

Summary
It remains largely unclear how stem cells regulate bioenergetics and genome integrity to ensure tissue homeostasis. Here, our integrative gene analyses suggest that metabolic and genotoxic stresses may underlie the common functional defects of both fetal and adult hematopoietic stem and progenitor cells (HSPCs) upon loss of DPY30, an epigenetic modulator that facilitates H3K4 methylation. DPY30 directly regulates expression of several key glycolytic genes, and its loss in HSPCs critically impaired energy metabolism, including both glycolytic and mitochondrial pathways. We also found significant increase in DNA breaks as a result of impaired DNA repair upon DPY30 loss, and inhibition of DNA damage response partially rescued clonogenicity of the DPY30-deficient HSPCs. Moreover, CDK inhibitor p21 was upregulated in DPY30-deficient HSPCs, and p21 deletion alleviated their functional defect. These results demonstrate that epigenetic mechanisms by H3K4 methylation play a crucial role in HSPC function through control of energy metabolism and protecting genome integrity.
Keywords: hematopoietic stem cells, SET1/MLL complexes, DPY30, H3K4 methylation, epigenetic regulation, energy metabolism, DNA damage response, p21, glycolysis, mitochondrion
Graphical Abstract
Highlights
-
•
DPY30-deficient fetal and adult HSCs are defective in maintenance and differentiation
-
•
Glycolytic and oxidative metabolism are dysregulated in DPY30-deficient HSCs
-
•
Increase in DNA damage response contributes to dysfunction of DPY30-deficient HSPCs
-
•
P21 increase partially mediates dysfunction of DPY30-deficient HSPCs
Jiang and colleagues show that HSCs deficient in DPY30 and thus histone H3K4 methylation are under metabolic and genotoxic stresses, as shown by reduction in both glycolytic and mitochondrial activities and increase in DNA damages. The authors show that dysregulated bioenergetics and increases in DNA damage response and CDK inhibitor P21 all functionally contribute to the loss of HSC activity.
Introduction
Adult tissue stem cells, including hematopoietic stem and progenitor cells (HSCs and HPCs, or HSPCs), usually remain in the quiescent state with limited cell divisions, but can enter rapid proliferation upon activation (Kondo et al., 2003). As one of the most fundamental processes of all life, metabolism profoundly influences the fate determination of stem and progenitor cells (Ito and Suda, 2014). Quiescent HSCs have relatively inactive mitochondria and mainly rely on glycolysis, but rapidly switch to mitochondrial oxidative phosphorylation as the major energy supply when they undergo activation and differentiation (Ito and Suda, 2014). Regulatory mechanisms in gene expression, including epigenetic mechanisms, can play an important role in stem and progenitor cell fate determination through impinging on the expression of rate-limiting enzymes in the metabolic pathways. Histone H3K4 methylation is one of the most prominent epigenetic marks associated with active gene expression (Shilatifard, 2008), but its role in regulation of metabolism, especially in stem and progenitor cell settings, remains unclear.
Maintaining the stability of the genetic material through the intricate network of DNA damage response (DDR) is particularly crucial for stem and progenitor cells to ensure the propagation of correct genetic information throughout the daughter cells in the tissue for the benefit of the whole organism (Blanpain et al., 2011). Mice deficient of components in either recognition (Ito et al., 2004) or repair (Nijnik et al., 2007, Rossi et al., 2007) of DNA lesions often have defective HSCs. As DDR occurs in the context of chromatin, it is thus governed by epigenetic mechanisms regulating chromatin structure and accessibility (Lukas et al., 2011). In addition to the well-established association with active gene expression, a role of H3K4 methylation in regulating genome stability is also gradually emerging (Burman et al., 2015, Faucher and Wellinger, 2010, Herbette et al., 2017, Higgs et al., 2018).
The most notable H3K4 methylation enzymes in mammals are the SET1/MLL complexes (Shilatifard, 2008), which comprise one of six different catalytic subunits and several shared core subunits including DPY30. DPY30 directly facilitates genome-wide H3K4 methylation (Jiang et al., 2011) and plays important roles in the fate transition between pluripotent and differentiated states (Jiang et al., 2011, Yang et al., 2015). We have further shown a crucial role of DPY30 in HSPCs and animal hematopoiesis (Yang et al., 2014). Ablation of Dpy30 in the hematopoietic system of the adult mouse bone marrow (BM) leads to loss of global H3K4 methylation, and disables differentiation and long-term maintenance of adult HSCs (Yang et al., 2016). This is shown in part by the striking accumulation of phenotypic HSCs and early HPCs at the expense of more downstream hematopoietic cells after Dpy30 ablation in BM (Yang et al., 2016), a phenotype shared by loss of other subunits of the SET1/MLL complexes (Arndt et al., 2018, Chen et al., 2014, Chun et al., 2014, Santos et al., 2014). Moreover, DPY30-deficient adult HSCs contribute poorly to all hematopoietic cell populations in the later stage after transplant. The normal transition of gene program between cell populations is also disrupted, and multiple genes important for HSC maintenance and differentiation are dysregulated following DPY30 loss (Yang et al., 2016). However, it is unclear what pathways are fundamentally important for DPY30's role in HSPC fate determination.
As fetal and adult HSCs differ in many aspects including cell-cycle regulation, self-renewal potential, surface marker expression, and reconstitution ability (Orkin and Zon, 2008), the function of DPY30 and H3K4 methylation in prenatal HSCs is still unknown. We reason that pathways fundamentally important for the activity of an epigenetic modulator are likely to be conserved in different biological systems. In this work, we first revealed a similar requirement of DPY30 in the activation and long-term maintenance of fetal HSPCs compared with adult HSPCs. Our dissection of targets shared in these two systems allowed us to identify energy metabolism and DDR as fundamentally important mediators of the H3K4 methylation pathway in hematopoiesis, as further supported by our rescue assays.
Results
DPY30 Deficiency in Fetal Liver Results in Anemia and Accumulation of Early HSPCs
We previously generated a Dpy30 conditional knockout (KO) mouse model where the Floxed (F) allele of Dpy30 is converted to a null allele upon Cre activity (Yang et al., 2016). Based on this and the Vav-Cre (Stadtfeld and Graf, 2005) mouse models, here we generated Dpy30F/+, Dpy30F/−, Vav-Cre; Dpy30F/+, and Vav-Cre; Dpy30F/− fetuses in the same litters. The Vav-Cre; Dpy30F/− fetuses developed normally until embryonic day 13.5 (E13.5), a time when Vav-Cre-mediated excision in HSCs becomes fully penetrant (Gan et al., 2010), but were never born and thus likely died at the late embryonic stage. The Vav-Cre; Dpy30F/− fetuses were anemic at E14.5, and anemia became more severe at E15.5 (Figure 1A). No difference was observed among the Dpy30F/+, Dpy30F/−, and Vav-Cre; Dpy30F/+ fetuses. We confirmed Dpy30 excision (Figure S1A) and dramatic reduction of Dpy30 mRNA (Figure 1B) in total and various HSPC populations (see Table S1 for cell surface markers) of the Vav-Cre; Dpy30F/− fetal liver (FL), and reduction of the DPY30 protein (Figure 1C) in total FL. DPY30 loss also resulted in marked reduction in H3K4 tri-methylation (H3K4me3) and mild reduction in H3K4 mono- and di-methylation (Figure 1C).
