

Abstract
Aims/Introduction
Type 2 diabetes mellitus is a risk factor of acute kidney injury after myocardial infarction (MI), a form of cardiorenal syndrome. Recent clinical trials have shown that a sodium–glucose cotransporter 2 (SGLT2) inhibitor improved both cardiac and renal outcomes in patients with type 2 diabetes mellitus, but effects of an SGLT2 inhibitor on cardiorenal syndrome remain unclear.
Materials and Methods
Type 2 diabetes mellitus (Otsuka Long‐Evans Tokushima Fatty rats [OLETF]) and control (Long‐Evans Tokushima Otsuka rats [LETO]) were treated with canagliflozin, an SGLT2 inhibitor, for 2 weeks. Renal tissues were analyzed at 12 h after MI with or without preoperative fasting.
Results
Canagliflozin reduced blood glucose levels in OLETF, and blood β‐hydroxybutyrate levels were increased by canagliflozin only with fasting. MI increased neutrophil gelatinase‐associated lipocalin and kidney injury molecule‐1 protein levels in the kidney by 3.2‐ and 1.6‐fold, respectively, in OLETF, but not in LETO. The renal messenger ribonucleic acid level of Toll‐like receptor 4 was higher in OLETF than in LETO after MI, whereas messenger ribonucleic acid levels of cytokines/chemokines were not significantly different. Levels of lipid peroxides, nicotinamide adenine dinucleotide phosphate oxidase (NOX)2 and NOX4 proteins after MI were significantly higher in OLETF than in LETO. Canagliflozin with pre‐MI fasting suppressed MI‐induced renal expression of neutrophil gelatinase‐associated lipocalin and kidney injury molecule‐1 in OLETF, together with reductions in lipid peroxides and NOX proteins in the kidney. Blood β‐hydroxybutyrate levels before MI were inversely correlated with neutrophil gelatinase‐associated lipocalin protein levels in OLETF. Pre‐incubation with β‐hydroxybutyrate attenuated angiotensin II‐induced upregulation of NOX4 in NRK‐52E cells.
Conclusions
The findings suggest that SGLT2 inhibitor treatment with a fasting period protects kidneys from MI‐induced cardiorenal syndrome, possibly by β‐hydroxybutyrate‐mediated reduction of NOXs and oxidative stress, in type 2 diabetic rats.
Keywords: Cardiorenal syndrome, Oxidative stress, Sodium–glucose cotransporter 2 inhibitor
Introduction
Cardiorenal syndromes (CRS) are defined as disorders of the heart and kidney in which acute or chronic dysfunction in one organ might induce acute or chronic dysfunction in another organ1. Of the five types of CRS, type 1 CRS is characterized by acute kidney injury (AKI) as a result of rapid worsening of cardiac function. Indeed, AKI often develops in patients with acute myocardial infarction (MI), and is strongly associated with mortality in patients with MI. Decreased renal blood flow as a result of cardiac dysfunction and elevated renal venous congestion have been suggested to induce type 1 CRS through activation of the sympathetic nervous system and the renin–angiotensin–aldosterone (RAA) system2. In addition, non‐hemodynamic mechanisms, such as increased oxidative stress and chronic inflammation, contribute to type 1 CRS3. Diabetes mellitus is an independent risk factor for the development of AKI in patients with MI4, 5, 6, and we recently reported that augmented activation of Toll‐like receptors (TLRs), germline‐encoded innate immune receptors7, plays roles in the susceptibility to AKI after MI in diabetic rats8. However, how pharmacological treatment of hyperglycemia modifies the diabetes mellitus‐induced increase in renal susceptibility to AKI remains unclear.
Sodium–glucose cotransporter 2 (SGLT2) inhibitors are newly developed glucose‐lowering agents that inhibit glucose reuptake at the renal proximal tubule where most of the glucose is reabsorbed. Recent clinical trials have shown that treatment with an SGLT2 inhibitor significantly reduced not only cardiovascular events, but also renal events, including decreases in estimated glomerular filtration rate, progression of albuminuria and onset of renal replacement therapy9, 10. Amelioration of renal hyperfiltration, high glucose‐induced increase in oxidative stress, inflammation and apoptosis are possible mechanisms of protection by SGLT2 inhibitors11. Indeed, an SGLT2 inhibitor suppressed oxidative stress and inflammation in the kidneys of animal models12, 13, 14. Interestingly, it has been reported that SGLT2 inhibitors increase the circulating level of β‐hydroxybutyrate (βOHB), a ketone body15, 16, and that βOHB attenuates oxidative stress by upregulating anti‐oxidative molecules through inhibition of histone deacetylases (HDACs)17. In contrast, the initial dip in estimated glomerular filtration rate by SGLT2 inhibitors treatment might increase the risk for AKI18, although there is no evidence to support such a possibility. The effects of SGLT2 inhibitors on CRS in diabetes mellitus have not been specifically examined.
In the present study, we examined the effect of canagliflozin, an SGLT2 inhibitor, on the increased susceptibility to AKI after MI in diabetes mellitus. As a model of diabetes mellitus, we used Otsuka Long‐Evans Tokushima Fatty rats (OLETF) at ages of 25–30 weeks. The reason for selection of OLETF in the present study is threefold. First, OLETF spontaneously develop diabetes mellitus primarily by hyperphagia as a result of a lack of cholecystokinin‐A receptor in the brain, and they show the typical phenotype of type 2 diabetes mellitus (i.e., obesity, hyperinsulinemia and hypertriglyceridemia)19, 20. Second, OLETF at the ages of 25–30 weeks show an early stage of diabetic nephropathy: lower serum creatinine level with protein urea than that in Long‐Evans Tokushima Otsuka rats (LETO), non‐diabetes mellitus control rats8, 21, 22. Third, we previously found that renal susceptibility to type 1 CRS was higher in OLETF than in LETO8. We postulated that a beneficial effect of canagliflozin on diabetes mellitus‐induced worsening of CRS, if any, should be detectable in OLETF.
Methods
This study was approved by the Animal Use Committee of Sapporo Medical University (16‐060_17‐055).
Animals and experimental protocol
Administration of canagliflozin and induction of MIProtocol 1
Male LETO and OLETF (Sankyo Lab Service, Tokyo, Japan) at the ages of 25–30 weeks were used in all experiments. Rats were subcutaneously pretreated with a vehicle (dimethyl sulfoxide and polyethylene glycol; 1:1 v/v) or canagliflozin (1 mg/kg per day) for 2 weeks using osmotic minipumps (Alzet, Cupertino, CA, USA). After measurement of blood pressure and pulse rate by a tail‐cuff method, rats were prepared for induction of MI or a sham operation, as in our previous studies8, 20, 23. In brief, the rats were anesthetized with isoflurane (1–2%), and a marginal branch of the left coronary artery was permanently ligated to induce MI. All rats were allowed ad libitum access to water, but were restricted from food for 12 h after surgery. The rats were divided into six groups: LETO‐sham, LETO‐MI, OLETF‐sham, OLETF‐MI, canagliflozin‐treated OLETF‐sham and canagliflozin‐treated OLETF‐MI groups.
Protocol 2
In this protocol, the effect of fasting on renal susceptibility to CRS in canagliflozin‐treated OLETF was examined. OLETF treated with the vehicle or canagliflozin were fasted for 12 h and then the coronary artery of each rat was permanently ligated.
Sampling of blood and kidney tissues was carried out at 12 h after MI, because our previous studies20, 23 showed that approximately two‐thirds of OLETF died during a period of 48 h after MI. After measurements of blood pressure and heart rate under anesthesia, blood samples were taken through a catheter placed in the carotid artery. Then the kidneys were excised, and one kidney was quickly frozen in liquid nitrogen and stored at −80°C, and the other kidney was fixed with 10% formaldehyde for histology.
