree with diabetes. Most of the previous studies identified the pathogenic GCK mutations through functional analysis, in which enzyme kinetics and thermal stability analysis were the most commonly used. For mutations with different function predictions by bioinformatic tools, laboratory functional analysis was especially important.
We identified 12 GCK mutations in the present study, and seven were functional analyzed. Among these GCK mutations, K169N has the worst enzyme activity. A study on the crystal structures of human glucokinase revealed that the K169 residue is one of the glucose binding sites15. In addition, K169 enhances the binding of glucokinase with both ATP and glucose, and directly participates in glucose phosphorylation16. Because of the important role of the K169 residue, it is reasonable that the K169N mutation would lead to a marked decrease in enzyme activity. The activity of the R191W mutation was just ~4% of the wild type. Studies have shown that the R191 residue is important for the transformation of glucokinase from the super‐open conformation (inactive) to the closed conformation (active)17. We hypothesized that the R191W mutation would lead to an obstacle in the transformation of the enzyme's spatial conformation and further cause abnormal glucose metabolism. Compared with other mutations, A379E mutation has the lowest affinity with ATP. This might be related to the spatial location of the A379 residue. Research has shown that A379 residue is located at the back of the ATP binding site18. It is interesting to find that mutations A379V18 and A379T19 have similar kinetics as A379E, which reflect the importance of the correct residue.
Compared with the wild type, R43C and E221K mutations have slightly increased thermal instability. However, it is important to note that the rapid decline of the enzyme activity of the two mutations occurred at temperatures >40°C, which suggests that in the physiological temperature range (~37°C), the slight changes in thermal stability are not the major molecular mechanism by which R43C and E221K causes hyperglycemia. For the two mutations, the changes in enzyme kinetics are more likely to be the cause of hyperglycemia.
Because the relative activity index of R250H and R275H mutations are both >70%13, whether they are pathogenic is still uncertain. However, near‐normal kinetics and protein thermal stability do not necessarily mean they are harmless. R250H mutation has been reported in another study and described as “likely pathogenic”20. The R275H (c.824G>A) variant is registered as rs767565869 in the dbSNP147 database. In the annotation of the variant, the allele frequency of T (A) is 0.001% (1/118,348). It is not a common single‐nucleotide polymorphism. Therefore, R275H should now be stated as a variant of uncertain significance. Another GCK mutation, R275C, which was found in a Pakistani family, also has near‐normal kinetics and protein stability. However, additional functional studies have shown that increased cellular degradation is the molecular mechanism by which R275C causes MODY221. Therefore, further research in a cellular model would be required for the two mutations. Islet‐specific glucokinase and liver‐specific glucokinase are isoforms from the GCK gene. The former is induced by glucose, whereas the latter is regulated by insulin and glucokinase regulator protein (GKRP)22, 23. When blood glucose decreases, GKRP inhibits and sequesters glucokinase into the nucleus, whereas when blood glucose increases, GKRP releases the enzyme into the cytoplasm to participate in glucose phosphorylation. The pathogenesis of MODY2 might be due to glucokinase deficiencies in pancreatic β‐cells, hepatocytes or both24. Previous studies have suggested that defects in the regulation of glucokinase by GKRP can lead to catalytic instability25, 26. In addition to GKRP, the activity of glucokinase in the cell is regulated by other substances, such as 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase27, 28and some small molecules29, 30. To clarify the interaction of the mutants with GKRP or 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase, and to better understand the cellular mechanism by which GCK mutation causes MODY2, further research is required.
In summary, we identified 11 GCK mutations and one variant of uncertain significance in Chinese families with hyperglycemia. Although the hyperglycemia of the GCK mutation carriers is similar, the biochemical defects caused by these mutations are uneven and range from full kinetic inactivation to almost normal activity.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 | Primers for amplification of the glucokinase gene.
Table S2 | Sequences of oligonucleotides used for site‐directed mutagenesis.
Figure S1 | Effect of temperature on the stability of the R250H and R275H mutants.
Acknowledgments
The authors thank the participants and families that graciously agreed to participate in the study. This work was supported by the National Natural Science Foundation of China (No. 81570715, No. 81170736); and the National Key Research and Development Program of China (No. 2016YFA010 1002), the National Natural Science Foundation of Young Scholars of China (No. 81300649), China Scholarship Council Foundation (201308110443), PUMC Youth Fund (33320140022) and Fundamental Research Funds for the Central Universities, and Scientific Activities Foundation for Selected Returned Overseas Professionals of Human Resources and Social Security Ministry.