Figure 1.

DPY30 Deficiency in the Fetal Hematopoietic System Results in Anemia and Defective HSC Function
Vav-Cre; Dpy30F/+ (F/+) and Vav-Cre; Dpy30F/− (F/−) littermate fetuses were used in (A–E and I).
(A) Image of littermate fetuses at E14.5 and E15.5.
(B) Relative Dpy30 mRNA levels in whole FL and sorted hematopoietic cell populations determined by qRT-PCR and normalized to Actb. n = 3–6 each for FL, n = 4 each for sorted cells.
(C) Immunoblotting for different levels of H3K4 methylation and other proteins in FL. Right, quantification from five embryos each.
(D) Absolute numbers of FL cell populations. n = 5 each.
(E) Colony formation assay using FL cells, showing representative images of colonies and quantification. n = 3 each.
(F) Scheme for the mixed chimera transplantation system using whole FL cells from Mx1-Cre; Dpy30F/+ or Mx1-Cre; Dpy30F/− fetuses as donors and whole BM cells from wild-type mice as competitors.
(G) Donor contribution to different cell populations in chimeras at indicated times after pIpC injections following scheme in (G). n = 4–6 each.
(H and I) Relative expression of CDK inhibitor genes in control (F/+) versus Dpy30 KO (F/−) HSCs in two independent BM chimeras (H) and three FLs at E14.5 (I), as analyzed by RNA sequencing (RNA-seq). The expression levels in F/+ cells were set as 1. The BM HSC data are based on RNA-seq results in donor (Mx1-Cre; Dpy30F/+ or Mx1-Cre; Dpy30F/−)-derived HSCs in the BM chimera recipients in two independent BM transplantations (TP1 and TP2) from our previous work (Yang et al., 2016).
Data are shown as mean ± SD for (C–E). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, by two-tailed Student's t test. See also Figures S1 and S2.
Although the number of total mononuclear FL cells was unaffected, we consistently detected a striking accumulation (Figures 1D, S1B, and S1C), in absolute number and frequency, of lineage-negative (Lin−) cells and early HSPCs including long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs), restricted lineage progenitors (RLPs) (Kiel et al., 2005), and Lin−Sca1+cKit+ (LSK) cells in the Vav-Cre; Dpy30F/− FLs. Conversely, the frequency of lineage-positive (Lin+) cells was decreased (Figure S1B). DPY30 loss in FL did not affect proliferation of multipotent stem cells (LT-HSCs and ST-HSCs), but significantly reduced that of oligopotent progenitors (RLP) and Lin+ cells (Figure S1D, left). Apoptosis was unaffected in all FL cell fractions upon DPY30 loss (Figure S1D, right).
DPY30-Deficient FL HSCs Are Defective in Differentiation and Long-Term Maintenance
Compared with Vav-Cre; Dpy30F/+, the Vav-Cre; Dpy30F/− FL cells were severely defective in forming any types of colonies (Figure 1E), and in reconstituting multiple peripheral blood (PB) lineages in competitive transplantation assays, with much lower contributions to hematopoietic cells at all stages (Figures S1E–S1G). We also established a mixed FL-BM chimeras assay and induced Dpy30 excision by injection of polyinosinic-polycytidylic acid (pIpC) into recipients after stable engraftment of the donor FLs carrying Mx1-Cre (Yang et al., 2016) (Figure 1F). At 2 weeks after pIpC injection, Dpy30 deletion had mild or no effects on donor contribution to early HSPCs, but significantly reduced that to oligopotent HPCs, developing T cells in the thymus and B cells in the spleen, and mature blood cells in the PB (Figure 1G), consistent with a multilineage differentiation defect. At 12 weeks after pIpC injection, we detected a dramatic reduction in the contribution of the KO donor cells to all cell populations including HSCs (Figure 1G). Taken together, these results demonstrated an essential role of DPY30 for FL HSC differentiation and long-term maintenance, similar to its role in adult hematopoiesis (Yang et al., 2016).
Differential and Common Regulation of Gene Expression in FL and Adult BM HSCs upon Dpy30 Ablation
The largely similar phenotypes of the Dpy30 KO FL and BM HSCs suggest that DPY30 and certain DPY30 targets are fundamentally important in regulating HSCs regardless of the developmental stages. However, we found that DPY30 loss in FLs did not reduce the expression of some key regulatory genes for adult HSC differentiation and maintenance (Yang et al., 2016) (Figure S2A). As shown by RNA sequencing analysis on FL LT-HSCs (Table S2), genes downregulated upon DPY30 loss in FLs were enriched in many different functions, and those upregulated were enriched in membrane proteins and immune functions (Figure S2B). In the 275 and 577 genes downregulated over 2-fold upon DPY30 loss in FL and BM HSCs, respectively, only 21 genes were shared, with no significant enrichment of pathways (Figure S2C; Table S2). The number of shared genes increased to 95 when we used 1.5-fold cutoff, with an enrichment of nucleotide excision repair genes and mitochondrial genes (Figure S2E; Table S2). Meanwhile, in the 340 and 669 genes that were upregulated over 2-fold upon DPY30 loss in FL and BM HSCs, respectively, 41 genes were shared (Figure S2D; Table S2). The number of shared genes increased to 138 when we used 1.5-fold cutoff, with the most significant enrichment in inflammation-related genes (Figures S2D and S2F). This suggests cellular stresses upon loss of DPY30 in both FL and BM HSCs, which is also reflected by the specific and dramatic upregulation of Cdkn1a (p21) in both types of HSCs as well as BM myeloid progenitors (Figures 1H, 1I, and S2G), a gene well-known to be induced by a wide spectrum of stress stimuli including genotoxic, metabolic, and oxidative insult (Gorospe et al., 1999). Indeed, several genes involved in redox homeostasis, such as Alox5, Cybb (Nox2), and Xdh, were commonly upregulated in both types of HSCs upon DPY30 loss (Table S2). Together with the enrichment of DNA repair and mitochondrial genes being commonly downregulated upon DPY30 loss, these results prompted us to further study the effect of DPY30 loss on the metabolic state and DNA integrity of HSCs.
