

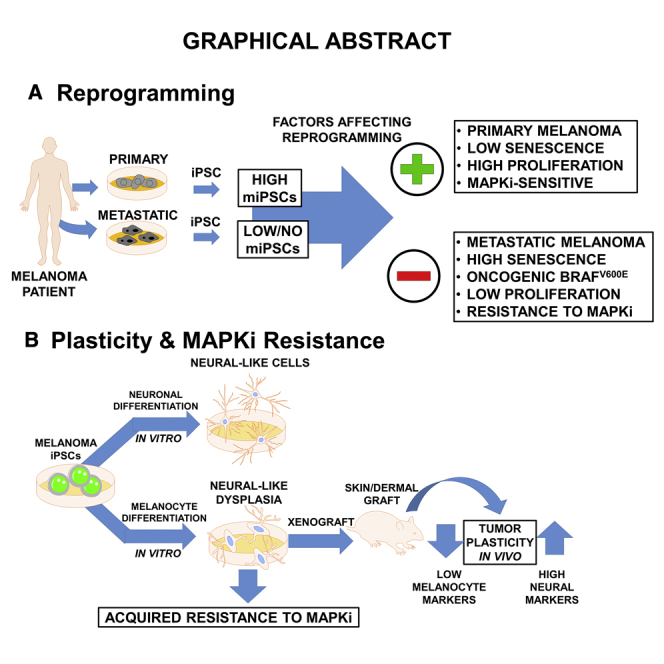
Summary
Melanomas are known to exhibit phenotypic plasticity. However, the role cellular plasticity plays in melanoma tumor progression and drug resistance is not fully understood. Here, we used reprogramming of melanocytes and melanoma cells to induced pluripotent stem cell (iPSCs) to investigate the relationship between cellular plasticity and melanoma progression and mitogen-activated protein kinase (MAPK) inhibitor resistance. We found that melanocyte reprogramming is prevented by the expression of oncogenic BRAF, and in melanoma cells harboring oncogenic BRAF and sensitive to MAPK inhibitors, reprogramming can be restored by inhibition of the activated oncogenic pathway. Our data also suggest that melanoma tumor progression acts as a barrier to reprogramming. Under conditions that promote melanocytic differentiation of fibroblast- and melanocyte-derived iPSCs, melanoma-derived iPSCs exhibited neural cell-like dysplasia and increased MAPK inhibitor resistance. These data suggest that iPSC-like reprogramming and drug resistance of differentiated cells can serve as a model to understand melanoma cell plasticity-dependent mechanisms in recurrence of aggressive drug-resistant melanoma.
Keywords: melanocytes, malignant transformation, BRAF oncogene, melanoma tumor progression, melanoma iPSC reprogramming, melanoma plasticity, melanocyte differentiation, neural-like dysplasia, MAPKi resistance
Graphical Abstract
Highlights
-
•
Metastatic melanoma exhibits less plasticity to reprogramming than primary melanoma
-
•
Oncogenic BRAFV600E and resistance to MAPKi inhibit reprogramming of melanoma
-
•
Differentiation of melanoma-iPSCs produces cells with mixed dysplastic phenotype
-
•
Melanoma-iPSC-differentiated cells exhibit acquired resistance to MAPKi
Setaluri and colleagues show that plasticity of melanocytes for reprogramming to iPSCs is inhibited by BRAFV600E oncogene and inhibition of BRAFV600E facilitates reprogramming of drug-sensitive, but not -resistant, metastatic melanoma cells. Melanoma-derived iPSCs (miPSCs) show limited pluripotency and tendency to differentiate along mixed neural and melanocyte lineage, but not to melanocytes, both in vitro and in vivo. Furthermore, miPSC-differentiated cells display acquired drug resistance.
Introduction
Melanoma is the most lethal form of skin disease. Current treatment modalities of metastatic melanoma with mitogen-activated protein kinase inhibitors (MAPKi) and immunotherapy are highly effective in the short term. Unfortunately, development of therapy resistance and recurrence of aggressive therapy-resistant tumors remains a major challenge. Resistance to MAPKi and aggressive recurrence have been associated with melanoma stem cells and stem cell pathways (Roesch et al., 2010). However, the relationship between melanoma plasticity and intrinsic and/or acquired MAPKi drug resistance are not well understood. Tumor cell-derived induced pluripotent stem cells (iPSCs) have been employed as model to study cancer cell plasticity in relation to the tissue/cell of tumor origin and differential response to therapy (Chao et al., 2017, Suknuntha et al., 2015). Attempts have been made to reprogram a limited number of melanoma cell lines into iPSCs (Bernhardt et al., 2017). However, systematic efforts to understand the plasticity of melanoma cells and their ability to generate iPSC-like cells have not been described. It is known, for instance, that cellular and molecular barriers, such as senescence and oncogenic mutations can either repress or enhance reprogramming of cells to iPSCs (Banito et al., 2009, Liu et al., 2015, Mosteiro et al., 2016). Much progress has been made in identifying mutations in melanoma that activate oncogenes, such as BRAF, NRAS, and KIT, and inactivate tumor suppressor genes, such as PTEN, CDKN2A, and TP53 (Hodis et al., 2012). The effect of these mutations on the plasticity of the malignant melanocytes and their ability to be reprogrammed is not well understood. Plasticity of cancers including melanoma to differentiate and transdifferentiate has been shown to influence tumor progression and drug sensitivity (Kemper et al., 2014, Roesch et al., 2016, Tsoi et al., 2018). Therefore, understanding the plasticity of malignant melanocytes, including their ability to generate pluripotent cells and differentiate might shed light on mechanisms of melanoma tumor progression and drug resistance. Such an approach was previously employed to understand drug resistance of chronic and acute myeloid leukemia (Chao et al., 2017, Suknuntha et al., 2015).