J Diabetes Investig 2019; 10: 963–971
References
- 1. Matschinsky FM. Regulation of pancreatic beta‐cell glucokinase: from basics to therapeutics. Diabetes 2002; 51(Suppl 3): S394–S404. [DOI] [PubMed] [Google Scholar]
- 2. Osbak KK, Colclough K, Saint‐Martin C, et al Update on mutations in glucokinase (GCK), which cause maturity‐onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 2009; 30: 1512–1526. [DOI] [PubMed] [Google Scholar]
- 3. Stride A, Vaxillaire M, Tuomi T, et al The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002; 45: 427–435. [DOI] [PubMed] [Google Scholar]
- 4. Steele AM, Shields BM, Wensley KJ, et al Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014; 311: 279–286. [DOI] [PubMed] [Google Scholar]
- 5. Wang Z, Ping F, Zhang Q, et al Preliminary screening of mutations in the glucokinase gene of Chinese patients with gestational diabetes. J Diabetes Investig 2018; 9: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin X, Tang G, Qiu L, et al A multicenter reference intervals study for specific proteins in China. Medicine 2015; 94: e2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gasperikova D, Tribble ND, Stanik J, et al Identification of a novel beta‐cell glucokinase (GCK) promoter mutation (‐71G>C) that modulates GCK gene expression through loss of allele‐specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes 2009; 58: 1929–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liang Y, Kesavan P, Wang LQ, et al Variable effects of maturity‐onset‐diabetes‐of‐youth (MODY)‐associated glucokinase mutations on substrate interactions and stability of the enzyme. Biochem J 1995; 309: 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miller SP, Anand GR, Karschnia EJ, et al Characterization of glucokinase mutations associated with maturity‐onset diabetes of the young type 2 (MODY‐2): different glucokinase defects lead to a common phenotype. Diabetes 1999; 48: 1645–1651. [DOI] [PubMed] [Google Scholar]
- 10. Ralph EC, Sun S. Biochemical characterization of MODY2 glucokinase variants V62M and G72R reveals reduced enzymatic activities relative to wild type. Biochemistry 2009; 48: 2514–2521. [DOI] [PubMed] [Google Scholar]
- 11. Galan M, Vincent O, Roncero I, et al Effects of novel maturity‐onset diabetes of the young (MODY)‐associated mutations on glucokinase activity and protein stability. Biochem J 2006; 393: 389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kesavan P, Wang L, Davis E, et al Structural instability of mutant beta‐cell glucokinase: implications for the molecular pathogenesis of maturity‐onset diabetes of the young (type‐2). Biochem J 1997; 322: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davis EA, Cuesta‐Munoz A, Raoul M, et al Mutants of glucokinase cause hypoglycaemia‐ and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia 1999; 42: 1175–1186. [DOI] [PubMed] [Google Scholar]
- 14. Ellard S, Bellanne‐Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetic diagnosis of maturity‐onset diabetes of the young. Diabetologia 2008; 51: 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamata K, Mitsuya M, Nishimura T, et al Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004; 12: 429–438. [DOI] [PubMed] [Google Scholar]
- 16. Zhang J, Li C, Shi T, et al Lys169 of human glucokinase is a determinant for glucose phosphorylation: implication for the atomic mechanism of glucokinase catalysis. PLoS ONE 2009; 4: e6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Molnes J, Bjorkhaug L, Sovik O, et al Catalytic activation of human glucokinase by substrate binding: residue contacts involved in the binding of D‐glucose to the super‐open form and conformational transitions. FEBS J 2008; 275: 2467–2481. [DOI] [PubMed] [Google Scholar]
- 18. Estalella I, Garcia‐Gimeno MA, Marina A, et al Biochemical characterization of novel glucokinase mutations isolated from Spanish maturity‐onset diabetes of the young (MODY2) patients. J Hum Genet 2008; 53: 460–466. [DOI] [PubMed] [Google Scholar]
- 19. Zelent B, Odili S, Buettger C, et al Mutational analysis of allosteric activation and inhibition of glucokinase. Biochem J 2011; 440: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santana LS, Caetano LA, Costa‐Riquetto AD, et al Clinical application of ACMG‐AMP guidelines inHNF1A and GCK variants in a cohort of MODY families. Clin Genet 2017; 92: 388–396. [DOI] [PubMed] [Google Scholar]
- 21. Negahdar M, Aukrust I, Molnes J, et al GCK‐MODY diabetes as a protein misfolding disease: the mutation R275C promotes protein misfolding, self‐association and cellular degradation. Mol Cell Endocrinol 2014; 382: 55–65. [DOI] [PubMed] [Google Scholar]