DPY30 Is Important for Glucose Metabolism in HSPCs through Direct Regulation of Key Glycolytic Genes
We efficiently deleted Dpy30 in various populations of hematopoietic cells (including HSCs) in the BM of Mx1-Cre; Dpy30F/− mice (Figure S3A). Consistent with metabolic stress, both immunoblotting and flow cytometry analyses showed a strong increase in phosphorylation of AMPK at Thr172, a well-established sensor and responder of low nutrient or energy (Lin and Hardie, 2018), in all different hematopoietic cell populations following DPY30 loss (Figures 2A and 2B). Moreover, phosphorylation of acetyl-CoA carboxylase (ACC) at Ser79, one of AMPK's substrates upon AMPK activation, also increased in DPY30-deficient BM (Figure 2A). These results suggest an activation of the cellular sensor of low nutrient or energy in response to DPY30 deficiency.
Figure 2.

DPY30 Deficiency Impairs Glucose Metabolism in BM HSPCs
(A) Immunoblotting in whole or Lin− BMs from pIpC-injected Mx1-Cre; Dpy30F/+ and Mx1-Cre; Dpy30F/− mice. Each lane represents an individual pool of cells. Bottom, quantification from three animals each.
(B) Representative fluorescence-activated cell sorting (FACS) analyses for phospho-AMPK (Thr172) in different cell populations in BMs from pIpC-injected Mx1-Cre; Dpy30F/+ and Mx1-Cre; Dpy30F/− mice.
(C) Relative expression of glycolytic genes, as analyzed by RNA-seq in LT-HSCs from two independent BM transplantations from previous work (Yang et al., 2016). Underlined genes are rate-limiting for glycolysis.
(D and E) Chromatin immunoprecipitation for DPY30 (D) and H3K4me3 (E) using Lin− BM cells from pIpC-injected Mx1-Cre; Dpy30F/+ and Mx1-Cre; Dpy30F/− mice. n = 3 mice each.
(F) Representative histogram showing reduced 2-NBDG uptake in vitro by sorted LSK cells from pIpC-injected Mx1-Cre; Dpy30F/+ and Mx1-Cre; Dpy30F/− mice. The gray line indicates isotype control. Each colored line represents an individual mouse out of three mice each.
(G) Scheme for the rescue assay using BMs from Mx1-Cre; Dpy30F/+ (F/+) and Mx1-Cre; Dpy30F/− (F/−) mice. Both the control and Pklr viral constructs expressed GFP.
(H) Donor contribution to indicated cell populations in the transplant recipients 2 weeks after pIpC-injections. n = 4 mice each.
Data are shown as mean ± SD for (A, D, E, and H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by two-tailed Student's t test for (A, D, and E), and by one-factor ANOVA with post hoc t test for (H). See also Figure S3.
We then examined the impact of deficiency of DPY30 and H3K4 methylation on glucose metabolism, a major constituent of cellular energy pathway. DPY30 loss in BM and FL HSCs resulted in downregulation of several genes encoding key glycolytic enzymes, including a number of rate-limiting ones, such as hexokinase (HK1 and HK2), phosphofructokinase muscle, and pyruvate kinase liver and RBC (PKLR) (Figures 2C, S3B, and S3C). DPY30 and H3K4me3 were enriched at the transcription start sites of these genes in the Lin− BM in the control mice, but both signals were significantly reduced in the KO littermates (Figures 2D and 2E). These results suggest that DPY30, likely through its activity in facilitating H3K4 methylation, directly regulates the expression of these key glycolytic genes.
Hexokinase phosphorylates glucose to form glucose 6-phosphate. This reaction is important in promoting continuous uptake of glucose into the cell, as it keeps the glucose concentration low and also prevents glucose from diffusing out of the cell due to the added charge of the reaction product. We thus predicted that downregulation of Hk1 and Hk2 would impair glucose uptake. Indeed, as measured by glucose analog (2-NBDG) uptake by sorted LSK cells, we found that glucose uptake was consistently impaired in HSPCs by DPY30 deficiency (Figure 2F). This impairment is probably not a result of reduced glucose transporter expression, because we did not find any significant downregulation of known glucose transporter genes (Table S2).
Transient and Modest Rescue of Hematopoiesis by Restoring Pklr Expression in Dpy30 KO Mice
PKLR catalyzes the transphosphorylation of phosphoenolpyruvate into ATP and pyruvate, a key intermediate in multiple metabolic pathways. Genetic alterations of PKLR are the common cause of chronic hereditary nonspherocytic hemolytic anemia (Zanella et al., 2005). To determine if the profound loss of H3K4me3 and expression of Pklr in the Dpy30 KO HSCs may contribute to the defective hematopoiesis, we attempted to rescue the in vivo HSC activity by overexpressing Pklr (Figures 2G, S3D, and S3E). Pklr overexpression did not affect the donor contribution to any hematopoietic cell populations before Dpy30 was deleted, but significantly (and partially) rescued the contribution of the Dpy30-deleted donors to early HSPCs (BM LSK) and B cells in PB 2 weeks after Dpy30 deletion (Figures 2H and S3F). However, at 12 weeks after Dpy30 deletion, we failed to observe any rescue for any cell populations (Figure S3F). These results suggest that regulation of Pklr expression by DPY30 functionally contributes to, but is far from the entirety of, the hematopoietic control by DPY30.
DPY30 Regulates Oxidative Energy Metabolism of the Stem Cells
Considering the dysregulation of genes involved in redox homeostasis, we next examined the impact of DPY30 deficiency on oxidative energy metabolism. Real-time measurement of oxygen consumption showed that DPY30 deficiency significantly reduced both the basal and the total oxidative capacity of Lin− progenitors (Figure 3A), as well as the extracellular acidification rate (Figure 3B). We also found that DPY30 deficiency in early HSPCs, including LT-HSCs, ST-HSCs, and RLPs, led to substantial reduction in mitochondrial membrane potential without affecting the mitochondrial mass or DNA level (Figures 3C–3F and S4A–S4C). Interestingly, DPY30 loss in the lineage-committed BM cells led to marked increase in mitochondrial membrane potential and mass (Figures S4A and S4B). Consistent with these results, the level of reactive oxygen species (ROS) was significantly reduced upon DPY30 loss in early HSPCs, including HSCs, but increased in more downstream cells (Figures 3F and S4C). Partially consistent with BM cells, FL cells showed significant reduction in ROS levels upon DPY30 loss in all different cell populations, including early HSPCs and Lin+ cells (Figure S4D), while primary mouse embryonic fibroblasts (MEFs) showed minimal alteration in ROS level upon DPY30 loss (Figure S4E). All these results suggest that DPY30 plays an important role in maintaining the activity of mitochondria and the oxidative energy metabolism. We found that the cellular ATP level was not significantly affected by DPY30 deficiency (Figure 3G). Whereas NADP/NADPH ratio was unaffected, NAD+/NADH ratio was modestly but significantly increased (Figure 3H), suggesting a dysregulated redox homeostasis in DPY30-deficient HSPCs.
