

Abstract
Aims/Introduction
The incidence of type 2 diabetes is increasing worldwide. Hepatic insulin resistance and liver lipid accumulation contributes to type 2 diabetes development. The aim of the present study was to investigate the key gene pathways and co‐expression networks in the livers of type 2 diabetes patients.
Materials and Methods
Dataset GSE15653 containing nine healthy individuals and nine type 2 diabetes patients was downloaded from the National Center for Biotechnology Information Gene Expression Omnibus database. Differentially expressed genes were obtained from the livers of type 2 diabetes patients, annotated pathway enrichment and protein–protein interaction network analysis. Next, functional modules and transcription factor networks were constructed. Gene co‐expression networks were analyzed by weighted correlation network analysis to identify key modules related to clinical traits, and the candidate key genes were validated in hepatic insulin resistance models in vitro.
Results
A total of 778 differentially expressed genes were filtered in the livers of type 2 diabetes patients, pathway enrichment analysis identified ke y pathways, such as the mitogen‐activated protein kinase signaling pathway, Hippo signaling pathway and hypoxia‐inducible factor‐1 signaling pathway, that were associated with type 2 diabetes. Several transcription factors of three functional modules identified from protein–protein interaction networks are likely to be implicated in type 2 diabetes. Furthermore, weighted correlation network analysis identified five modules that were shown to be highly correlated with type 2 diabetes and other clinical traits. Functional annotation showed that these modules were mainly enriched in pathways such as metabolic pathways, phosphoinositide 3‐kinase‐protein kinase B signaling pathway and natural killer cell‐mediated cytotoxicity. UBE2M and GPER were upregulated in L02 and HepG2 models, whereas P2RY11 only upregulated in L02 model, and UBE2N only downregulated in HepG2 model at a significant level.
Conclusions
These results would offer new insights into hepatic insulin resistance, type 2 diabetes pathogenesis, development and drug discovery.
Keywords: Co‐expression network, Type 2 diabetes, Weighted gene correlation network analysis
Introduction
In recent years, the incidence of diabetes has been increasing worldwide, and the first World Health Organization Global Report on Diabetes showed that approximately 422 million people worldwide had diabetes in 20141. Type 2 diabetes accounts for approximately 90% of all diabetes, and the prevalence of type 2 diabetes has risen dramatically in the past three decades (https://www.who.int/diabetes/en/). Therefore, elucidating the underlying mechanisms in type 2 diabetes is urgently required for disease progression and treatment.
Although the pathogenesis for type 2 diabetes is still controversial, it is well known that insulin resistance in various tissues, such as the liver and muscle, contributes to the generation and development of type 2 diabetes2. Lipid accumulation in tissues leads to organ‐specific insulin resistance3. Research has shown that fat in the liver rather than muscle is associated with features of the metabolic syndrome and occurs close to the time of type 2 diabetes development2, 4, 5. Furthermore, non‐alcoholic fatty liver disease has recently been reported to be a major predisposing factor for type 2 diabetes6, 7. The liver produces glucose during fasting and takes a central role in glucose homeostasis. Insulin normally inhibits the hepatic gluconeogenesis and promotes hepatic glycogen synthesis. Under the condition of type 2 diabetes, insulin resistance increases glucose production from the liver, and reduces glucose uptake by target tissues, which leads to hyperglycemia8. Despite the critical role of the liver in type 2 diabetes, the underlying molecular genes and pathways in the livers of type 2 diabetes patients are still incompletely elucidated.
Gene expression profile analysis has been widely applied in disease diagnosis, pathological mechanism exploration and treatment9, 10, 11. Several studies using microarray analysis have focused on the liver molecular genetics of type 2 diabetes pathogenesis8, 12, 13. However, the differentially expressed genes (DEGs), the underlying functional pathways, and the co‐expression networks between the livers of type 2 diabetes patients and non‐diabetic individuals remain to be clarified.
In the present study, we obtained the dataset GSE15653 from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database. We filtered the DEGs from the livers of type 2 diabetes patients, annotated functional pathway enrichment and protein–protein interaction (PPI) network analyses for DEGs. The correlated modules and transcription factor networks were also constructed. The co‐expression network of GSE15653 was then analyzed to identify the key gene modules related to clinical traits. Furthermore, key genes identified in this work were further validated in hepatic insulin resistance models in vitro. These results would offer new insights into type 2 diabetes pathogenesis, development and drug discovery.
Methods
Microarray data collection
Dataset GSE15653 contributed by Pihlajamäki et al. was downloaded from NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15653)12. The dataset contained 18 liver samples, including five lean samples, four obese without type 2 diabetes samples and nine obese with type 2 diabetes samples. We defined the five lean and four obese without type 2 diabetes samples as the non‐diabetes group, whereas nine obese with type 2 diabetes samples were defined as the type 2 diabetes group. The dataset was based on platform GPL96, and the microarray was carried out using Affymetrix Human Genome U133A Array. As the dataset was obtained from a public database, the present study did not involve any human or animal experiments, so ethics approval was unnecessary for our study.
DEGs screening
The raw data of GSE15653 were analyzed by the interactive web tool, GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/). The GEO2R tool carried out comparisons using the limma R packages from the Bioconductor project. Statistically significant DEGs were defined with P < 0.05 and |log2fold change (FC)| >1 as the cut‐off criteria.
Go and KEGG pathway enrichment analysis
GO analysis and KEGG pathway were analyzed using the DAVID online tool (https://david.ncifcrf.gov/) at the functional level. P < 0.05 was set as the cut‐off criterion. The top 10 enriched pathways were listed by the GO enrichment plot using a web online tool (http://www.ehbio.com/ImageGP/).