Blood levels of glucose, βOHB and angiotensin II were measured using a Glutest‐mint (Sanwa Kagaku Kenkyusho, Nagoya, Japan), Precision Xceed (Abbott, Chicago, IL, USA) and Angiotensin II EIA Kit (Sigma‐Aldrich, St. Louis, MO, USA), respectively.
Histological analyses
Formaldehyde‐fixed paraffin sections (3 μm) were stained using primary antibodies against neutrophil gelatinase‐associated lipocalin (NGAL; sc‐50531, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and kidney injury molecule‐1 (KIM‐1; AF3689, 1:20; R&D Systems, Minneapolis, MN, USA). The NGAL‐ and KIM‐1‐positive areas were determined in 10 randomly selected fields from six kidneys in each group. The glomerular area was excluded for analyses of NGAL and KIM‐1.
Cell culture and treatment
NRK‐52E cells, a rat renal proximal tubular cell line, were cultured with Dulbecco's modified Eagle's medium containing 10% bovine serum. After serum deprivation for 24 h, cells were pretreated with βOHB (Sigma‐Aldrich) for 1 h followed by 24 h‐incubation with 3 μmol/L of angiotensin II (Alomone Labs, Jerusalem, Israel). After 24 h, cells were harvested for immunoblotting.
Messenger ribonucleic acid quantification
Total ribonucleic acid (RNA) was isolated from frozen tissues by using an RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA, USA). First‐strand complementary deoxyribonucleic acid (DNA) was synthesized using a SuperScript VILO™ cDNA Synthesis Kit (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). DNA amplification was carried out in ABI PRISM7500 (Life Technologies) by using Taqman Universal PCR Master Mix. Taqman gene expression assays are shown in Table S1. All assays were carried out in duplicate and by the standard curve method using serial complementary DNA dilution. β‐Actin served as an internal control.
Immunoblotting
Immunoblot analysis was carried out as in our previous studies8, 20, 22. Equal amounts of protein were analyzed by immunoblot assays using specific antibodies (Table S1).
Measurements of lipid peroxidation and xanthine oxidase activity
Levels of malondialdehyde (MDA) and 4‐hydroxynonenal (4HNE), indicators of lipid peroxidation, and xanthine oxidase activity in renal tissues were measured using a Lipid Peroxidation Microplate Assay Kit (FR12; Oxford Biomedical Research, Oxford, MI, USA) and a Xanthine Oxidase Activity Assay Kit (ab102522; Abcam, Cambridge, UK), respectively.
Statistical analysis
Data are presented as the mean ± standard error of the mean. One‐way ANOVA and Student–Newman–Keuls post‐hoc test were used to analyze differences in data among multiple groups. Unpaired Student's two‐tailed t‐test was used to determine statistical significance of two data sets. Differences in mortalities in treatment groups were examined by the χ2‐test. For all tests, a difference was considered to be statistically significant if P < 0.05. All analyses were carried out with SigmaStat (Systat, San Jose, CA, USA).
Results
Mortality after MI
In protocol 1, none of the sham‐operated rats in the LETO (n = 10), OLETF (n = 10) or canagliflozin‐treated OLETF groups (n = 10) died after surgery. In contrast, approximately 30% of the rats died within 12 h after MI: three of 14 rats in the LETO‐MI, five of 16 rats in the OLETF‐MI and six of 19 rats in the canagliflozin‐treated OLETF‐MI groups. In protocol 2, five of 15 rats in the OLETF‐MI and five of 14 rats in the canagliflozin‐treated OLETF‐MI groups died within 12 h after MI, suggesting that a fasting condition did not influence the mortality rate in OLETF. Mortality rates did not differ in the study groups with MI.
Metabolic and hemodynamic profiles
OLETF had higher bodyweight and higher casual blood glucose levels than those of LETO, and treatment with canagliflozin reduced the blood glucose level without affecting bodyweight in OLETF (Table 1). Blood βOHB levels were similar in LETO, OLETF and canagliflozin‐treated OLETF when blood was sampled without fasting. Mean blood pressure, but not heart rate, in a conscious state was higher by ~5 mmHg in OLETF than in LETO, and canagliflozin did not change these parameters. In protocol 2, 12 h of fasting reduced blood glucose levels in OLETF, and there was no statistical difference between glucose levels in OLETF and canagliflozin‐treated OLETF. However, blood βOHB level was significantly elevated after 12 h of fasting in canagliflozin‐treated OLETF.
Table 1.
Physiological parameters before induction of myocardial infarction
| n | Bodyweight (g) | Blood glucose (mg/dL) | β‐hydroxubutyrate (mmol/L) | Heart rate (b.p.m.) | Mean blood pressure (mmHg) | |
|---|---|---|---|---|---|---|
| Protocol 1 (without fasting before surgery) | ||||||
| LETO | 21 | 578 ± 15 | 139 ± 4 | 0.42 ± 0.03 | 337 ± 5 | 114 ± 2 |
| OLETF | 21 | 646 ± 10† | 343 ± 28† | 0.35 ± 0.03 | 343 ± 4 | 119 ± 1† |
| OLETF + canagliflozin | 23 | 640 ± 8† | 184 ± 9‡ | 0.39 ± 0.02 | 350 ± 2 | 119 ± 1† |
| Protocol 2 (fasted before myocardial infarction) | ||||||
| OLETF | 10 | 622 ± 10 | 164 ± 17 | 0.54 ± 0.05 | 337 ± 9 | 110 ± 4 |
| OLETF + canagliflozin | 9 | 621 ± 5 | 149 ± 9 | 0.79 ± 0.06§ | 336 ± 12 | 111 ± 4 |
Values are mean ± standard error of the mean. † P < 0.05 versus Long‐Evans Tokushima Otsuka rats (LETO). ‡ P < 0.05 versus Otsuka Long‐Evans Tokushima Fatty rats (OLETF). § P < 0.05 versus OLETF with fasting.
At 12 h after the operation (and fasting), blood glucose levels were still higher in OLETF than in LETO, and treatment with canagliflozin significantly reduced the blood glucose level in OLETF (Table 2). The level of βOHB after MI was slightly increased in OLETF and markedly increased in canagliflozin‐treated OLETF. Although blood urea nitrogen levels were comparable in the sham‐operated LETO and OLETF, serum creatinine level was lower in OLETF than in LETO, reflecting glomerular hyperfiltration associated with diabetes in this model8, 22, 24. MI did not affect these indices of glomerular function in either LETO or OLETF. Canagliflozin treatment increased blood urea nitrogen level, but not serum creatinine level after MI. Serum angiotensin II level after MI was similar in LETO and OLETF, and canagliflozin treatment with or without fasting did not change the angiotensin II level in OLETF. Acidemia was not induced by canagliflozin, even after MI and 12 h of fasting. After anesthesia, heart rate was lower in OLETF than in LETO, regardless of induction of MI and canagliflozin treatment, although mean blood pressures were comparable among the six groups. In protocol 2, continuation of fasting for 24 h reduced the blood glucose level and increased βOHB level in canagliflozin‐treated OLETF. Other parameters were not significantly affected by longer fasting (Table 2).
Table 2.