Here, we describe studies on reprogramming of melanocytes and primary and metastatic melanoma cells into iPSC-like cells and their ability to retain melanocytic differentiation. We show that (1) compared with skin fibroblasts and melanocytes, reprogramming of melanoma cells to iPSCs is less efficient, and metastatic melanoma cells are more resistant to reprogramming than primary melanoma cells derived from the same patient, (2) expression of BRAFV600E inhibits reprogramming of melanocytes, and inhibition of BRAFV600E facilitates reprogramming of BRAFV600E mutant, BRAF inhibitor-sensitive metastatic melanoma cells, (3) although melanoma-derived iPSCs (miPSCs) are able to differentiate into cells of the three germ layers, they failed to (re)differentiate into melanocytes, but displayed a neuronal-like dysplastic phenotype in vitro and in vivo, and (4) miPSC-differentiated cells exhibit increased resistance to MAPKi. We propose that iPSC reprogramming of melanoma cells and differentiation of miPSCs can serve as a model to understand the mechanisms of recurrence of aggressive MAPKi-resistant tumors.
Results
Melanoma Cells Exhibit Resistance to Reprogramming to iPSCs
First, we asked whether melanoma cells retain the plasticity to be reprogrammed to iPSC-like cells. To test this, we transduced four primary melanoma cell lines WM75, WM1552C, WM1361A, and WM1862, and five patient-derived early passage (less than 30 passages) metastatic melanoma cell lines MRA2, MRA4, MRA5, MRA6, and MRA9 (Table S1) with lentiviruses for reprogramming factors, OCT4-SOX2, NANOG-LIN28, and KLF4-cMYC, and cultured them in reprogramming medium on mouse embryonic fibroblast (MEF) feeders (Figure 1A). As controls, we identically performed reprogramming of human neonatal foreskin-derived fibroblasts and melanocytes. After 1 week, transduced fibroblasts and melanocytes, but not melanoma cells, produced granular colonies, which are early indicators of iPSC induction.
Figure 1.

Reprogramming of Primary and Metastatic Melanoma Cells into miPSCs
(A) Schematic of the protocol for reprogramming melanoma cells to miPSCs.
(B and C) Reprograming progress of primary (B) and metastatic (C) cells at 3 weeks and passage 1 and expression of AP at each time point. All data shown are for colonies generated using pluripotency enhancing agents.
(D) Quantification of miPSC colony formation shown as number of colonies/1,000 plated cells. Data (mean ± SD; n = 6 replicate wells for each cell line) from a representative experiment (n ≥ 3) are shown.
(E) Aggregate number of miPSC colonies generated from primary and metastatic melanoma cell lines. Pooled data for one experiment with four primary and five metastatic melanoma cell lines (n = 6 replicate wells for each cell line) are shown. Student's t test, ∗∗∗∗p < 0.0001.
(F) Reprogramming of primary and metastatic cell lines established from the same patient. Data (mean ± SD; n = 6 replicate wells for each cell line) from one experiment are shown.
(G) Western blot analysis of reprogramming factors OCT4, NANOG, and c-Myc during reprogramming: parental cells baseline, nontransduced control (C), 1 and 5 days after transduction; and miPSCs at passage 3. GAPDH shows equal loading.
We asked whether addition of chemical agents that are known to enhance reprogramming could improve the reprograming of melanoma cells (Hou et al., 2013). These chemical agents include valproic acid (VPA), an HDAC inhibitor; CHIR-99021, a GSK-3α/β inhibitor, forskolin (FSK), an activator of adenylyl cyclase/cAMP pathway, tranylcypromine (TCP), a histone lysine-specific demethylase 1 inhibitor; and RepSox, an inhibitor of the TGF-βR-1/ALK5 pathway. After an additional 2 weeks of culture containing these compounds, fibroblasts and melanocytes generated colonies (Figure S1A). At this time (3 weeks from induction), all primary melanoma cell lines also generated iPSC-like colonies without the addition of chemicals (data not shown), but the presence of chemical boosters enhanced their ability to form such colonies. In contrast, among the metastatic melanoma cell lines, only MRA2 and MRA5 generated colonies. However, neither primary nor metastatic miPSCs exhibited alkaline phosphatase (AP) expression (Figures 1B and 1C, left panels), in contrast to the strong AP activity in fibroblast- and melanocyte-derived iPSC colonies (Figure S1A). Metastatic cells did not exhibit reprogramming in the absence of chemical enhancers (data not shown). These data suggest that malignant melanocytes, specifically metastatic melanoma cells, are resistant (less plastic) to reprogramming into iPSC-like state compared with normal melanocytes.