PPI and module analysis
The PPI network of DEGs was analyzed by the STRING database (http://string-db.org). A confidence score >0.9 was set as significant. The PPI network was then visualized by Cytoscape software (version 3.6.1; The Cytoscape Consortium, New York City, NY, USA). The modules of the PPI network were analyzed by the Molecular Complex Detection (MCODE) plugin of Cytoscape. MCODE plugin detects densely connected modules in PPI networks that might represent molecular complexes14. Significant modules were screened using the following plugin cut‐off criteria: degree cut off: 5; node score cut off: 0.2; k‐core: 2; and max. depth: 100. Resulting modules from the algorithm are scored and ranked. The top three significant modules were ranked and obtained by scores. A gene with the highest weighted vertex was defined as a seed gene by MCODE.
Transcription factor analysis
Transcription factors of identified modules were analyzed by the iRegulon plugin of Cytoscape15. The iRegulon plugin was set as the default. Transcription factors with normalized enrichment score (NES) >5 were identified as predicted transcription factors. The top three transcription factors of each module with higher NES were listed.
Construction of WGCNA
Weighted gene correlation network analysis (WGCNA) is applied to reveal the correlation patterns among genes in microarray samples as previous described16, 17. We used the WGCNA package in R software (https://www.r-project.org/) to carry out weighted correlation network analysis of all genes in GSE15653. We selected the power of β = 14 to ensure a scale‐free network. Gene modules differing by colors were summarized by a hierarchical clustering dendrogram, and module structure was visualized by a heatmap and topological overlap matrix plot. Furthermore, the relationships between each module and clinical traits were analyzed to identify the highly correlated modules. Finally, highly correlated modules with clinical traits were then analyzed using the DAVID online tool (https://david.ncifcrf.gov/) for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment. P < 0.05 was set as the cut‐off criterion. The top five enriched pathways were listed.
Establishment of human hepatic insulin resistance models
Human liver L02 cells18 and HepG2 cells19 were stimulated with palmitic acid (PA) to mimic hepatic insulin resistance models in vitro. L02 cells and HepG2 cells were cultured in Dulbecco's Modified Eagle's Medium (Sigma, St Louis, MO, USA) and Minimum Essential Medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco). PA (Sigma) was added to a 10% fatty acid‐free bovine serum albumin and dissolved by shaking gently overnight at 37°C to yield an 8 mmol/L solution20. L02 cells were treated with 250 μmol/L PA for 24 h. HepG2 cells were treated with 300 μmol/L PA for 24 h. Control cells were treated with bovine serum albumin alone. Cells then were collected for key gene validation.
Validation of key genes with quantitative reverse transcription polymerase chain reaction
Key gene validation using quantitative reverse transcription polymerase chain reaction. Total ribonucleic acids were extracted by RNAsimple Total RNA Kit (Tian Gen, Beijing, China). Complementary deoxyribonucleic acids (DNAs) were then synthesized using PrimeScript™ RT reagent Kit (Takara Bio, Shiga, Japan). A quantitative reverse transcription polymerase chain reaction procedure using TB Green™ Premix Ex Taq™ II (TaKaRa Bio) was applied for amplifications. Each measurement was normalized to Gapdh for each sample. All primer pairs are listed in Table S1. The relative gene expression was presented by the comparative C T method. Statistical analysis was carried out by the Student's t‐test when two groups were compared. P < 0.05 was considered to be significant.
Results
DEGs identification in the livers of type 2 diabetes patients
Dataset GSE15653 contained 18 liver samples, including five lean samples, four obese without type 2 diabetes samples and nine obese with type 2 diabetes samples. In order to lessen the influence of the difference between lean and obese individuals without type 2 diabetes, we combined the five lean and four obese without type 2 diabetes samples as the non‐diabetes group and compared the DEGs in nine obese individuals with type 2 diabetes to the normal group. A false discovery rate <0.05 is always used to filter DEGs from gene expression profiles. However, in the present study, we found that only one DEG met the criterion using the GEO2R web tool. In order to obtain more useful DEGs, we applied a nominal P < 0.05 and fold change >2 criteria for DEG identification. As a result, a total of 778 DEGs including 136 upregulated DEGs and 642 downregulated DEGs were filtered by GEO2R (Table S2). The expressions of the top 10 upregulated and downregulated DEGs were displayed in a heatmap (Figure 1). The top 10 upregulated DEGs were TRBC1, IL17RC, CDHR5, ALDOA, CLDN3, IGKV1OR2‐108, CHI3L1, EMILIN1, SCD and JUND. The top 10 downregulated DEGs were MYOT, BTRC, PDE4DIP, GART, RPGRIP1L, EEA1, GRIK2, TLX3, IGF2BP3 and GTSE1.
Figure 1.
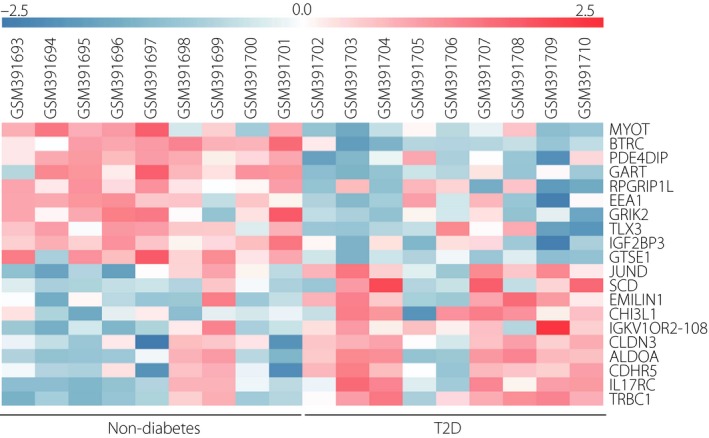
Heatmap of the top 10 upregulated and downregulated differentially expressed genes in GSE15653 Red, upregulation; blue, downregulation. T2D, type 2 diabetes.