Blood analyses and hemodynamic parameters 12 h after surgery
| n | Blood glucose (mg/dL) | β‐hydroxubutyrate (mmol/L) | Blood urea nitrogen (mg/dL) | Serum creatinine (mg/dL) | Angiotensin II (pg/mL) | Arterial pH | Heart rate (b.p.m.) | Mean blood pressure (mmHg) | |
|---|---|---|---|---|---|---|---|---|---|
| Protocol 1 (without fasting before surgery) | |||||||||
| LETO | |||||||||
| Sham | 10 | 129 ± 4 | 0.54 ± 0.05 | 15.6 ± 0.9 | 0.49 ± 0.02 | ‐ | 7.431 ± 0.013 | 350 ± 18 | 92 ± 4 |
| MI | 11 | 137 ± 8 | 0.60 ± 0.06 | 19.0 ± 1.5 | 0.48 ± 0.03 | 50.5 ± 19.1 | 7.412 ± 0.012 | 384 ± 14†† | 84 ± 5 |
| OLETF | |||||||||
| Sham | 10 | 196 ± 26 | 0.31 ± 0.04 | 15.6 ± 0.7 | 0.35 ± 0.01† | ‐ | 7.426 ± 0.016 | 280 ± 12† | 79 ± 5 |
| MI | 11 | 249 ± 23‡ | 1.09 ± 0.08 | 20.7 ± 1.2 | 0.39 ± 0.03‡ | 36.9 ± 9.9 | 7.401 ± 0.008 | 295 ± 10‡ | 87 ± 3 |
| OLETF + canagliflozin | |||||||||
| Sham | 10 | 130 ± 9§ | 0.70 ± 0.05 | 17.2 ± 1.0 | 0.35 ± 0.02† | ‐ | 7.395 ± 0.008 | 264 ± 7† | 80 ± 4 |
| MI | 13 | 176 ± 15¶ | 4.56 ± 0.45‡ , ¶ , †† | 31.8 ± 1.7‡ , ¶ , †† | 0.39 ± 0.02‡ | 86.1 ± 20.2 | 7.377 ± 0.014 | 316 ± 8‡ , †† | 79 ± 4 |
| Protocol 2 (fasted before myocardial infarction) | |||||||||
| OLETF | |||||||||
| MI | 10 | 166 ± 15 | 1.37 ± 0.17 | 19.4 ± 0.7 | 0.40 ± 0.02 | 87.2 ± 37.0 | 7.409 ± 0.017 | 317 ± 11 | 85 ± 4 |
| OLETF + canagliflozin | |||||||||
| MI | 9 | 123 ± 7‡‡ | 2.99 ± 0.34‡‡ | 23.0 ± 0.8‡‡ | 0.37 ± 0.02 | 48.4 ± 35.0 | 7.407 ± 0.021 | 298 ± 10 | 76 ± 3 |
Values are mean ± standard error of the mean. † P < 0.05 versus Long‐Evans Tokushima Otsuka rats (LETO)‐Sham. ‡ P < 0.05 versus LETO‐myocardial infarction (MI). § P < 0.05 versus Otsuka Long‐Evans Tokushima Fatty rats (OLETF)‐Sham. ¶ P < 0.05 versus OLETF‐MI. †† P < 0.05 versus corresponding sham‐operated rats. ‡‡ P < 0.05 versus OLETF with fasting. Measurement of angiotensin II was carried out from seven to eight samples.
AKI after MI in OLETF
As in our previous study8, periodic acid–Schiff staining in kidney sections did not show detectable abnormalities in tubular cells, such as loss of the brush border, tubular dilation, cast formation and cell lysis, in LETO and OLETF regardless of MI, although sizes of glomeruli were larger in OLETF (data not shown).
In protocol 1, NGAL signals in immunohistochemistry were barely detected in LETO regardless of MI (Figure 1a,c). The extent of NGAL staining of renal tubules in sham‐operated OLETF was not significantly different from that in LETO. However, OLETF with MI showed focal NGAL‐positive areas in tubular epithelial cells, and the NGAL‐positive area was significantly larger than that in sham‐operated OLETF (Figure 1a,c). Treatment with canagliflozin without fasting did not change the NGAL staining in OLETF. NGAL messenger RNA (mRNA) level in the kidney was higher in sham‐operated OLETF than in sham‐operated LETO (Figure 1d). The expression level of NGAL was not affected by canagliflozin treatment. In protocol 2, although fasting alone did not reduce NGAL levels, the extent of the NGAL‐positive areas and the level of NGAL mRNA after MI in OLETF were both significantly decreased in canagliflozin‐treated OLETF (Figure 1b‐d).
Figure 1.
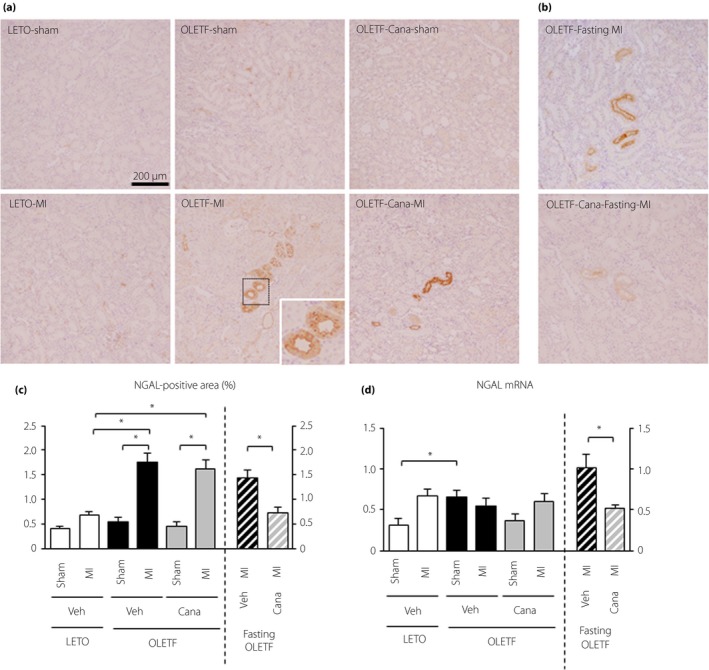
Analysis of for neutrophil gelatinase‐associated lipocalin (NGAL) expression in the kidney after myocardial infarction. (a,b) Representative images of immunohistochemistry for NGAL in kidney sections from Long‐Evans Tokushima Otsuka rats (LETO) and Otsuka Long‐Evans Tokushima Fatty rats (OLETF) treated with a vehicle (Veh) or canagliflozin (Cana) for 2 weeks that were sampled 12 h after a sham operation (Sham) or myocardial infarction (MI) (a) without fasting (protocol 1) and (b) with fasting (protocol 2) before surgery. (c) Summary data of NGAL‐positive areas. In each group, 10 randomly selected fields from 9 to 13 kidneys were analyzed. (d) Quantification of NGAL messenger ribonucleic acid (mRNA) levels in the kidney; n = 8–12 in each group. *P < 0.05.
Immunohistochemical staining of KIM‐1 showed few KIM‐1 signals in LETO regardless of MI in protocol 1 (Figure 2a). KIM‐1‐positive cells were detected mostly as proximal tubular epithelial cells in OLETF, and the extent of KIM‐1 positive areas was increased in sham‐operated OLETF compared with that in LETO, and was further increased after MI. The extent of KIM‐1 positive areas in OLETF was not affected by treatment with canagliflozin (Figure 2a,c). In protocol 2, however, canagliflozin with pre‐MI fasting decreased the extent of the KIM‐1‐positive area after MI in OLETF (Figure 2b,c). The expression pattern of KIM‐1 mRNA was similar to that of KIM‐1 staining (Figure 2d).
Figure 2.