Figure 3.

DPY30 Deficiency Impairs Mitochondrial Function and Alters Quiescence State of the BM HSCs
BMs from pIpC-injected Mx1-Cre; Dpy30F/+ (F/+) and Mx1-Cre; Dpy30F/− (F/−) mice were used in this figure.
(A and B) Oxygen consumption rate (OCR) (A) and basal extracellular acidification rates (B) of live Lin− BM cells. n = 3 mice each.
(C, E, and F) Analyses (left) and representative histograms (right) for mitochondrial membrane potential (C), mitochondrial mass (E) and ROS level (F) of indicated BM cell populations. n = 3 mice each.
(D) Mitochondrial DNA levels from BM LSK cells were determined by qPCR on mt-Co2 and normalized to Hbb for genomic DNA levels. n = 3 mice each.
(G and H) Total cellular ATP levels (G), and NAD+/NADH and NADP+/NADPH ratios (H) in BM LSK cells were determined. n = 3 mice each.
(I) Representative FACS plots of Ki-67 staining on LT-HSCs.
(J) G0, G1, and S/G2/M phases of LT-HSCs were quantified. n = 5 mice each. Data are shown as mean ± SD for all panels. n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by two-tailed Student's t test.
See also Figure S4.
To examine the functional relevance of ROS increase in the lineage-committed BM cells to pancytopenia of the DPY30-deficient animals, we administered N-acetyl-cysteine (NAC), a potent antioxidant, into these animals. We found that, while NAC only slightly but insignificantly improved the survival of KO mice (Figure S4F), it significantly (yet partially) and selectively rescued the concentration of the red blood cells in PB of the KO mice (Figure S4G). We also induced ex vivo Dpy30 deletion in isolated Lin+ BM, and found that NAC treatment in cultured BM ameliorated the apoptotic response of erythroid cells that was increased by Dpy30 deletion, but did not affect the proliferation of Lin+ cells that was reduced by Dpy30 depletion (Figure S4H). These results suggest that increase of ROS level in the lineage-committed cells functionally contributes to the reduced erythroid cell production upon DPY30 loss.
DPY30 Regulates the Quiescent State of the Stem Cells
Considering the intimate connection of HSC energy state and its cell-cycle regulation, we further studied how DPY30 deficiency might affect the HSC cell cycle. We previously showed that DPY30 deficiency did not significantly affect the proliferative capacity of HSCs or LSK cells as measured by short-time incorporation of nucleotide analog bromodeoxyuridine (Yang et al., 2016). Here we analyzed the proliferative state of the HSCs by staining of Ki-67, a nuclear protein not only strictly associated with cell proliferation (Scholzen and Gerdes, 2000), but also functionally crucial for successful cell division (Cuylen et al., 2016). DPY30 deficiency resulted in significant increase in the percentage of HSCs in G0 stage but not in the S/G2/M of the cell cycle (Figures 3I and 3J), suggesting that DPY30-deficient HSCs were in an aberrant and more deeply quiescent state of cell cycle compared with the control HSCs.
DPY30 and H3K4 Methylation Is Important for Genome Integrity of Cells
We then focused on genotoxic stress upon DPY30 loss. We found a significant increase in the γ-H2AX level in the FL and Lin− BM cells upon DPY30 loss in vivo (Figure 4A), suggesting breach of genome integrity and increase in DDR. We also directly demonstrated a significant increase in DNA breaks following DPY30 loss in early BM HSPCs using the comet assay (Figures S5A and S5B). Because the major source of DNA damage, ROS, was not increased in Dpy30 KO HSCs (Figure 3F), we sought to determine if the increase in DNA damage could be a result of poor resolution of damage in the absence of DPY30. To avoid complication by non-cell-autonomous and cumulative effects for cells derived from the KO mice, we first used primary MEFs from Dpy30F/F and CAG-CreER; Dpy30F/F mice. Treatment of the latter cells with 4-OH tamoxifen induced Dpy30 excision and led to great reduction in Dpy30 mRNA level (Figure S5C) and global H3K4 methylation (Figure S5E, and immunoblotting results shown previously [Yang et al., 2018]). A high level of nuclear γ-H2AX foci was observed in all cells at 15 min after ionizing radiation and then gradually reduced to the background level by 24 h, reflecting the process of DNA repair after damage (Figures S5D and S5E). We found that the Dpy30-deleted MEFs accumulated similar levels of DNA damage as the control MEFs at 15 min after radiation, but exhibited significantly higher levels of DNA damage than the control at 2 and 24 h after radiation (Figures S5D and S5E). We then performed similar assays on HSPCs (Figures 4B–4G, S5F, and S5G), and monitored DDR and DNA damage levels using nuclear γ-H2AX foci and comet assays (Figure 4B). Alternatively, we also irradiated the Dpy30-deleted Lin− BM cells and monitored ATM activation reflected by the level of the phosphorylated KAP1 (an ATM substrate) (Figure 4F). Compared with control HSPCs, the Dpy30-deleted HSPCs had significantly higher levels of DNA breaks (by comet assays, Figures 4E and S5G) as well as DDR (by nuclear γ-H2AX foci, Figures 4D and S5F, and phosphorylated KAP1, Figure 4G) both before and after irradiation. These results suggest that DNA repair capacity is significantly impaired in cells deficient of DPY30 and H3K4 methylation, thus leading to sustained DNA damage and DDR.
Figure 4.

DPY30 Deficiency Results in Increase in DNA Damage and Impairs DNA Repair
(A) Immunoblotting of γ-H2AX and H3 in Vav-Cre; Dpy30F/+ and Vav-Cre; Dpy30F/− FL and Lin− BM cells from pIpC-injected Mx1-Cre; Dpy30F/+ and Mx1-Cre; Dpy30F/− mice. Bottom, quantification from five animals each.
(B) Scheme for DNA damage assays and the mouse genotype legend for (C–E). LSK cells sorted from tamoxifen-treated Lin− BM were irradiated for assays at different time points. n = 3 mice each.
(C) Relative Dpy30 mRNA levels were determined by qRT-PCR and normalized to Actb.
(D) γ-H2AX foci numbers per cell from 20 to 30 LSK cells each. Representative images are in Figure S5F.
(E) Comet assay on LSK cells. We used four classes of cell morphology to show increasing DNA damage severity, and quantified (bottom) the percentages of each class before and after irradiation.
(F) Assay scheme and the mouse genotype legend for (G). Tamoxifen-treated Lin− BM were irradiated, followed by FACS assays gated on LSK cells at different time points. n = 3 mice each.