KEGG pathway enrichment and GO analysis in DEGs of type 2 diabetes
All DEGs were uploaded to the DAVID online tool pathway enrichment analysis. For KEGG pathway enrichment (Figure 2a), the DEGs mainly enriched in pathways in cancer, mitogen‐activated protein kinase signaling pathway, Hippo signaling pathway, prostate cancer, hypoxia‐inducible factor‐1 signaling pathway, bacterial invasion of epithelial cells, regulation of actin cytoskeleton, phosphoinositide 3‐kinase‐protein kinase B (PI3K–Akt) signaling pathway, insulin resistance and transcriptional misregulation in cancer. For the GO term biological process analysis (Figure 2b), the DEGs enriched in positive regulation of cell proliferation, positive regulation of cell migration, positive regulation of ERK1 and ERK2 cascade, cell–cell signaling, cellular response to hormone stimulus, cell division, positive regulation of DNA biosynthetic process, lens fiber cell differentiation, cell adhesion and ossification.
Figure 2.
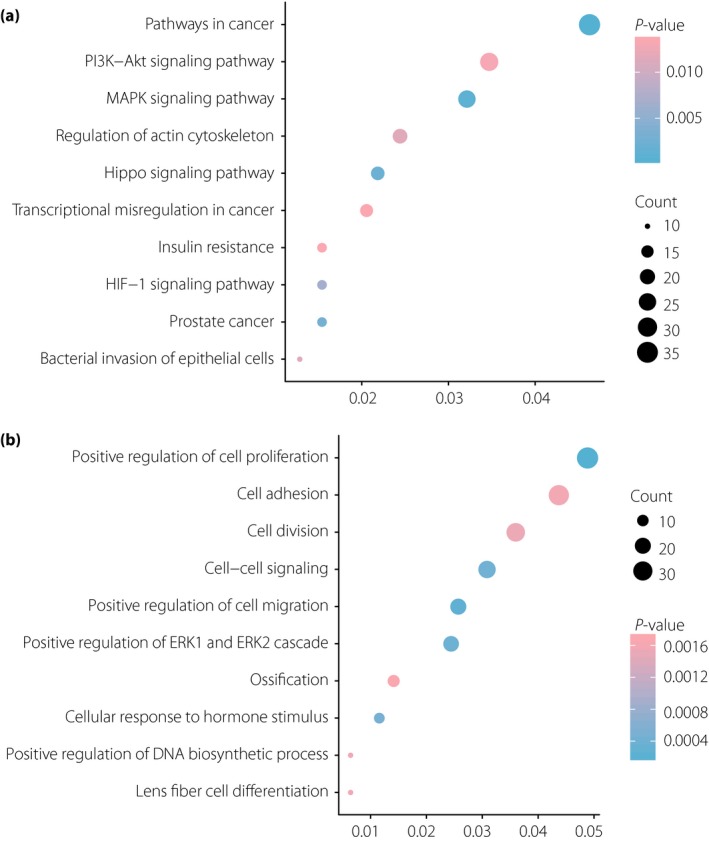
Enrichment analyses for (a) Kyoto Encyclopedia of Genes and Genomes pathways and (b) GO terms of differentially expressed genes in liver samples of type 2 diabetes patients. DNA, deoxyribonucleic acid; HIF‐1, hypoxia‐inducible factor; MAPK, mitogen‐activated protein kinase; PI3K–Akt, phosphoinositide 3‐kinase‐protein kinase B.
PPI and module analysis of livers of type 2 diabetes patients
To explore the functional connection of all DEGs, we constructed the PPI network by the STRING database. A total of 778 DEGs were analyzed and then visualized by Cytoscape. As a result, there were 315 nodes and 670 edges in the PPI network, which represented proteins and functional interactions (Figure 3a). Furthermore, functional modules were verified from the PPI network by the MCODE plugin. The plugin detected 19 significant modules that meet the cut‐off criteria, and the top three significant modules ranked by score were listed. Module 1 (score: 12) consisted of 12 nodes and 66 edges, and the seed gene was TRIM36 (Figure 3b). Module 2 (score: 10) consisted of 10 nodes and 45 edges, and the seed gene was P2RY11 (Figure 3c). Module 3 (score: 8) consisted of eight nodes and 28 edges, and the seed gene was GPER (Figure 3d).
Figure 3.

Protein–protein interaction network and module analysis of differentially expressed genes (DEGs) in GSE15653. (a) Protein–protein interaction network based on 778 DEGs constructed by Cytoscape. Red indicated the upregulated DEGs, and blue indicated the downregulated DEGs. (b) The top three modules identified from the protein–protein interaction network.
Transcription factor networks construction of modules
Transcription factors bind to a specific DNA sequence to regulate gene expression and function. Here, we predicted the transcription factors in the three modules using iRegulon plugin, respectively. Predicted transcription factors with NES >5 are shown in Table S3. The top three transcription factors with the highest NES are shown in Figure 4. In module 1, we predicted that IRX6 would regulate SMURF1, UBE2N, TRIM36 and NEDD4L; RUNX1 would regulate NEDD4L and RNF126; and NFATC3 would regulate SMURF1, UBE2N, TRIM36, NEDD4L, ANAPC10 and KLHL22 (Figure 4a). In module 2, we predicted that SIX1 would regulate PLCB1, HRH1, GHSR, HTR2C and EDNRA; SIX6 would regulate PLCB1, HRH1, PIK3R3, GHSR and HTR2C; and STAT5A would regulate PLCB1, HRH1, PIK3R3, EDNRA and HTR2C (Figure 4b). In module 3, we predicted that BDP1 would regulate GPER, GABBR2 and GRM3; ESR1 would regulate GPER, GRM8, PNOC, CCR3, CXCL11, and GRM3; and GATA2 would regulate GABBR2 and PNOC (Figure 4c).
Figure 4.