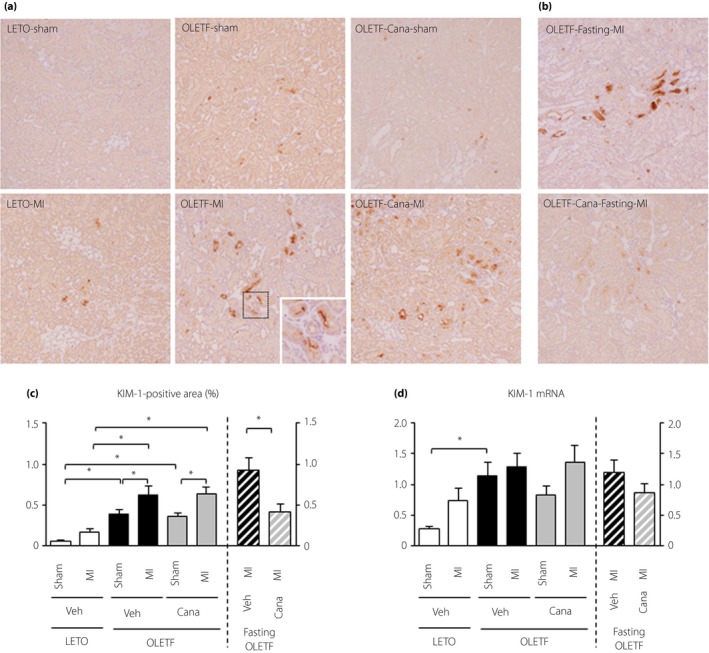
Analysis of kidney injury molecule‐1 (KIM‐1) expression in the kidney after myocardial infarction. (a,b) Representative images of immunohistochemistry for KIM‐1 in kidney sections from Long‐Evans Tokushima Otsuka rats (LETO) and Otsuka Long‐Evans Tokushima Fatty rats (OLETF) pretreated with a vehicle (Veh) or canagliflozin (Cana) 12 h after a sham operation (Sham) or myocardial infarction (MI) (a) without fasting and (b) with fasting before surgery. (c) Summary data of KIM‐1‐positive areas. In each group, 10 randomly selected fields from 9 to 12 kidneys were analyzed. (d) Quantification of KIM‐1 messenger ribonucleic acid (mRNA) levels in the kidney. n = 8–12 in each group. *P < 0.05.
Involvement of renal TLR activation, RAA system and 5´ adenosine monophosphate‐activated protein kinase activation
As we recently found involvement of TLR activation in CRS in OLETF8, we assessed changes in TLR‐mediated signaling after MI by canagliflozin. Expression levels of TLR2, TLR4, interleukin‐6, interleukin‐1β and chemokine (C‐C motif) ligand 3 were higher in OLETF than in LETO, although most of the differences did not reach statistical significance (Figure 3). Treatment with canagliflozin significantly reduced the mRNA level of TLR4, but not those of TLR2 or cytokines and chemokines. The mRNA levels of these molecules involved in TLR‐mediated signaling were comparable in OLETF and canagliflozin‐treated OLETF in the fasting protocol.
Figure 3.
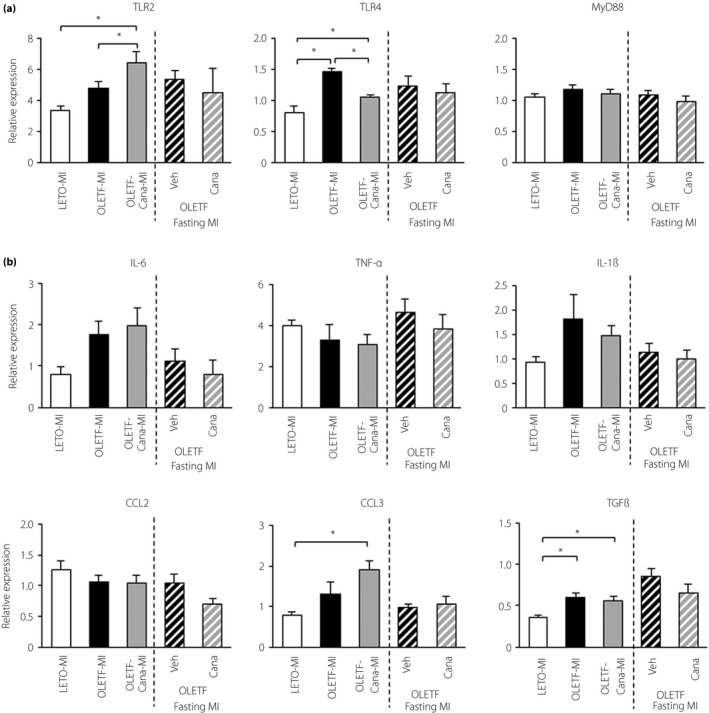
Expression levels of Toll‐like receptors, cytokines and chemokines in the kidney. Expression levels of genes related to (a) Toll‐like receptors, and (b) cytokines and chemokines in the kidney; n = 6–11 in each group. *P < 0.05. Cana, canagliflozin; MI, myocardial infarction; Veh, vehicle.
Renal mRNA level of transforming growth factor (TGF)‐β was higher in OLETF than LETO after MI, suggesting that renal RAA activity was increased in diabetic rats. However, the increased renal TGF‐β mRNA level in OLETF was not altered by canagliflozin treatment with or without fasting (Figure 3b). In contrast, levels of TGF‐β mRNA in the non‐infarcted myocardium after MI were similar in LETO and OLETF (data not shown).
The phosphorylation level of 5´ adenosine monophosphate‐activated protein kinase (AMPK)α at Thr172 in the kidney at 12 h after MI was unchanged in OLETF compared with that in LETO. The level of phospho‐AMPKα was increased in canagliflozin‐treated OLETF, although there was no further increase in phospho‐AMPKα level by fasting in canagliflozin‐treated OLETF (Figure 4b).
Figure 4.
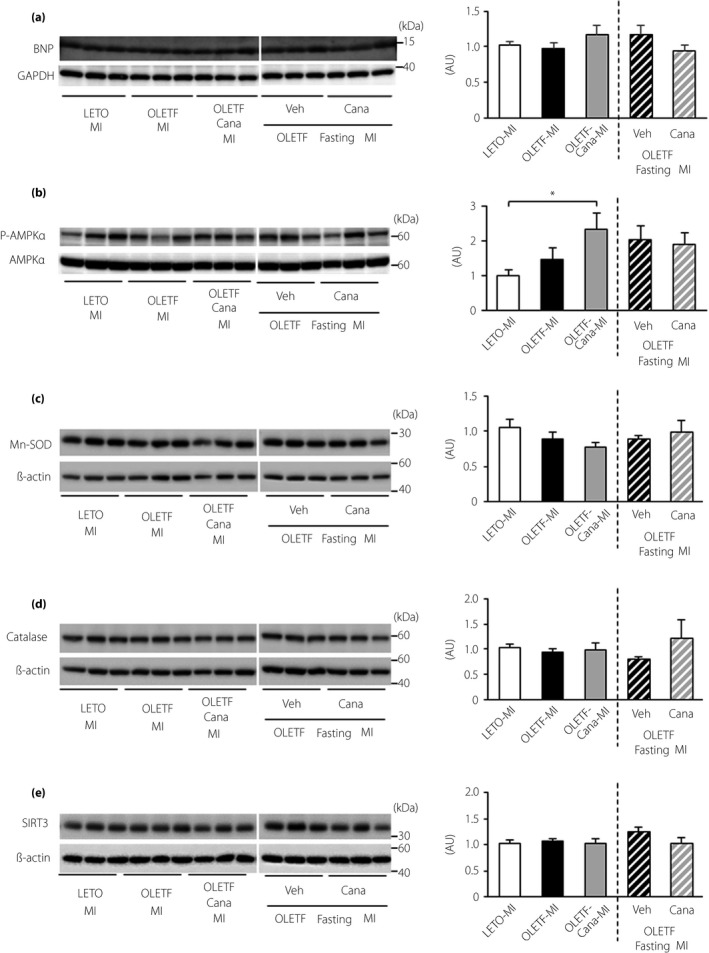
Effects of canagliflozin and fasting on expression levels of B‐type natriuretic peptide (BNP), 5´ adenosine monophosphate‐activated protein kinase (AMPK) and anti‐oxidant proteins. Representative immunoblots (left) and summary data (right) of protein levels are shown. (a) Protein levels for BNP in the non‐infarct myocardium and (b) protein levels for phospho‐AMPKα at Thr172, (c) manganese superoxide dismutase (Mn‐SOD), (d) catalase and (e) sirtuin 3 (SIRT3) in the kidney. Samples were run on the same gel, but were not contiguous; n = 6 in each group. AU, arbitrary unit.