(G) FACS analyses for phosphorylated Kap-1 in LSK cells. Left, representative plots. Right, quantification based on the demarcation of the vertical line shown in the left plots.
Data are shown as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by two-tailed Student's t test. Scale bars, 10 μm. See also Figure S5.
Inhibition of DDR Partially Rescues the Function of the DPY30-Deficienct HSPCs
We first confirmed that 4-OH-tamoxifen treatment of CAG-CreER; Dpy30F/F Lin− BM cells greatly reduced their Dpy30 expression (Figure 5A) and capacity in forming all different types of colonies (Figures 5B and 5C). We then showed that, while the ATM inhibitor (KU55933) treatment had no effect on the clonogenicity of the control cells, it partially but significantly rescued the ability of the Dpy30-deleted cells in forming all different types of colonies (Figures 5C–5E). Moreover, pharmacologic inhibition of ATR and CHK1, but not CHK2, also significantly enhanced the clonogenicity of the Dpy30-deleted Lin− BM cells in a dose-dependent manner (Figure S6). These results suggest that the sustained DDR functionally affects HSPC activities.
Figure 5.

Inhibition of DDR Partially Rescues the Function of the DPY30-Deficient Hematopoietic Cells
(A–E) Lin− BM cells from CAG-CreER; Dpy30F/F mice were treated with indicated agents in liquid culture for 2 days and subjected to colony formation assays.
(A) Relative Dpy30 mRNA levels were determined by qRT-PCR and normalized to Actb. n = 3 each.
(B and C) All colonies (B) (three independent biological repeats) and different types of colonies (C) (two independent biological repeats) were quantified.
(D and E) Representative image of the plates (D) and colonies (E) are shown.
(F–H) Mx1-Cre; Dpy30F/+ or Mx1-Cre; Dpy30F/− mice were injected with pIpC and either vehicle (DMSO) or ATM inhibitor KU55933 (F), followed by determination of mitochondrial membrane potential of HSCs by FACS. The FACS plots with the gating for the effect of KU55933 in the KO mice are shown in (G) where individual mouse is represented by the individual line, and the results are quantified in (H), n = 3 for each.
Data are shown as mean ± SD for all bar graphs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by one-factor ANOVA with post hoc t test. Scale bars, 400 μm. See also Figure S6.
We also attempted to rescue the function of the DPY30-deficient HSC in vivo by suppressing DDR. To this end, we have not been able to rescue the hematopoietic defects of DPY30-deficient mice by in vivo administration of ATM inhibitor based on PB profiling. However, the mitochondrial membrane potential, which was reduced by DPY30 loss, was consistently (although not reaching statistical significance) increased by DDR suppression in the DPY30-deficient mice, although such an increase was mainly mediated by an apparently new population of cells among the phenotypic HSCs in these mice (Figures 5F–5H). These results suggest a connection of the DDR, energy metabolism, and HSPC function, all under the control of the epigenetic regulator DPY30.
Induction of p21 Is Partially Responsible for the Functional Defects of the DPY30-Deficient HSPCs
Considering the established role of P21 in regulation of cell cycle and stem cell quiescence in response to metabolic and genotoxic stresses, we next sought to determine if the specific and dramatic elevation of p21 expression played a role in HSC defect upon DPY30 loss. We generated Dpy30F/F; p21−/−; Mx1-Cre mice, which, upon pIpC injection, became Dpy30 and p21 double KO in the hematopoietic system. p21 KO had no effect on the numbers or frequencies of any hematopoietic cell populations when DPY30 was present. However, upon Dpy30 deletion, which caused severe accumulation of early HSPCs, p21 KO significantly alleviated the aberrant accumulation of phenotypic LSK cells (Figures 6A and S7A).
Figure 6.

P21 Deletion Partially Rescues the Function of the Dpy30 KO Hematopoietic Cells
(A) Absolute numbers of BM cell populations in pIpC-injected mice of indicated genotypes. n = 3 each.
(B) Scheme for the mixed chimera transplantation system using whole BM cells from Dpy30F/F; p21+/−; Mx1-Cre or Dpy30F/F; p21−/−; Mx1-Cre mice as donors and whole BM cells from wild-type mice as competitors.
(C) Representative whole BMs flushed from long bones (top) and spleens (middle) in recipient mice (that received indicated donor cells) at 2 weeks after pIpC injection. Spleen weights are quantified (bottom). n = 4 for each.
(D) Contribution of indicated donors to different cell populations in BM chimeras at 2 and 12 weeks after pIpC injection. n = 4 for each.
Data are shown as mean ± SD for all bar graphs. ∗p < 0.05, ∗∗p < 0.01, by one-factor ANOVA with post hoc t test (A) and two-tailed Student's t test (C and D). See also Figure S7.
We then transplanted donor BM from the Dpy30F/F; p21−/−; Mx1-Cre and Dpy30F/F; p21+/−; Mx1-Cre mice into irradiated recipient mice (Figure 6B). Two weeks after pIpC injection, we found that the recipients transplanted with the Dpy30F/F; p21−/−; Mx1-Cre BM had noticeably more red blood cells than those with the Dpy30F/F; p21+/−; Mx1-Cre BM (Figure 6C). Because the hematopoietic system of the irradiated recipient mice was mainly contributed by the donor BM, this result indicates that the hematopoietic functionality damaged by DPY30 deficiency was partially rescued by the compound deficiency of P21. Consistent with the improved red blood cell generation, the recipients with double KO donor BM had significantly smaller spleens than those with Dpy30-KO donor BM (Figure 6C), suggesting that P21 deficiency alleviated splenomegaly induced by DPY30 loss. Moreover, at 2 weeks after Dpy30 deletion, the double KO donors contributed significantly more than the Dpy30 KO donors to a subset of hematopoietic cell populations, including granulocyte-macrophage progenitor and common lymphoid progenitor cells in the BM, B cells in the spleen, and B cells and Gr/Mac cells in the PB. At 12 weeks after Dpy30 deletion, this effect was sustained for the splenic and PB cell populations (Figures 6D and S7B). These results support a functional role of P21 in mediating the regulation of HSPCs by DPY30.