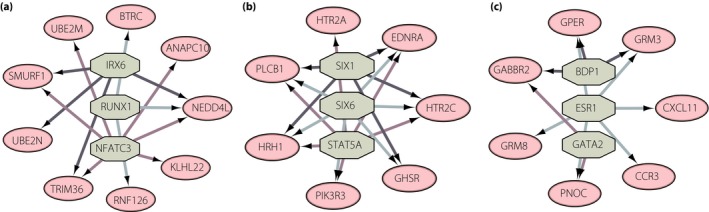
Transcription factor target networks of the three modules. Green octagon nodes represent the predicted transcription factor. Pink oval nodes represent transcription factor regulated genes.
Co‐expression network analyzed by WGCNA of the livers of type 2 diabetes patients
Here, WGCNA was used to reveal the highly correlated genes and co‐expression networks of type 2 diabetes patients. Altogether, six types of clinical data including body mass index (BMI), hemoglobin A1c (HbA1c), fasting glucose, fasting insulin, obese and type 2 diabetes were found in all samples of dataset GSE15653 (Figure 5a). Gene modules were analyzed; the grey module represents genes that cannot be clustered into other modules. In total, the hierarchical clustering dendrogram based on gene expressions identified 10 distinct gene modules (Figure 5b). Genes in different modules are listed in Table S4. Then, we generated the topological overlap matrix plot of a gene network with the corresponding hierarchical clustering dendrograms and the resulting modules (Figure 5c). Eigengene adjacency heatmap was carried out for the identification of module correlation (Figure 5d). Furthermore, in order to show the relationships between modules and clinical traits, we carried out a correlation analysis of the module–trait relationships (Figure 6). We found that BMI and obese traits were positively associated with the turquoise module, and negatively associated with the blue module. The HbA1c trait was positively related with the red module, and negatively related with the blue and brown modules. Fasting glucose and fasting insulin were positively associated with the magenta module, but negatively associated with the brown or blue module, respectively. Finally, the type 2 diabetes trait was found to be positively related to the turquoise module, and negatively related to the blue trait.
Figure 5.

Samples clustering and module detection. (a) Sample dendrogram and trait indicator. In the obese row and type 2 diabetes (T2D) row, the red color represents obese and type 2 diabetes samples. In other rows, the red color intensity is proportional to higher body mass index (BMI), fasting glucose, fasting insulin and hemoglobin A1c (HbA1c). The gray color represents no data in clinical traits. (b) Cluster dendrogram based on 1‐topological overlap matrix. Gene dendrogram acquired by average linkage hierarchical clustering. The color row under the gene dendrogram represents the module assignment determined by the dynamic tree cut. Each colored branch indicates a module of highly connected genes. (c) Network heatmap plot of genes in GSE15653. The gene dendrogram and module assignment are shown along the left and top. The heatmap colors in the matrix present the degree of topological overlap. (d) Eigengene adjacency heatmap indicates the correlation between modules. Red represents positive correlation and blue represents negative correlation. The color intensity is proportional to a higher association.
Figure 6.
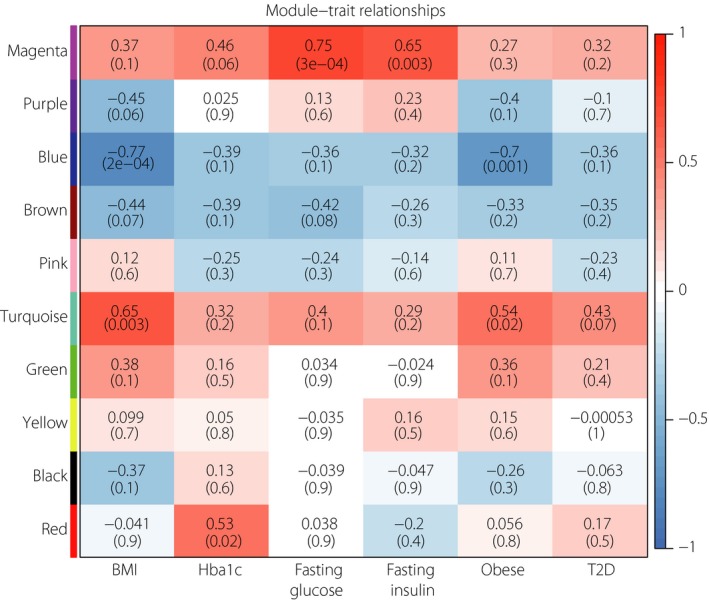
Correlation between modules and clinical traits. Red represents positive correlation and blue represents negative correlation. The number in the top line of each cell indicates the robust correlation, and the number in the parentheses of each cell indicates the corresponding asymptotic P‐value. BMI, body mass index; HbA1c, hemoglobin A1c, T2D, type 2 diabetes.
Functional annotation of highly correlated modules
Finally, the highly correlated modules with BMI, HbA1c, fasting glucose, fasting insulin, obese and type 2 diabetes were selected for functional annotation, respectively (Figure 7). The genes in the magenta module mainly enriched in focal adhesion, extracellular matrix‐receptor interaction, protein digestion and absorption, amoebiasis, and the PI3K–Akt signaling pathway. The genes in the red module mainly enriched in metabolic pathways; valine, leucine and isoleucine degradation; butanoate metabolism; complement and coagulation cascades; and bile secretion, whereas the genes in the turquoise module mainly enriched in metabolic pathways, oxidative phosphorylation, carbon metabolism, non‐alcoholic fatty liver disease and Parkinson's disease. Just four pathways met the criterion of P < 0.05. The genes in the blue module mainly enriched in ubiquitin‐mediated proteolysis, folate biosynthesis, spliceosome and metabolic pathways, whereas the genes in the brown module mainly enriched in Alzheimer's disease, tuberculosis, vascular smooth muscle contraction, natural killer cell mediated cytotoxicity and measles.