Oxidant stress and its regulatory proteins in the kidney
The level of MDA + 4HNE in the kidney after MI was higher in OLETF than in LETO, and this increase in OLETF was blocked by canagliflozin only when rats were fasted for 12 h before induction of MI (Figure 5a).
Figure 5.
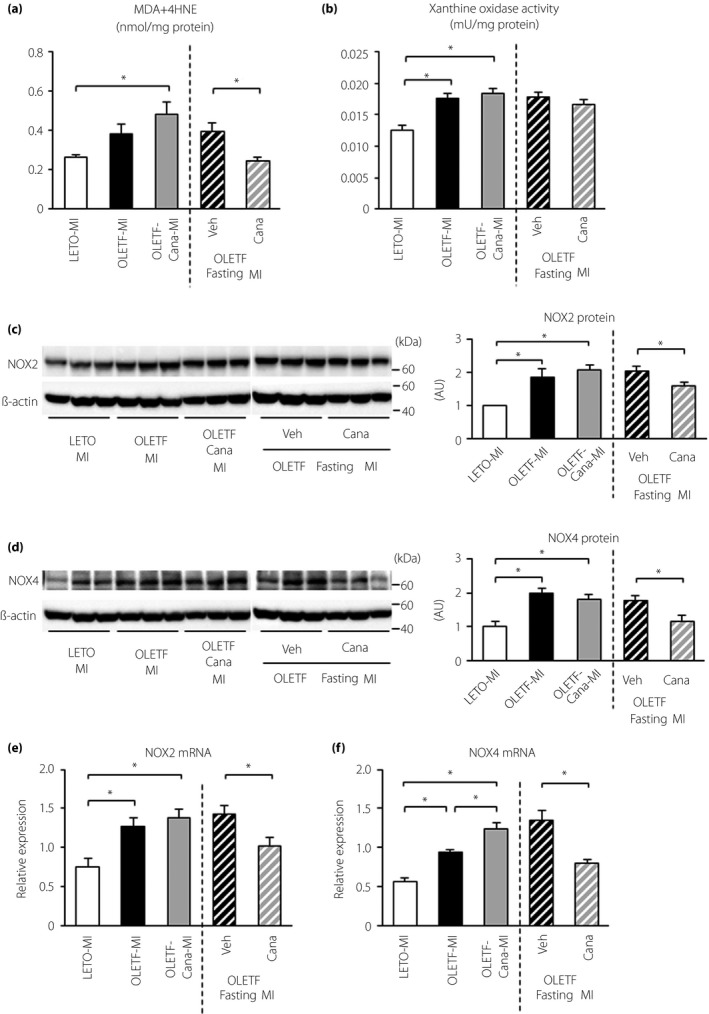
Effects of canagliflozin and fasting on oxidative stress in the kidney after myocardial infarction. (a) Malondialdehyde (MDA) + 4‐hydroxynonenal (4HNE) levels in the kidney. Kidney tissues were sampled 12 h after myocardial infarction (MI) in Long‐Evans Tokushima Otsuka rats (LETO), Otsuka Long‐Evans Tokushima Fatty rats (OLETF) and canagliflozin (Cana)‐treated OLETF. Vehicle (Veh)‐ or canagliflozin‐treated OLETF with 12 h fasting before MI were also analyzed; n = 5–6 in each group. (b) Xanthine oxidase activity in the kidney; n = 6 in each group. (c) Representative immunoblots for nicotinamide adenine dinucleotide phosphate oxidase (NOX)2 (left) and summary data of NOX2 protein level normalized by β‐actin level (right) in the kidney; n = 6 in each group. (d) Representative immunoblots for NOX4 (left) and summary data of NOX4 protein level normalized by β‐actin level (right) in the kidney; n = 6 in each group. (c,d) Samples were run on the same gel, but were not contiguous. (e,f) Levels of (e) NOX2 and (f) NOX4 messenger ribonucleic acid (mRNA) in the kidney; n = 6–8 in each group. *P < 0.05. AU, arbitrary unit.
Xanthine oxidase activity was significantly increased in OLETF compared with that in LETO after MI. This higher activity in OLETF was not modified by canagliflozin regardless of fasting (Figure 5b). Protein levels of both nicotinamide adenine dinucleotide phosphate oxidase (NOX)2 and NOX4 were significantly increased in OLETF compared with those in LETO. These increases in NOX2 and NOX4 proteins were significantly suppressed by canagliflozin only when rats were fasted for 12 h before MI (Figure 5c,d). The mRNA levels of NOX2 and NOX4 were higher in OLETF than in LETO, and the increased expression levels were also reduced by canagliflozin with fasting (Figure 5e,f).
As shown in Figure 4, protein levels of manganese superoxide dismutase, catalase and SIRT3, a mitochondrial sirtuin, in the kidney after MI were similar in OLETF and LETO, and canagliflozin treatment did not change these levels in OLETF.
Effects of exogenous βOHB on NOX4 induction
Angiotensin II (3 μmol/L) increased protein levels of NOX4. Pre‐incubation of 1 and 2 mmol/L of βOHB attenuated the increase in NOX4 proteins by 50 and 62%, respectively (Figure 6).
Figure 6.
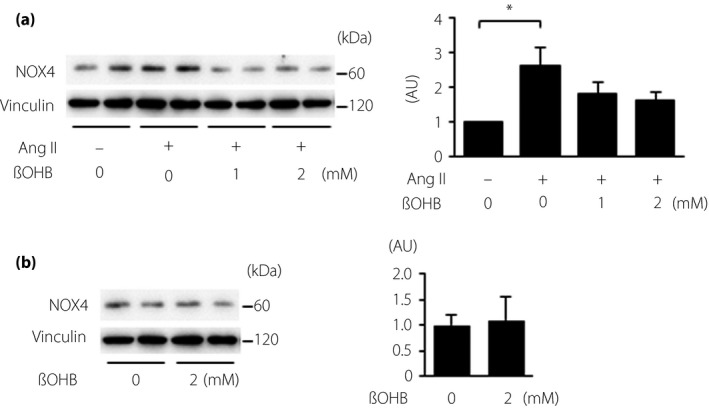
Effects of exogenous β‐hydroxybutyrate (βOHB) on nicotinamide adenine dinucleotide phosphate oxidase (NOX)4 protein in vitro. Representative immunoblots (left) and summary data (right) of NOX4 protein level normalized by vinculin are shown. (a) NRK‐52E cells were treated with angiotensin II (Ang II) in the presence or absence of βOHB. (b) NRK‐52E cells were treated with vehicle or βOHB; n = 6 in each treatment. *P < 0.05. AU, arbitrary unit.