Discussion
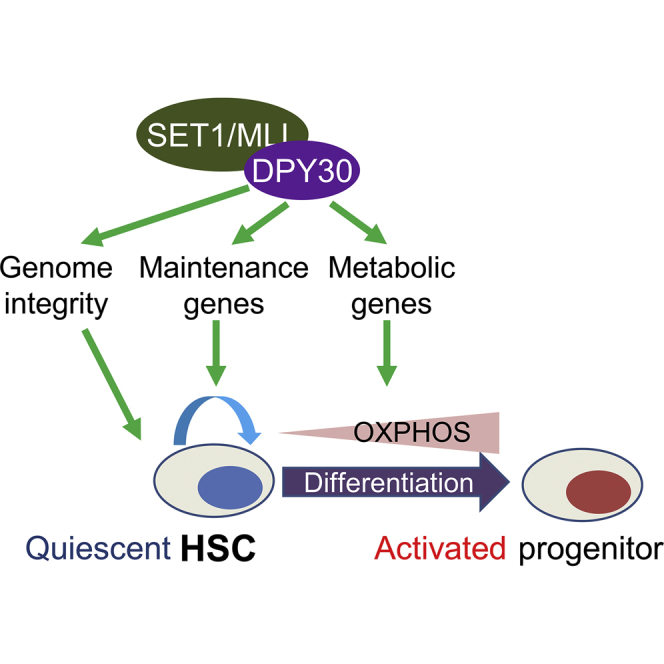
Together with our previous results (Yang et al., 2016), the data in this work support a model for the role of DPY30 in fetal and adult HSPCs through regulation of energy metabolism and genome integrity (Figure 7). In normal HSPCs, DPY30, via its activity in facilitating H3K4 methylation, ensures the HSPC functionality through multiple pathways. These pathways include expression of key regulatory genes for adult HSC maintenance (Yang et al., 2016) and likely a different set of regulatory genes for fetal HSC maintenance, expression of key metabolic genes to maintain appropriate bioenergetic pathways, and safeguarding genome integrity through promoting efficient DNA repair. In the absence of DPY30 and efficient H3K4 methylation, expression of genes in many of these pathways is dysregulated, leading to dysregulation of energy metabolism and increase in DNA damage. The impairment in glycolysis may affect HSC maintenance, and the impairment in mitochondrial function may disable HSC activation. These stresses induce responses, including P21 and other inflammatory pathways, which will suppress the activity of HSPCs and lead to attrition of stem cells in the long term. Considering that loss of SETD1A, one of the catalytic subunits responsible for the bulk level of cellular H3K4 methylation, increases the sensitivity of HSCs to inflammatory challenges (Arndt et al., 2018), we cannot formally exclude the potential complications by the inflammation-driven Dpy30 ablation by the Mx1-Cre system. However, our conclusions were supported by alternative methods of Dpy30 ablation in this and previous studies (Yang et al., 2016).
Figure 7.

A Model for the Role of DPY30 in HSPC through Regulation of Energy Metabolism and Genome Integrity
(A) DPY30 normally ensures the functionality of HSPC through guarding genome integrity and regulating key maintenance genes. It also enables HSC activation by regulating genes in energy metabolism.
(B) DPY30 loss results in dysregulation of metabolism and key maintenance gene expression, as well as breach of genome integrity. These stresses induce P21 and other responses that lead to stem cell loss and activation defect.
We note that our gene analyses for BM used cells from donor-derived cells in the transplant recipients (Yang et al., 2016), whereas those for FL here used cells in a homeostatic setting without transplant. As a result, the commonly affected genes by DPY30 deficiency in both FL and BM HSCs identified in this work are likely underestimated, but represent genes truly responsive to DPY30 loss regardless of developmental stage or homeostatic/transplant settings. These genes are likely important in mediating the effect of DPY30, but may alternatively reflect a compensatory effect following DPY30 loss and alteration of certain important biological pathways. The common upregulation of a number of enzymes in oxidative metabolism in both FL and BM HSCs following DPY30 deficiency is likely a compensatory response to the impaired mitochondrial function. The overall level of ATP was not significantly perturbed in DPY30-deficient HSCs, likely due to compensatory responses from other pathways, such as potential increase in fatty acid catabolism reflected by the increased phosphorylation (Figure 2A) and inhibition of its negative regulator, ACC (Viollet et al., 2009). However, the unaffected ATP level makes it unlikely that the HSC dysfunction is a result of reduced total cellular energy level. Rather, we favor the possibility that the altered usage of bioenergetics pathways affects the HSC functionality. DPY30 deficiency appears to lead to a deeper quiescence of the HSCs based on the cell-cycle entry. As quiescent HSCs are known to rely on glycolysis but increase oxidative phosphorylation and energy production upon activation (Kohli and Passegue, 2014, Yu et al., 2013), our results suggest that DPY30 plays an important role in enabling HSC activation by ensuring metabolic reprogramming at the chromatin level.
Several mouse models with deleted chromatin modulators (Liu et al., 2009, Santos et al., 2014, Tasdogan et al., 2016) and consequent HSPC defects exhibit increases in DDR and ROS levels, and the excessive ROS level was shown to be a major contributor to the altered HSPC activity. Consistent with an increase in ROS upon depletion of DPY30 in human fibroblast cells (Simboeck et al., 2013), our results here demonstrate that the ROS level was increased in differentiated adult cells. However, we found a marked reduction of ROS in early HSPCs in the adult stage, and in all hematopoietic cells in the prenatal stage upon depletion of DPY30 and H3K4 methylation, consistent with the reduced mitochondrial activity. Loss of the Setd1a also leads to decrease in ROS in HSCs, although the effect on mitochondrial function was not examined (Arndt et al., 2018). Our data show that the differential increase of ROS in lineage-committed cells functionally contributes to the reduced erythropoiesis in the DPY30-deficient animals, as it can be partially rescued by antioxidant treatment, but we do not know the functional impact of the abnormally low level of ROS on HSPC activity. It appeared that the DPY30-deficient HSCs in both FL and BM made an effort to increase ROS level by upregulating expression of ROS-producing enzymes, suggesting a need to maintain a certain level of ROS for normal HSC function. This is consistent with previous findings that moderate levels of intracellular ROS are needed to activate DNA repair pathway and maintain genomic stability in stem cells (Li and Marban, 2010), and also to support HSC proliferation, differentiation, and mobilization (Chaudhari et al., 2014).
The important function of DPY30 and H3K4 methylation in efficient DNA repair is likely mediated through two nonexclusive mechanisms. First, DPY30 and H3K4 methylation are important for appropriate expression of certain DNA repair genes. This echoes the finding that SETD1A loss also impairs DNA repair capacity and results in increased DNA breaks, probably due to reduced H3K4 methylation and expression of multiple DNA repair genes (Arndt et al., 2018). Interestingly, efficient DDR gene expression in leukemia cells does not require the H3K4 methylation activity, but a novel region of SETD1A that binds to cyclin K, and H3K4 methylation at the DDR genes was sustained by other methylases in the absence of SETD1A in leukemia cells (Hoshii et al., 2018). Second, H3K4 methylation may directly regulate DNA repair through impact on the chromatin setting at the damaged sites. H3K4 methylation plays an important role in efficient DNA repair in yeast (Faucher and Wellinger, 2010) and C. elegans (Herbette et al., 2017), but may also predispose chromatin for DNA double-stranded breaks through decondensing local chromatin (Burman et al., 2015). As DPY30-associated H3K4 methylation is functionally important for chromatin accessibility (Yang et al., 2018), it may regulate the efficiency of DNA repair by promoting the accessibility of DNA repair proteins. In addition to facilitating DNA repair, H3K4 methylation may also protect nascent DNA from degradation in replication stress (Higgs et al., 2018), thereby contributing to the maintenance of genome stability. Our results also suggest that it is the response to DNA damage, rather than the damage itself, that results in the inactivity of HSPCs. Although suppression of DDR can temporarily rescue the function of HSPCs, we speculate that it would have a long-term consequence on the system with uncleansed HSPCs containing damaged genetic information.