Figure 7.
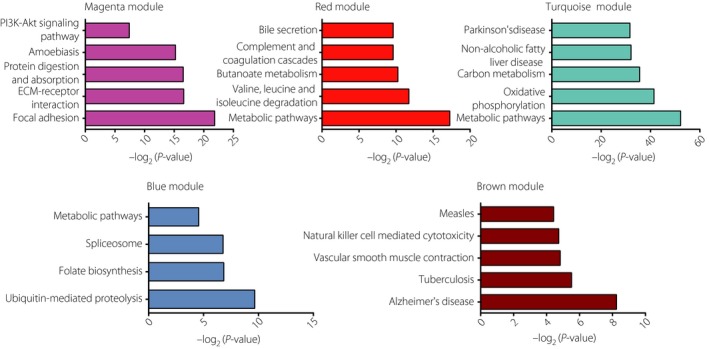
Gene‐annotation enrichment analysis of Kyoto Encyclopedia of Genes and Genomes pathway in each module. ECM, extracellular membrane; PI3K–Akt, phosphoinositide 3‐kinase‐protein kinase B.
Validation of key genes correlated with the livers of type 2 diabetes patients
Here, we screened the possible key genes based on type 2 diabetes highly‐correlated modules (turquoise and blue; Figure 6), and three functional modules from PPI networks (Figure 3b–d). One gene, UBE2M, was included in the functional modules and the turquoise module, and two genes, ANAPC13 and UBE2N, were included in the functional modules and the blue modules (Figure 8a). In addition, three seed genes in three functional modules, including GPER, P2RY11 and TRIM36 (Figure 3b–d), were also regarded as key genes. As shown in Table S2, ANAPC13, UBE2N and TRIM36 were downregulated, and UBE2M, GPER and P2RY11 were upregulated in dataset GSE15653. In order to further confirm the role of six key genes in the livers of type 2 diabetes patients, we next detected the key gene expressions in hepatic insulin resistance models in vitro. PA, a saturated fatty acid, is reported to be closely associated with the development of insulin resistance21. Here, we established two hepatic insulin resistance models using human liver L02 cells and HepG2 cells under the stimulation of PA in vitro 18, 19. As a result, UBE2M and GPER were upregulated in both L02 and HepG2 cell models, whereas P2RY11 only upregulated in L02 model, and UBE2N only downregulated in the HepG2 model at a significant level (Figure 8b,c).
Figure 8.
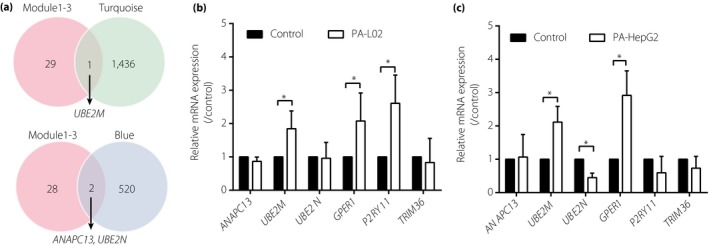
Validation of potential key genes in hepatic insulin resistance models in vitro. (a) Venn diagram shows the key genes in functional module 1–3 and turquoise or blue module identified from weighted gene correlation network analysis. Validation of key genes in the (b) L02 cell model (n = 5) and (c) HepG2 cell model (n = 3) by quantitative reverse transcription polymerase chain reaction. *P < 0.05 versus control. mRNA, messenger ribonucleic acid.
Discussion
The liver plays a key role in glucose homeostasis, and fatty liver is a major predisposition for insulin resistance contributing to type 2 diabetes. Several gene profiles of the livers of type 2 diabetes patients have been uploaded to NCBI GEO public databases, such as GSE1565312, GSE233438 and GSE6499813. Here, we attempted to explore the molecular functional pathways and co‐expression networks instead of individual gene function in the livers of type 2 diabetes patients. As the dataset GSE15653 provided more patient features, such as BMI, HbA1c, fasting glucose and fasting insulin, we believed that analyzing the gene profile of GSE15653 would help us to reveal the relationships between genes and clinical traits. As a consequence, we screened 778 DEGs, including 136 upregulated DEGs and 642 downregulated DEGs, between the type 2 diabetes and non‐diabetic group from GSE15653. Among the top 10 upregulated DEGs, CHI3L1, also known as YKL40, has been shown to be increased in the serum of type 2 diabetes patients22, 23. Similarly, SCD is positively associated with type 2 diabetes24, 25. Among the top 10 downregulated DEGs, BTRC is significantly associated with type 2 diabetes in a Chinese Han population26. RPGRIP1L is one of the type 2 diabetes loci analyzed by the genome‐wide association study27. The EEA1 gene mutation is found in the Japanese population with type 2 diabetes28. GRIK2 is associated with diabetic retinopathy29. However, little is known about their expression levels in the livers of type 2 diabetes patients. Except for the genes mentioned above, the remaining 14 novel genes have not been specifically implicated in type 2 diabetes, and might be involved in hepatic insulin resistance.
For functional annotation, KEGG pathway enrichment and GO analysis were carried out to elucidate the underlying pathways in upregulated and downregulated DEGs. We found that DEGs mainly enriched in mitogen‐activated protein kinase signaling pathway, Hippo signaling pathway, hypoxia‐inducible factor‐1 signaling pathway, regulation of actin cytoskeleton, PI3K–Akt signaling pathway, insulin resistance, positive regulation of cell proliferation, positive regulation of cell migration, positive regulation of the ERK1 and ERK2 cascade, cellular response to hormone stimulus, cell division, positive regulation of DNA biosynthetic process, and cell adhesion (Figure 2). Insulin resistance is well known as the major feature and cause of type 2 diabetes. Meanwhile, this is consistent with the knowledge that mitogen‐activated protein kinase/ERK signaling pathway activation is involved in hepatic insulin resistance and type 2 diabetes development30, 31, 32, 33. Several diabetes‐related pathways were associated with Hippo signaling34. Hippo was reported as a modulator of metabolism in the liver, disruption of the Hippo pathway might be associated with development of type 2 diabetes35. Hypoxia‐inducible factor‐1 signaling pathway contributed to insulin resistance36, 37, and might be involved in hepatic insulin sensitivity in HepG2 cells38. Activation of the PI3K–Akt pathway would attenuate hepatic insulin resistance39, 40, and reduce gluconeogenesis in type 2 diabetes mice41. Actin cytoskeleton participates in muscle insulin resistance of type 2 diabetes42, but the role in the livers of type 2 diabetes patients remains unclear. The role of the remaining enriched pathways in the livers of type 2 diabetes patients still requires further exploration.