Discussion
As in our previous studies8, 22, OLETF at the ages of 25–30 weeks showed phenotypes of an early stage of diabetic nephropathy: lower serum creatinine level, indicating glomerular hyperfiltration (Table 2); no histological abnormalities in the kidney, except for larger sizes of glomeruli; and higher level of KIM‐1 protein without change in NGAL level (Figures 1,2) compared with those in LETO. After acute MI, protein levels of NGAL and KIM‐1 in the kidney were significantly increased by 3.2‐ and by 1.6‐fold, respectively, in OLETF, although blood urea nitrogen and serum creatinine levels were not changed. The impact of MI on the kidney appears modest, and an unchanged level of serum creatinine indicates that renal injury in OLETF after MI is a model of “subclinical AKI” in patients with CRS. Subclinical AKI is not a trivial injury, as it has a significant impact on clinical outcomes25, 26. Haase et al.25 showed that subclinical AKI, defined as elevated urine or plasma NGAL level without elevation of serum creatinine level, increases the risk of adverse outcomes, including death and renal replacement therapy, by 3.6‐fold. The findings support the notion that the present model of MI‐induced AKI is a clinically relevant model of type 1 CRS.
Compared with the findings in a previous study using the same animal model of diabetes mellitus8, differences in the expression levels of TLRs and inflammatory cytokines after MI between OLETF and LETO were slightly smaller in the present study. The reason for the difference is unclear, but the possible difference in sizes of MI might have been involved, as the amount of damage‐associated molecular patterns released from the infarcted myocardium depends on the size of MI27. The mean blood pressure drop after MI in OLETF was larger in our previous study (64 mmHg in the MI group vs 78 mmHg in the sham group)8 than in the present study (87 mmHg in the MI group vs 79 mmHg in the sham group; Table 2), suggesting that the extent of ventricular damage after MI was smaller in the present study.
In addition to activation of TLR‐mediated signaling, increased oxidative stress and RAA system contributes to type 1 CRS2, 3, 28. NOX4 is highly expressed in renal tubules, renal fibroblasts, glomerular mesangial cells and podocytes, and upregulation of NOXs together with increased generation of reactive oxygen species has been reported in the diabetic kidney29, 30. Inhibition of NOX4 in experimental models of diabetes mellitus afforded marked protection from both structural and functional kidney damage31, 32. The contribution of xanthine oxidase to reactive oxygen species‐mediated renal injury in diabetes mellitus has also been indicated by the findings that inhibition of xanthine oxidase activity significantly suppressed kidney injury in diabetes mellitus 33, 34. In the present study, renal expression levels of MDA + 4HNE, NOX2 and NOX4, and xanthine oxidase activity were significantly higher in OLETF than in LETO (Figure 5). As the renal TGF‐β mRNA level was higher in OLETF than in LETO (Figure 3b), the increases in NOXs in OLETF might be partly explained by activation of the renal RAA system35. Canagliflozin significantly reduced levels of NOX2 and NOX4, but not xanthine oxidase activity (Figure 5), whereas canagliflozin did not significantly change the TGF‐β mRNA level. Renal activity of RAA system after an SGLT2 inhibitor was reported to be increased, decreased or unchanged depending on the reports36, 37, 38. The differences in study participants, duration of SGLT2 inhibitor treatment or volume change after the treatment might explain the inconsistent results of the previous reports and the present study. Nevertheless, the present findings suggest that NOX‐mediated oxidative stress plays a major role in AKI in OLETF and is a target of canagliflozin treatment.
Of interest, protection against AKI by canagliflozin was detected only when the rats were fasted before induction of MI. Protective effects of canagliflozin on tissue MDA + 4HNE, and on NOX2 and NOX4 proteins were also observed when combined with fasting before MI (Figure 5a,c‐f). Hemodynamic parameters, including blood pressure and heart rate, before and 12 h after MI were comparable in canagliflozin‐treated OLETF with or without fasting (Tables 1,2). In contrast, the blood βOHB level was twofold higher in canagliflozin‐treated OLETF with pre‐MI fasting than in canagliflozin‐treated OLETF without fasting (0.79 ± 0.06 vs 0.39 ± 0.02 mmol/L; Table 1), whereas blood βOHB levels after MI were increased to 3–4 mmol/L in canagliflozin‐treated OLETF regardless of pre‐MI fasting (Table 2). As shown in Figure 7, the level of renal NGAL expression was inversely correlated with blood βOHB levels before MI, but not with those at 12 h after MI, suggesting a significant association of an increase in βOHB before MI and renal protection from CRS by canagliflozin.
Figure 7.
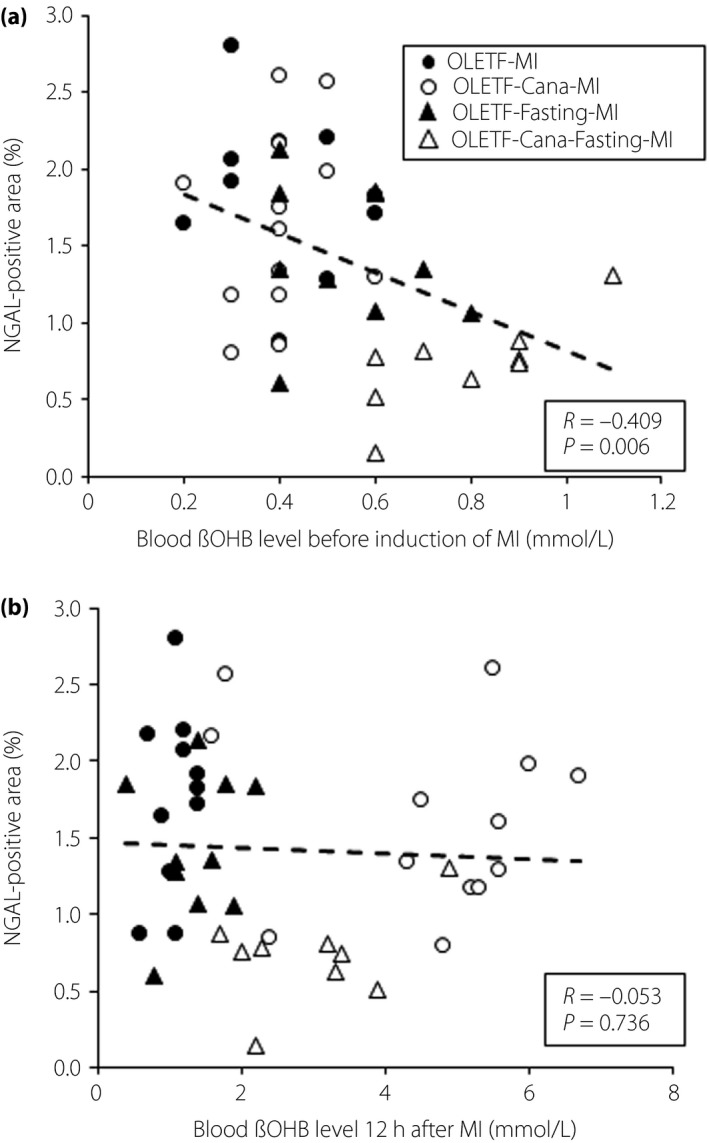
Correlation between blood β‐hydroxybutyrate (βOHB) levels and neutrophil gelatinase‐associated lipocalin (NGAL)‐positive areas in Otsuka Long‐Evans Tokushima Fatty rats (OLETF) after myocardial infarction (MI). The relationship between blood βOHB levels (a) before or (b) 12 h after induction of MI and NGAL‐positive areas in OLETF (closed circle), canagliflozin (Cana)‐treated OLETF (open circle), OLETF with 12 h of fasting before MI (closed triangle) and Cana‐treated OLETF with fasting (open triangle).
As the correlation between blood βOHB and renal NGAL expression was modest (r = −0.41), mechanisms other than increased βOHB are possibly involved in renoprotection by canagliflozin with pre‐MI fasting. Activation of AMPK is one of the candidates22, 39, 40, 41, but the present results showed that the contribution of AMPK to renoprotection against diabetes mellitus‐induced worsening of CRS was limited. It has been suggested that the reduction of oxygen consumption in the proximal tubules and prevention of cellular senescence are the mechanisms of renoprotection by an SGLT2 inhibitor42, 43, 44, 45, but whether these mechanisms are involved in amelioration of CRS should be examined in future studies.