In line with P21 being important in maintaining HSC quiescence (Cheng et al., 2000), its level is uniquely (among all cyclin-dependent kinase [CDK] inhibitors) and greatly elevated in the DPY30-deficient HSCs in both FL and BM, keeping the cells in aberrantly deep quiescence regarding cell-cycle entry. Removing this quiescence maintenance factor helps release the DPY30-deficient HSCs from the deep quiescence and partially enables them to be activated and contribute to the hematopoietic reconstitution after transplant. Similar to the DDR inhibition, we speculate that such release would be mutagenic in the long term during the animals' lifespan. In another study (Shah et al., 2019), we have shown upregulation of p21 and p57 (Cdkn1c) upon DPY30 loss in the neural stem cell-enriched brain regions, suggesting that upregulation of CDK inhibitors as a general pathway in limiting the activity of tissue-specific stem and progenitor cells upon impaired epigenetic modifications following DPY30 loss. The incomplete rescue by p21 KO suggests that the metabolic and genotoxic stresses upon DPY30 loss may lead to HSPC dysfunction through other effectors. For example, mitochondrial inactivation can lead to HSC defects through metabolite imbalances and epigenetic dysregulation (Anso et al., 2017).
Our results demonstrate a profound control of HSPC fate determination by a key chromatin modulator via regulating energy metabolism and genome integrity. The functional interplay among the metabolic regulation, ROS level, DDR, and the HSPC activities warrants further studies. Considering a critical role of DPY30 in MLL-rearranged leukemogenesis (Yang et al., 2014) and MYC-driven lymphomagenesis (Yang et al., 2018), it would be of great interest to investigate whether and how inhibition of this key epigenetic modulator affects cellular metabolism and genome integrity as part of mechanisms underlying cancer suppression.
Experimental Procedures
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. All mice were maintained under specific pathogen-free conditions and housed in individually ventilated cages. Dpy30+/− mice were generated in our laboratory as reported previously (Yang et al., 2016) and were crossed to Mx1-Cre (Jackson Laboratory, JAX 003556) and Vav-Cre (Jackson Laboratory, JAX 008610) mice to produce Cre; Dpy30+/− Mice. These mice were further crossed with Dpy30F/F mice to produce Cre; Dpy30F/+ and Cre; Dpy30F/− littermates for experimental use. All transplant recipient mice were C57Bl/6J and CD45.1+, and purchased from Charles River Laboratories. P21−/− (129S2-Cdkn1atm1Tyj/J) mice (Jackson Laboratory, JAX 003263) were further crossed with Mx1-Cre and Dpy30F/F to generate the littermates of Dpy30F/F; P21+/−; Mx1-Cre and Dpy30F/F; P21−/−; Mx1-Cre.
Measurement of Oxygen Consumption, NAD+/NADH, NADP+/NADPH, and Total ATP Level
Measurement of intact cellular respiration was performed using the Seahorse XF24 analyzers. In brief, the respiration of BM Lin− cells was measured under basal conditions, in the presence of mitochondrial inhibitor oligomycin (500 nM), mitochondrial uncoupling compound carbonylcyanide-4-trifluoromethoxyphenylhydrazone (4 μM), and respiratory chain inhibitor rotenone (1 μM). Sorted BM LSK cells were suspended in PBS, and 10,000 cells were added to each well. The ratios of NAD+/NADH, NADP/NADPH, and GSH/GSSG were measured with NAD+/NADH-Glo Assay, NADP/NADPH-Glo Assay, and GSH/GSSG-Glo Assay Luminescent Cell Viability Assay kits (all from Promega, Madison, WI), respectively, according to the manufacturer's instructions. To determine total cellular ATP levels, BM LSK cells were sorted and assessed using a CellTiter-Glo Luminescent Cell Viability Assay kit (Promega).
Flow Cytometry for Analysis and Cell Isolation
Single cell suspensions were prepared from FL, BM, thymus, spleen, or PB and stained with antibodies as described previously (Yang et al., 2016).
Statistics
Unless indicated in the figure legends, the unpaired two-tailed Student's t test was used to calculate p values and evaluate the statistical significance of the difference between indicated samples. A p value < 0.05 was considered significant. Four groups comparison was analyzed by one-factor ANOVA as indicated in figure legends. If ANOVA was overall significant, post hoc t test was used for pairwise comparisons of interest.
Author Contributions
Z.Y. and H.J. conceived the project and designed the experiments. Z.Y. conducted most of the experiments and analyzed the results. K.S. conducted some of the experiments and analyzed the results. A.K.-J. conducted bioinformatic analyses. Z.Y. and H.J. wrote the paper.
Acknowledgments
We thank the Comprehensive Flow Cytometry Core (CFCC) at UAB, which are supported by NIH core grants P30 AR048311 and P30 AI027767. The X-RAD 320 irradiator was purchased by the UAB animal facility using NIH grant G20RR022807. We thank Ying Gai Tusing and Yanfang Zhao for technical assistance with mice work. This work was supported by NIH grant R01DK105531, start-up funds from the State of Alabama and University of Virginia. H.J. is a recipient of the American Society of Hematology Scholar Award, the American Cancer Society Research Scholar Award 128609-RSG-15-166-01-DMC, and the Leukemia and Lymphoma Society Scholar Award 1354-19.
Published: June 20, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.05.023.
Contributor Information
Zhenhua Yang, Email: zhenhua@hust.edu.cn.
Hao Jiang, Email: hj8d@virginia.edu.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is GEO: GSE101856.