Furthermore, we constructed the PPI network by all DEGs for functional interactions (Figure 3a). The most significant three functional modules were filtered (Figure 3b–d). We found the seed gene of these three modules was TRIM36, P2RY11 and GPER, respectively, and we validated these three seed genes expressions in hepatic insulin resistance models in vitro. GPER has been shown to be involved in obesity, insulin resistance and type 2 diabetes43, 44. Consistently, the present results showed that GPER was upregulated in both L02 cells and HepG2 cells stimulated by PA (Figure 8b,c). Borno et al.45 found that both type 2 diabetes patients and healthy controls showed similar distribution of P2RY11 in skeletal muscles. We found that P2RY11 was upregulated in PA‐treated L02 cells (Figure 8b), but not in HepG2 cells (Figure 8c).Little is known about the relationship between TRIM36 and type 2 diabetes, and we did not observe a significant alteration in TRIM36 expression in hepatic insulin resistance models. The present results showed that GPER is probably associated with type 2 diabetes insulin resistance in the liver, and P2RY11 might have a potential role in the development of type 2 diabetes, and the interactions between these seed genes and type 2 diabetes hepatic insulin resistance still require further investigation. Our transcription factor analysis identified that several transcription factors are likely to be implicated in type 2 diabetes (Figure 4; Table S2). A recent study showed that mineralocorticoid receptor directly regulated ESR1 in macrophages, and then was implicated to play important roles in non‐alcoholic fatty liver disease and type 2 diabetes46, but the role of ESR1 in the liver is unknown. Meanwhile, the association between other transcription factors and type 2 diabetes, especially hepatic insulin resistance, still needs to be fully elucidated.
WGCNA was applied to identify the significantly enriched gene co‐expression network of the livers of type 2 diabetes patients. A total of 10 gene modules were identified in GSE15653 (Figure 5). We investigated the highly correlated gene modules with six types of clinical characteristics, including BMI, HbA1c, fasting glucose, fasting insulin, obese and type 2 diabetes. As a result, we found that the turquoise module, magenta module and red module were positively correlated with the clinical traits, whereas the blue module and brown module were negatively associated with the clinical traits (Figure 6). The red module, turquoise module and blue module were all involved in metabolic pathways. The red and blue modules were associated with HbA1c, namely the key marker of type 2 diabetes, whereas the turquoise and blue module were associated with type 2 diabetes and obesity. Type 2 diabetes involves the complex interplay of multiple metabolic pathways, and the metabolites contributed to the hepatic insulin resistance47. We inferred the metabolic pathway played a critical role in the livers of type 2 diabetes patients and liver lipid accumulation. Metabolomics has been used to predict potential and novel treatment for type 2 diabetes48, but the role of many metabolites in hepatic insulin resistance and type 2 diabetes is still known. In addition, the magenta module, which is associated with fasting glucose and insulin, was also involved in the PI3K–Akt signaling pathway in accord with Figure 2a. The PI3K–Akt pathway activation by a long non‐coding ribonucleic acid suppressor has been proven to regulate the hepatic gluconeogenesis and lipogenesis, and might be a potential strategy for type 2 diabetes treatment49. The brown module was associated with fasting glucose and HbA1c, and was involved in natural killer cell‐mediated cytotoxicity. Natural killer cell‐mediated cytotoxicity was reported to be related to gestational diabetes mellitus by microarray analysis50. Therefore, we confer the pathway might be an important role in the development of diabetes.
For further investigation, we validated the expressions of three key genes, ANAPC13, UBE2M, UBE2N, obtained from functional modules of the PPI network and highly correlated modules of WGCNA (Figure 8a). Little is known about the effects of ANAPC13 in type 2 diabetes, and ANAPC13 did not change in either hepatic insulin resistance model (Figure 8b,c). UBE2M and UBE2N are ubiquitin‐conjugating enzymes; the modification of proteins with ubiquitin is an important cellular mechanism for targeting abnormal or short‐lived proteins for degradation. The ubiquitin‐proteasome system was reported to have a role in type 2 diabetes51, 52, 53. UBE2M antibody had a higher signal in type 2 diabetes patients’ plasma with the lower insulin‐secretion human leukocyte antigen background54. We observed that UBE2M was increased in two hepatic insulin resistance models, indicating that UBE2M possibly plays a key role in type 2 diabetes hepatic insulin resistance. UBE2N haploinsufficiency in female mice protected against high‐fat diet‐induced obesity, hepatic steatosis and insulin resistance55. In contrast, UBE2N was decreased in type 2 diabetes samples of GSE15653 and PA‐induced HepG2 cells, further study is required to elucidate the role of UBE2N in the livers of type 2 diabetes patients.
In our present work, the results offer potential genes and underlying pathways in the livers of type 2 diabetes patients. The results provide new strategies for type 2 diabetes pathogenesis, development and drug discovery. However, further and deep experiments are still required for the candidate gene and pathway in type 2 diabetes.
Disclosure
The authors declare no conflict of interest.