As it is actually difficult to maintain the blood βOHB level by administration of exogenous βOHB to a level achieved by SGLT2 inhibitor treatment and fasting in OLETF46, we examined the effects of βOHB on NOX4 protein level in vitro. Pre‐incubation with βOHB attenuated angiotensin II‐induced upregulation of NOX4 protein expression in NRK‐52E cells (Figure 6). Recently, it was reported that βOHB functions as an endogenous HDACs inhibitor, and suppresses oxidative stress by upregulation of anti‐oxidative proteins, including manganese superoxide dismutase and catalase, in the kidney17. Kong et al.47 reported that exposure of PC12 cells to βOHB attenuated H2O2‐induced upregulation of NOX2 and NOX4 proteins. They also showed that knockdown of either HDAC1 or HDAC2 reduced NOX2 and NO4 protein levels47. Taken together, the findings suggest that elevation of blood βOHB level before MI is causally related to suppressed expression of NOXs and oxidant stress in the kidney after MI in canagliflozin‐treated OLETF with pre‐MI fasting. Interestingly, in contrast to suppressed expression of NOXs, upregulation of anti‐oxidative stress proteins was not accompanied by an elevation of blood βOHB levels by canagliflozin and fasting. These results suggest that increased βOHB by canagliflozin treatment protected the kidney from injury downstream of the RAA pathway, but independent from the HDAC pathway. Precise mechanisms of how βOHB attenuate NOXs protein and subsequent renal injury are uncertain and warrant further investigation.
Several reports have shown that treatment with an SGLT2 inhibitor ameliorated diabetic nephropathy by suppression of NOX4 expression and subsequent oxidative stress in diabetes mellitus mice12, 48, 49. Thus, the reason why treatment with canagliflozin without fasting failed to suppress NOX4 expression and oxidative stress in the present study is uncertain. However, there are a few possible explanations. First, the duration of SGLT2 inhibitor treatment was shorter in this study than in earlier studies (2 vs 8–12 weeks), which possibly resulted in a lower level of blood ketone bodies16, 50. In fact, blood βOHB levels after 12 h fasting were higher after 8 weeks of canagliflozin treatment than that of 2 weeks of treatment (1.08 ± 0.12 vs 0.79 ± 0.06 mmol/L, P < 0.05). Second, as OLETF are hyperphagic due to a lack of cholecystokinin‐A receptor in the brain19, time periods of fasting might be shorter in OLETF under the condition of ad libitum feeding, leading to less elevation of blood ketone level, compared with those in db/db and Akita mice. In a clinical setting, chronic treatment with an SGLT2 inhibitor increased the blood ketone level throughout the day in patients with diabetes mellitus16, 51. Therefore, βOHB‐mediated renoprotection would appear in diabetes mellitus patients treated with an SGLT2 inhibitor, but the protection might be attenuated after meal.
There were limitations to the present study. First, AKI was assessed only at 12 h after MI, and the time courses of AKI after MI and impact of AKI on the long‐term function of diabetic kidneys were not examined. The high mortality rate (i.e., approximately 60%) of OLETF within 48 h after MI precludes tissue sampling at later time points after MI in OLETF without selection bias in data20, 23. Second, treatment with canagliflozin might have affected cardiac function after MI, as SGLT2 inhibitors reportedly reduced heart failure in clinical and experimental studies52, 53. However, it is unlikely that canagliflozin had a large impact on the cardiac function 12 h after MI, as myocardial B‐type natriuretic peptide levels (Figure 4a) and mortality rates after MI were not different in canagliflozin‐treated and ‐untreated OLETF.
In conclusion, the results of the present study suggest that increased susceptibility to MI‐induced AKI in diabetes mellitus rats is mediated by a NOX‐induced increase in oxidative stress in the kidney, and is improved by treatment with canagliflozin with an appropriate fasting period.
Disclosure
MT, TS, TaM and TeM received lecture honoraria from Mitsubishi Tanabe Pharma Corporation.
Supporting information
Table S1 ¦ Antibodies and Taqman gene expression assays.
Acknowledgment
This study was supported by a grant from Mitsubishi Tanabe Pharma Corporation, and a Research and Education Grant 2017‐2018 from Sapporo Medical University.
J Diabetes Investig 2019; 10: 933–946
References
- 1. Ronco C, Haapio M, House AA, et al Cardiorenal syndrome. J Am Coll Cardiol 2008; 52: 1527–1539. [DOI] [PubMed] [Google Scholar]
- 2. Thind GS, Loehrke M, Wilt JL. Acute cardiorenal syndrome: mechanisms and clinical implications. Cleve Clin J Med 2018; 85: 231–239. [DOI] [PubMed] [Google Scholar]
- 3. Di Lullo L, Bellasi A, Barbera V, et al Pathophysiology of the cardio‐renal syndromes types 1‐5: an uptodate. Indian Heart J 2017; 69: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fox CS, Muntner P, Chen AY, et al Short‐term outcomes of acute myocardial infarction in patients with acute kidney injury: a report from the national cardiovascular data registry. Circulation 2012; 125: 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goldberg A, Kogan E, Hammerman H, et al The impact of transient and persistent acute kidney injury on long‐term outcomes after acute myocardial infarction. Kidney Int 2009; 76: 900–906. [DOI] [PubMed] [Google Scholar]
- 6. Kim JH, Lee JH, Jang SY, et al Prognostic value of early acute kidney injury after primary percutaneous coronary intervention in patients with ST‐segment elevation myocardial infarction. Am J Cardiol 2014; 114: 1174–1178. [DOI] [PubMed] [Google Scholar]
- 7. Panchapakesan U, Pollock C. The role of toll‐like receptors in diabetic kidney disease. Curr Opin Nephrol Hypertens 2018; 27: 30–34. [DOI] [PubMed] [Google Scholar]
- 8. Ohno K, Kuno A, Murase H, et al Diabetes increases the susceptibility to acute kidney injury after myocardial infarction through augmented activation of renal Toll‐like receptors in rats. Am J Physiol Heart Circ Physiol 2017; 313: H1130–H1142. [DOI] [PubMed] [Google Scholar]
- 9. Wanner C, Inzucchi SE, Lachin JM, et al Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375: 323–334. [DOI] [PubMed] [Google Scholar]
- 10. Neal B, Perkovic V, Mahaffey KW, et al Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377: 644–657. [DOI] [PubMed] [Google Scholar]
- 11. Jaikumkao K, Pongchaidecha A, Chatsudthipong V, et al The roles of sodium‐glucose cotransporter 2 inhibitors in preventing kidney injury in diabetes. Biomed Pharmacother 2017; 94: 176–187. [DOI] [PubMed] [Google Scholar]
- 12. Terami N, Ogawa D, Tachibana H, et al Long‐term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One 2014; 9: e100777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yao D, Wang S, Wang M, et al Renoprotection of dapagliflozin in human renal proximal tubular cells via the inhibition of the high mobility group box 1‐receptor for advanced glycation end products‐nuclear factor‐κB signaling pathway. Mol Med Rep 2018; 18: 3625–3630. [DOI] [PubMed] [Google Scholar]
- 14. Dekkers CCJ, Petrykiv S, Laverman GD, et al Effects of the SGLT‐2 inhibitor dapagliflozin on glomerular and tubular injury markers. Diabetes Obes Metab 2018; 20: 1988–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Min SH, Oh TJ, Baek SI, et al Degree of ketonaemia and its association with insulin resistance after dapagliflozin treatment in type 2 diabetes. Diabetes Metab 2018; 44: 73–76. [DOI] [PubMed] [Google Scholar]
- 16. Ferrannini E, Baldi S, Frascerra S, et al Shift to fatty substrate utilization in response to sodium‐glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 2016; 65: 1190–1195. [DOI] [PubMed] [Google Scholar]