Supplemental Information
References
- Anso E., Weinberg S.E., Diebold L.P., Thompson B.J., Malinge S., Schumacker P.T., Liu X., Zhang Y., Shao Z., Steadman M. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017;19:614–625. doi: 10.1038/ncb3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt K., Kranz A., Fohgrub J., Jolly A., Bledau A.S., Di Virgilio M., Lesche M., Dahl A., Hofer T., Stewart A.F. SETD1A protects HSCs from activation-induced functional decline in vivo. Blood. 2018;131:1311–1324. doi: 10.1182/blood-2017-09-806844. [DOI] [PubMed] [Google Scholar]
- Blanpain C., Mohrin M., Sotiropoulou P.A., Passegue E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell. 2011;8:16–29. doi: 10.1016/j.stem.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Burman B., Zhang Z.Z., Pegoraro G., Lieb J.D., Misteli T. Histone modifications predispose genome regions to breakage and translocation. Genes Dev. 2015;29:1393–1402. doi: 10.1101/gad.262170.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari P., Ye Z., Jang Y.Y. Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 2014;20:1881–1890. doi: 10.1089/ars.2012.4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Liu Y., Rappaport A.R., Kitzing T., Schultz N., Zhao Z., Shroff A.S., Dickins R.A., Vakoc C.R., Bradner J.E. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–665. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., Scadden D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Chun K.T., Li B., Dobrota E., Tate C., Lee J.H., Khan S., Haneline L., HogenEsch H., Skalnik D.G. The epigenetic regulator CXXC finger protein 1 is essential for murine hematopoiesis. PLoS One. 2014;9:e113745. doi: 10.1371/journal.pone.0113745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuylen S., Blaukopf C., Politi A.Z., Muller-Reichert T., Neumann B., Poser I., Ellenberg J., Hyman A.A., Gerlich D.W. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature. 2016;535:308–312. doi: 10.1038/nature18610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucher D., Wellinger R.J. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan T., Jude C.D., Zaffuto K., Ernst P. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24:1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorospe M., Wang X., Holbrook N.J. Functional role of p21 during the cellular response to stress. Gene Expr. 1999;7:377–385. [PMC free article] [PubMed] [Google Scholar]
- Herbette M., Mercier M.G., Michal F., Cluet D., Burny C., Yvert G., Robert V.J., Palladino F. The C. elegans SET-2/SET1 histone H3 Lys4 (H3K4) methyltransferase preserves genome stability in the germline. DNA Repair. 2017;57:139–150. doi: 10.1016/j.dnarep.2017.07.007. [DOI] [PubMed] [Google Scholar]
- Higgs M.R., Sato K., Reynolds J.J., Begum S., Bayley R., Goula A., Vernet A., Paquin K.L., Skalnik D.G., Kobayashi W. Histone methylation by SETD1A protects nascent DNA through the nucleosome chaperone activity of FANCD2. Mol. Cell. 2018;71:25–41.e6. doi: 10.1016/j.molcel.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshii T., Cifani P., Feng Z., Huang C.H., Koche R., Chen C.W., Delaney C.D., Lowe S.W., Kentsis A., Armstrong S.A. A non-catalytic function of SETD1A regulates cyclin K and the DNA damage response. Cell. 2018;172:1007–1021.e17. doi: 10.1016/j.cell.2018.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I., Nomiyama K., Hosokawa K., Sakurada K., Nakagata N. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- Jiang H., Shukla A., Wang X., Chen W.Y., Bernstein B.E., Roeder R.G. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kohli L., Passegue E. Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014;24:479–487. doi: 10.1016/j.tcb.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M., Wagers A.J., Manz M.G., Prohaska S.S., Scherer D.C., Beilhack G.F., Shizuru J.A., Weissman I.L. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu. Rev. Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- Li T.S., Marban E. Physiological levels of reactive oxygen species are required to maintain genomic stability in stem cells. Stem Cells. 2010;28:1178–1185. doi: 10.1002/stem.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.C., Hardie D.G. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299–313. doi: 10.1016/j.cmet.2017.10.009. [DOI] [PubMed] [Google Scholar]
- Liu J., Cao L., Chen J., Song S., Lee I.H., Quijano C., Liu H., Keyvanfar K., Chen H., Cao L.Y. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J., Lukas C., Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- Nijnik A., Woodbine L., Marchetti C., Dawson S., Lambe T., Liu C., Rodrigues N.P., Crockford T.L., Cabuy E., Vindigni A. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447:686–690. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- Orkin S.H., Zon L.I. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D.J., Bryder D., Seita J., Nussenzweig A., Hoeijmakers J., Weissman I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- Santos M.A., Faryabi R.B., Ergen A.V., Day A.M., Malhowski A., Canela A., Onozawa M., Lee J.E., Callen E., Gutierrez-Martinez P. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514:107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T., Gerdes J. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shah K., King G., Jiang H. A chromatin modulator sustains self-renewal and enables differentiation of postnatal neural stem and progenitor cells. J. Mol. Cell Biol. 2019 doi: 10.1093/jmcb/mjz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simboeck E., Gutierrez A., Cozzuto L., Beringer M., Caizzi L., Keyes W.M., Di Croce L. DPY30 regulates pathways in cellular senescence through ID protein expression. EMBO J. 2013;32:2217–2230. doi: 10.1038/emboj.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development. 2005;132:203–213. doi: 10.1242/dev.01558. [DOI] [PubMed] [Google Scholar]
- Tasdogan A., Kumar S., Allies G., Bausinger J., Beckel F., Hofemeister H., Mulaw M., Madan V., Scharfetter-Kochanek K., Feuring-Buske M. DNA damage-induced HSPC malfunction depends on ROS accumulation downstream of IFN-1 signaling and Bid mobilization. Cell Stem Cell. 2016;19:752–767. doi: 10.1016/j.stem.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Viollet B., Guigas B., Leclerc J., Hebrard S., Lantier L., Mounier R., Andreelli F., Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol. 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Augustin J., Chang C., Hu J., Shah K., Chang C.W., Townes T., Jiang H. The DPY30 subunit in SET1/MLL complexes regulates the proliferation and differentiation of hematopoietic progenitor cells. Blood. 2014;124:2025–2033. doi: 10.1182/blood-2014-01-549220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Augustin J., Hu J., Jiang H. Physical interactions and functional coordination between the core subunits of set1/Mll complexes and the reprogramming factors. PLoS One. 2015;10:e0145336. doi: 10.1371/journal.pone.0145336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Shah K., Khodadadi-Jamayran A., Jiang H. Dpy30 is critical for maintaining the identity and function of adult hematopoietic stem cells. J. Exp. Med. 2016;213:2349–2364. doi: 10.1084/jem.20160185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Shah K., Busby T., Giles K., Khodadadi-Jamayran A., Li W., Jiang H. Hijacking a key chromatin modulator creates epigenetic vulnerability for MYC-driven cancer. J. Clin. Invest. 2018;128:3605–3618. doi: 10.1172/JCI97072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.M., Liu X., Shen J., Jovanovic O., Pohl E.E., Gerson S.L., Finkel T., Broxmeyer H.E., Qu C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella A., Fermo E., Bianchi P., Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br. J. Haematol. 2005;130:11–25. doi: 10.1111/j.1365-2141.2005.05527.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.