Supporting information
Table S1 Primers and conditions for quantitative reverse transcription polymerase chain reaction.
Table S2 All differentially expressed genes of GSE15653 identified by GEO2R.
Table S3 Predicted transcription factor with normalized enrichment score >5.
Table S4 Genes in different modules identified by weighted gene correlation network analysis.
Acknowledgment
This work was supported by The National Natural Science Foundation of China (No. 81703578) and the foundation of Zhejiang Traditional Chinese Medicine (No. 2018ZA062).
J Diabetes Investig 2019; 10: 951–962
References
- 1. World Health Organization . Global Report on Diabetes. Geneva, Switzerland: World Health Organization, 2016. [Google Scholar]
- 2. Sattar N, Gill JM. Type 2 diabetes as a disease of ectopic fat? BMC Med 2014; 12: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cusi K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Curr Diab Rep 2010; 10: 306–315. [DOI] [PubMed] [Google Scholar]
- 4. Taylor R, Al‐Mrabeh A, Zhyzhneuskaya S, et al Remission of human Type 2 Diabetes requires decrease in liver and pancreas fat content but is dependent upon capacity for beta cell recovery. Cell Metab 2018; 28: 547–556. [DOI] [PubMed] [Google Scholar]
- 5. Kotronen A, Seppala‐Lindroos A, Bergholm R, et al Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia 2008; 51: 130–138. [DOI] [PubMed] [Google Scholar]
- 6. Bril F, Cusi K. Management of nonalcoholic fatty liver disease in patients with type 2 diabetes: a call to action. Diabetes Care 2017; 40: 419–430. [DOI] [PubMed] [Google Scholar]
- 7. Perry RJ, Kim T, Zhang XM, et al Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver‐targeted mitochondrial uncoupler. Cell Metab 2013; 18: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Misu H, Takamura T, Takayama H, et al A liver‐derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab 2010; 12: 483–495. [DOI] [PubMed] [Google Scholar]
- 9. Barrett T, Wilhite SE, Ledoux P, et al NCBI GEO: archive for functional genomics data sets‐update. Nucleic Acids Res 2013; 41: D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parkinson H, Kapushesky M, Shojatalab M, et al ArrayExpress – a public database of microarray experiments and gene expression profiles. Nucleic Acids Res 2007; 35: D747–D750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. GTEx Consortium . Human genomics. The Genotype‐Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pihlajamaki J, Boes T, Kim EY, et al Thyroid hormone‐related regulation of gene expression in human fatty liver. J Clin Endocrinol Metab 2009; 94: 3521–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kirchner H, Sinha I, Gao H, et al Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol Metab 2016; 5: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003; 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janky R, Verfaillie A, Imrichova H, et al iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol 2014; 10: e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Pan Z, Yang S, et al Identification of key gene pathways and coexpression networks of islets in human type 2 diabetes. Diabetes Metab Syndr Obes 2018; 11: 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wan XD, Yang WB, Xia YZ, et al Disruption of glucose homeostasis and induction of insulin resistance by elevated free fatty acids in human L02 hepatocytes. J Endocrinol Invest 2009; 32: 454–459. [DOI] [PubMed] [Google Scholar]
- 19. Heo JI, Yoon DW, Yu JH, et al Melatonin improves insulin resistance and hepatic steatosis through attenuation of alpha‐2‐HS‐glycoprotein. J Pineal Res 2018; 65: e12493. [DOI] [PubMed] [Google Scholar]
- 20. Joshi‐Barve S, Barve SS, Amancherla K, et al Palmitic acid induces production of proinflammatory cytokine interleukin‐8 from hepatocytes. Hepatology 2007; 46: 823–830. [DOI] [PubMed] [Google Scholar]
- 21. Reynoso R, Salgado LM, Calderon V. High levels of palmitic acid lead to insulin resistance due to changes in the level of phosphorylation of the insulin receptor and insulin receptor substrate‐1. Mol Cell Biochem 2003; 246: 155–162. [PubMed] [Google Scholar]
- 22. Nielsen AR, Erikstrup C, Johansen JS, et al Plasma YKL‐40: a BMI‐independent marker of type 2 diabetes. Diabetes 2008; 57: 3078–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rondbjerg AK, Omerovic E, Vestergaard H. YKL‐40 levels are independently associated with albuminuria in type 2 diabetes. Cardiovasc Diabetol 2011; 10: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liew CF, Groves CJ, Wiltshire S, et al Analysis of the contribution to type 2 diabetes susceptibility of sequence variation in the gene encoding stearoyl‐CoA desaturase, a key regulator of lipid and carbohydrate metabolism. Diabetologia 2004; 47: 2168–2175. [DOI] [PubMed] [Google Scholar]
- 25. Jacobs S, Schiller K, Jansen EH, et al Evaluation of various biomarkers as potential mediators of the association between Delta5 desaturase, Delta6 desaturase, and stearoyl‐CoA desaturase activity and incidenttype 2 diabetes in the European Prospective Investigation into Cancer and Nutrition‐Potsdam Study. Am J Clin Nutr 2015; 102: 155–164. [DOI] [PubMed] [Google Scholar]
- 26. Zhou JB, Yang JK, Zhang BH, et al Interaction of Wnt pathway related variants with type 2 diabetes in a Chinese Han population. PeerJ 2015; 3: e1304. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Qu HQ, Grant SF, Bradfield JP, et al Association analysis of type 2 diabetes Loci in type 1 diabetes. Diabetes 2008; 57: 1983–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanaka D, Nagashima K, Sasaki M, et al Exome sequencing identifies a new candidate mutation for susceptibility to diabetes in a family with highly aggregated type 2 diabetes. Mol Genet Metab 2013; 109: 112–117. [DOI] [PubMed] [Google Scholar]