- 22. Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta‐cells and hepatocytes. Diabetes 1990; 39: 647–652. [DOI] [PubMed] [Google Scholar]
- 23. Choi JM, Seo MH, Kyeong HH, et al Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci USA 2013; 110: 10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakera AJ, Steele AM, Gloyn AL, et al Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 2015; 38: 1383–1392. [DOI] [PubMed] [Google Scholar]
- 25. Heredia VV, Carlson TJ, Garcia E, et al Biochemical basis of glucokinase activation and the regulation by glucokinase regulatory protein in naturally occurring mutations. J Biol Chem 2006; 281: 40201–40207. [DOI] [PubMed] [Google Scholar]
- 26. Garcia‐Herrero CM, Galan M, Vincent O, et al Functional analysis of human glucokinase gene mutations causing MODY2: exploring the regulatory mechanisms of glucokinase activity. Diabetologia 2007; 50: 325–333. [DOI] [PubMed] [Google Scholar]
- 27. Okar DA, Lange AJ, Wu C. Interaction with PFK‐/FBP‐2 is essential to glucokinase molecular physiology. Cell Mol Life Sci 2009; 66: 731–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Payne VA, Arden C, Wu C, et al Dual role of phosphofructokinase‐2/fructose bisphosphatase‐2 in regulating the compartmentation and expression of glucokinase in hepatocytes. Diabetes 2005; 54: 1949–1957. [DOI] [PubMed] [Google Scholar]
- 29. Hofmeister‐Brix A, Kollmann K, Langer S, et al Identification of the ubiquitin‐like domain of midnolin as a new glucokinase interaction partner. J Biol Chem 2013; 288: 35824–35839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cullen KS, Al‐Oanzi ZH, O'Harte FP, et al Glucagon induces translocation of glucokinase from the cytoplasm to the nucleus of hepatocytes by transfer between 6‐phosphofructo 2‐kinase/fructose 2,6‐bisphosphatase‐2 and the glucokinase regulatory protein. Biochim Biophys Acta 2014; 1843: 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borowiec M, Antosik K, Fendler W, et al Novel glucokinase mutations in patients with monogenic diabetes ‐ clinical outline of GCK‐MD and potential for founder effect in Slavic population. Clin Genet 2012; 81: 278–283. [DOI] [PubMed] [Google Scholar]
- 32. Turkkahraman D, Bircan I, Tribble ND, et al Permanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. J Pediatr 2008; 153: 122–126. [DOI] [PubMed] [Google Scholar]
- 33. Durmaz E, Flanagan S, Berdeli A, et al Variability in the age at diagnosis of diabetes in two unrelated patients with a homozygous glucokinase gene mutation. J Pediatr Endocrinol Metab 2012; 25: 805–808. [DOI] [PubMed] [Google Scholar]
- 34. Gloyn AL. Glucokinase (GCK) mutations in hyper‐ and hypoglycemia: maturity‐onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat 2003; 22: 353–362. [DOI] [PubMed] [Google Scholar]
- 35. Massa O, Meschi F, Cuesta‐Munoz A, et al High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first‐phase insulin response, insulin sensitivity and BMI. Diabetologia 2001; 44: 898–905. [DOI] [PubMed] [Google Scholar]
- 36. Ellard S, Beards F, Allen LI, et al A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia 2000; 43: 250–253. [DOI] [PubMed] [Google Scholar]
- 37. Hwang JS, Shin CH, Yang SW, et al Genetic and clinical characteristics of Korean maturity‐onset diabetes of the young (MODY) patients. Diabetes Res Clin Pract 2006; 74: 75–81. [DOI] [PubMed] [Google Scholar]
- 38. Sagen JV, Bjorkhaug L, Molnes J, et al Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY Registry. Pediatr Diabetes 2008; 9: 442–449. [DOI] [PubMed] [Google Scholar]
- 39. Sagen JV, Odili S, Bjorkhaug L, et al From clinicogenetic studies of maturity‐onset diabetes of the young to unraveling complex mechanisms of glucokinase regulation. Diabetes 2006; 55: 1713–1722. [DOI] [PubMed] [Google Scholar]
- 40. Guazzini B, Gaffi D, Mainieri D, et al Three novel missense mutations in the glucokinase gene (G80S; E221K; G227C) in Italian subjects with maturity‐onset diabetes of the young (MODY). Mutations in brief no. 162. Online. Hum Mutat 1998; 12: 136. [DOI] [PubMed] [Google Scholar]
- 41. Gidh‐Jain M, Takeda J, Xu LZ, et al Glucokinase mutations associated with non‐insulin‐dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci USA 1993; 90: 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | Primers for amplification of the glucokinase gene.
Table S2 | Sequences of oligonucleotides used for site‐directed mutagenesis.
Figure S1 | Effect of temperature on the stability of the R250H and R275H mutants.