- 17. Shimazu T, Hirschey MD, Newman J, et al Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013; 339: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perlman A, Heyman SN, Matok I, et al Acute renal failure with sodium‐glucose‐cotransporter‐2 inhibitors: analysis of the FDA adverse event report system database. Nutr Metab Cardiovasc Dis 2017; 27: 1108–1113. [DOI] [PubMed] [Google Scholar]
- 19. Bi S, Moran TH. Obesity in the Otsuka Long Evans Tokushima Fatty rat: mechanisms and discoveries. Front Nutr 2016; 3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murase H, Kuno A, Miki T, et al Inhibition of DPP‐4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc Diabetol 2015; 14: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yagi K, Kim S, Wanibuchi H, et al Characteristics of diabetes, blood pressure, and cardiac and renal complications in Otsuka Long‐Evans Tokushima Fatty rats. Hypertension 1997; 29: 728–735. [DOI] [PubMed] [Google Scholar]
- 22. Muratsubaki S, Kuno A, Tanno M, et al Suppressed autophagic response underlies augmentation of renal ischemia/reperfusion injury by type 2 diabetes. Sci Rep 2017; 7: 5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takada A, Miki T, Kuno A, et al Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PLoS One 2012; 7: e39893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawano K, Mori S, Hirashima T, et al Examination of the pathogenesis of diabetic nephropathy in OLETF rats. J Vet Med Sci 1999; 61: 1219–1228. [DOI] [PubMed] [Google Scholar]
- 25. Haase M, Devarajan P, Haase‐Fielitz A, et al The outcome of neutrophil gelatinase‐associated lipocalin‐positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol 2011; 57: 1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Albert C, Albert A, Kube J, et al Urinary biomarkers may provide prognostic information for subclinical acute kidney injury after cardiac surgery. J Thorac Cardiovasc Surg 2018; 155: 2441–2452. [DOI] [PubMed] [Google Scholar]
- 27. Andrassy M, Volz HC, Riedle N, et al HMGB1 as a predictor of infarct transmurality and functional recovery in patients with myocardial infarction. J Intern Med 2011; 270: 245–253. [DOI] [PubMed] [Google Scholar]
- 28. Virzì GM, Clementi A, de Cal M, et al Oxidative stress: dual pathway induction in cardiorenal syndrome type 1 pathogenesis. Oxid Med Cell Longev 2015; 2015: 391790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shiose A, Kuroda J, Tsuruya K, et al A novel superoxide‐producing NAD(P)H oxidase in kidney. J Biol Chem 2001; 276: 1417–1423. [DOI] [PubMed] [Google Scholar]
- 30. Gorin Y, Block K. Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 2013; 61: 130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jha JC, Gray SP, Barit D, et al Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long‐term diabetic nephropathy. J Am Soc Nephrol 2014; 25: 1237–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gorin Y, Cavaglieri RC, Khazim K, et al Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 2015; 308: F1276–F1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu J, Wang C, Liu F, et al Metabonomics revealed xanthine oxidase‐induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal Bioanal Chem 2015; 407: 2569–2579. [DOI] [PubMed] [Google Scholar]
- 34. Komers R, Xu B, Schneider J, et al Effects of xanthine oxidase inhibition with febuxostat on the development of nephropathy in experimental type 2 diabetes. Br J Pharmacol 2016; 173: 2573–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nguyen Dinh Cat A, Montezano AC, Burger D, et al Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal 2013; 19: 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cherney DZ, Perkins BA, Soleymanlou N, et al Renal hemodynamic effect of sodium‐glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014; 129: 587–597. [DOI] [PubMed] [Google Scholar]
- 37. Shin SJ, Chung S, Kim SJ, et al Effect of sodium‐glucose co‐transporter 2 inhibitor, dapagliflozin, on renal renin‐angiotensin system in an animal model of type 2 diabetes. PLoS One 2016; 11: e0165703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li L, Konishi Y, Morikawa T, et al Effect of a SGLT2 inhibitor on the systemic and intrarenal renin‐angiotensin system in subtotally nephrectomized rats. J Pharmacol Sci 2018; 137: 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lieberthal W, Tang M, Lusco M, et al Preconditioning mice with activators of AMPK ameliorates ischemic acute kidney injury in vivo . Am J Physiol Renal Physiol 2016; 31: F731–F739. [DOI] [PubMed] [Google Scholar]
- 40. Hawley SA, Ford RJ, Smith BK, et al The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes 2016; 65: 2784–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mancini SJ, Boyd D, Katwan OJ, et al Canagliflozin inhibits interleukin‐1β‐stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP‐activated protein kinase‐dependent and ‐independent mechanisms. Sci Rep 2018; 8: 5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Neill J, Fasching A, Pihl L, et al Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am J Physiol Renal Physiol 2015; 309: F227–F234. [DOI] [PubMed] [Google Scholar]
- 43. Layton AT, Vallon V, Edwards A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Renal Physiol 2016; 310: F1269–F1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kitada K, Nakano D, Ohsaki H, et al Hyperglycemia causes cellular senescence via a SGLT2‐ and p21‐dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J Diabetes Complications 2014; 28: 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Umino H, Hasegawa K, Minakuchi H, et al High basolateral glucose increases sodium‐glucose cotransporter 2 and reduces sirtuin‐1 in renal tubules through glucose transporter‐2 detection. Sci Rep 2018; 8: 6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oshima H, Miki T, Kuno A, et al Empagliflozin, an SGLT2 inhibitor, reduced the mortality rate after acute myocardial infarction with modification of cardiac metabolomes and anti‐oxidants in diabetic rats. J Pharmacol Exp Ther 2018. 10.1124/jpet.118.253666. [DOI] [PubMed] [Google Scholar]
- 47. Kong G, Huang Z, Ji W, et al The ketone metabolite β‐hydroxybutyrate attenuates oxidative stress in spinal cord injury by suppression of class I histone deacetylases. J Neurotrauma 2017; 34: 2645–2655. [DOI] [PubMed] [Google Scholar]
- 48. Hatanaka T, Ogawa D, Tachibana H, et al Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose‐induced oxidative stress in type 1 diabetic mice. Pharmacol Res Perspect. 2016; 4: e00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamezaki M, Kusaba T, Komaki K, et al Comprehensive renoprotective effects of ipragliflozin on early diabetic nephropathy in mice. Sci Rep 2018; 8: 4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yabe D, Iwasaki M, Kuwata H, et al Sodium‐glucose co‐transporter‐2 inhibitor use and dietary carbohydrate intake in Japanese individuals with type 2 diabetes: a randomized, open‐label, 3‐arm parallel comparative, exploratory study. Diabetes Obes Metab 2017; 19: 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Biester T, Aschemeier B, Fath M, et al Effects of dapagliflozin on insulin‐requirement, glucose excretion and β‐hydroxybutyrate levels are not related to baseline HbA1c in youth with type 1 diabetes. Diabetes Obes Metab 2017; 19: 1635–1639. [DOI] [PubMed] [Google Scholar]
- 52. Rådholm K, Figtree G, Perkovic V, et al Canagliflozin and heart failure in type 2 diabetes mellitus: results from the CANVAS program. Circulation 2018; 138: 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Byrne NJ, Parajuli N, Levasseur JL, et al Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload‐induced heart failure. JACC Basic Transl Sci 2017; 2: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 ¦ Antibodies and Taqman gene expression assays.