- 29. Lin HJ, Huang YC, Lin JM, et al Association of genes on chromosome 6, GRIK2, TMEM217 and TMEM63B (linked to MRPL14) with diabetic retinopathy. Ophthalmologica 2013; 229: 54–60. [DOI] [PubMed] [Google Scholar]
- 30. Lejnev K, Khomsky L, Bokvist K, et al Thioredoxin‐mimetic peptides (TXM) inhibit inflammatory pathways associated with high‐glucose and oxidative stress. Free Radic Biol Med 2016; 99: 557–571. [DOI] [PubMed] [Google Scholar]
- 31. Zhao Y, Tang Z, Zhu X, et al TAB 3 involves in hepatic insulin resistance through activation of MAPK pathway. Gen Comp Endocrinol 2015; 224: 228–234. [DOI] [PubMed] [Google Scholar]
- 32. Bak EJ, Choi KC, Jang S, et al Licochalcone F alleviates glucose tolerance and chronic inflammation in diet‐induced obese mice through Akt and p38 MAPK. Clin Nutr 2016; 35: 414–421. [DOI] [PubMed] [Google Scholar]
- 33. Manowsky J, Camargo RG, Kipp AP, et al Insulin‐induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am J Physiol Endocrinol Metab 2016; 310: E938–E946. [DOI] [PubMed] [Google Scholar]
- 34. Ardestani A, Maedler K. MST1: a promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia 2016; 59: 1843–1849. [DOI] [PubMed] [Google Scholar]
- 35. Ardestani A, Lupse B, Maedler K. Hippo signaling: key emerging pathway in cellular and whole‐body metabolism. Trends Endocrinol Metab 2018; 29: 492–509. [DOI] [PubMed] [Google Scholar]
- 36. Yang XD, Xiang DX, Yang YY. Role of E3 ubiquitin ligases in insulin resistance. Diabetes Obes Metab 2016; 18: 747–754. [DOI] [PubMed] [Google Scholar]
- 37. Chen S, Okahara F, Osaki N, et al Increased GIP signaling induces adipose inflammation via a HIF‐1alpha‐dependent pathway and impairs insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2015; 308: E414–E425. [DOI] [PubMed] [Google Scholar]
- 38. Xia N, Tang Z, Wang C, et al PCBP2 regulates hepatic insulin sensitivity via HIF‐1alpha and STAT3 pathway in HepG2 cells. Biochem Biophys Res Commun 2015; 463: 116–122. [DOI] [PubMed] [Google Scholar]
- 39. Pang M, de la Monte SM, Longato L, et al PPARdelta agonist attenuates alcohol‐induced hepatic insulin resistance and improves liver injury and repair. J Hepatol 2009; 50: 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu H, Zhou Y, Liu Y, et al Metformin improves hepatic IRS2/PI3K/Akt signaling in insulin‐resistant rats of NASH and cirrhosis. J Endocrinol 2016; 229: 133–144. [DOI] [PubMed] [Google Scholar]
- 41. Liu TY, Shi CX, Gao R, et al Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin Sci (Lond) 2015; 129: 839–850. [DOI] [PubMed] [Google Scholar]
- 42. Jin W, Goldfine AB, Boes T, et al Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest 2011; 121: 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meyer MR, Clegg DJ, Prossnitz ER, et al Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) 2011; 203: 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuente‐Martin E, Garcia‐Caceres C, Morselli E, et al Estrogen, astrocytes and the neuroendocrine control of metabolism. Rev Endocr Metab Disord 2013; 14: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Borno A, Ploug T, Bune LT, et al Purinergic receptors expressed in human skeletal muscle fibres. Purinergic Signal 2012; 8: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang YY, Li C, Yao GF, et al Deletion of macrophage mineralocorticoid receptor protects hepatic steatosis and insulin resistance through ERalpha/HGF/Met pathway. Diabetes 2017; 66: 1535–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol 2018; 10: 654–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guasch‐Ferre M, Hruby A, Toledo E, et al Metabolomics in prediabetes and diabetes: a systematic review and meta‐analysis. Diabetes Care 2016; 39: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J, Yang W, Chen Z, et al Long noncoding RNA lncSHGL recruits hnRNPA1 to suppress hepatic gluconeogenesis and lipogenesis. Diabetes 2018; 67: 581–593. [DOI] [PubMed] [Google Scholar]
- 50. Qian Y, Sun H, Xiao H, et al Microarray analysis of differentially expressed genes and their functions in omental visceral adipose tissues of pregnant women with vs. without gestational diabetes mellitus. Biomed Rep 2017; 6: 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jaisson S, Gillery P. Impaired proteostasis: role in the pathogenesis of diabetes mellitus. Diabetologia 2014; 57: 1517–1527. [DOI] [PubMed] [Google Scholar]
- 52. Hartley T, Brumell J, Volchuk A. Emerging roles for the ubiquitin‐proteasome system and autophagy in pancreatic beta‐cells. Am J Physiol Endocrinol Metab 2009; 296: E1–E10. [DOI] [PubMed] [Google Scholar]
- 53. Costes S, Huang CJ, Gurlo T, et al Beta‐cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide‐induced UCH‐L1 deficiency. Diabetes 2011; 60: 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang DC, Piaggi P, Hanson RL, et al Use of a high‐density protein microarray to identify autoantibodies in subjects with type 2 diabetes mellitus and an HLA background associated with reduced insulin secretion. PLoS ONE 2015; 10: e143551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Joo E, Fukushima T, Harada N, et al Ubc13 haploinsufficiency protects against age‐related insulin resistance and high‐fat diet‐induced obesity. Sci Rep 2016; 6: 35983. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers and conditions for quantitative reverse transcription polymerase chain reaction.
Table S2 All differentially expressed genes of GSE15653 identified by GEO2R.
Table S3 Predicted transcription factor with normalized enrichment score >5.
Table S4 Genes in different modules identified by weighted gene correlation network analysis.