After first passage, miPSC colonies expressed AP and were comparable with fibroblast-iPSCs (Figures 1B and 1C, right panels). We quantitated the reprogramming efficiency by counting the colonies generated after first passage (no. of colonies/1,000 cells plated). As shown in Figures 1D and 1E, primary melanoma cell lines generated significantly higher number of iPSC-like colonies than metastatic melanoma cell lines. We verified the time course expression of the reprogramming factors by western blotting. Data in Figure 1G show that melanoma cell lines, including those that did not generate iPSC colonies, showed expression of the reprogramming factors before culture on MEF, demonstrating that failure to generate iPSCs is not due to the lack of expression of the reprogramming factors.
Loss of Plasticity Is Associated with Melanoma Tumor Progression
To test whether the loss of plasticity of metastatic cells for reprogramming to stem cell state is related to melanoma tumor progression, we performed reprogramming of a set of primary and metastatic melanoma cell lines derived from the same patient. WM115 is a vertical growth phase primary melanoma cell line and WM266-4, WM165-1, and WM239A are cell lines derived from lymph node metastatic lesions in the same patient (Herlyn et al., 1985). This matched set of primary and metastatic cell lines were subjected to iPSC reprogramming (Figures 1F and S1B). After 2 weeks in passage 1, primary cell line WM115, but none of the three metastatic lines, generated iPSC-like colonies. Even addition of chemical enhancers did not improve the reprogramming of the matched metastatic cells suggesting that loss of plasticity for reprogramming to iPSC-like state is associated with melanoma tumor progression.
Senescence and Cell Death Are Inversely Related with Reprogramming
We asked which factors might be responsible for the limited plasticity of metastatic melanoma cells. Senescence and cell death has been reported to influence the efficiency of reprogramming of cells to iPSCs in vitro and in vivo (Banito et al., 2009, Mosteiro et al., 2016). We asked if senescence induction on reprogramming could be a barrier for iPSC generation by metastatic melanoma cells. We evaluated the effect of transduction with the reprogramming factors on senescence and proliferation of melanoma cells. We scanned the wells (using an EVOS FL Auto microscope) on days 1 and 5 posttransduction with the reprogramming factors, and estimated cell number and percent senescent cells (senescence-associated β-galactosidase [SA-β-gal] stained) in each well (ImageJ analysis of acquired microscope images) (Figures 2A, 2B, and S2). Data showed that metastatic melanoma cells lines MRA4 and MRA6 transduced with the reprogramming factors failed to survive, suggesting that decreased cell survival affected their reprogramming. Quantitation of SA-β-gal staining showed that there was little or no induction of senescence in most primary melanoma cells, whereas transduction with the reprogramming factors induced senescence in metastatic melanoma cells. Activation of senescence was confirmed by expression of p21 (Figures 2C and 2D), a commonly used marker to evaluate senescence during iPSC reprogramming in vitro and in vivo (Banito et al., 2009, Mosteiro et al., 2016). There was higher expression of the senescence marker p21 in metastatic than in primary cells (Figures 2C and 2D) and it remained relatively high up to 5 days. When miPSCs were generated, p21 expression was not detected in primary- or metastatic-derived miPSCs. In primary melanoma cells, p21 expression was not significantly altered on transduction. Importantly, double staining for SA-β-gal and reprogramming factor OCT4 showed that the SA-β-gal-positive senescent cells had no expression of the reprogramming factor OCT4 (Figures S2C and S2D, arrows), whereas cells with low/no SA-β-gal staining exhibited high OCT expression. These data show mutually exclusive expression of the reprogramming factors and the senescence marker, thus correlating with reprogramming efficiency.
Figure 2.

Effect of Transduction with Reprogramming Factors on Senescence and Cell Proliferation
(A and B) Primary (A) and metastatic (B) melanoma cells senescence (red lines) and survival/proliferation (green lines). Data (mean ± SD; n = 3 replicate wells/cell line for each time point) are shown. Approximately 5,000 cells/well of 24-well plates were seeded and transduced with reprogramming factor lentiviruses (day 0) and all wells were scanned using an EVOS FL Auto microscope, and cell number and percent SA-β-gal-stained cells were estimated using ImageJ analysis of the scanned images.
(C and D) Western blot analysis of p21 expression at 1 and 5 days after transduction in primary (C) and metastatic cells (D) and at miPSC stage. GAPDH shows equal loading.
Expression of Oncogenic BRAFV600E Inhibits Reprogramming
In melanocytes, mutations in BRAF lead to the activation of oncogene-induced senescence (Dhomen et al., 2009, Michaloglou et al., 2005). In addition, oncogene activation such as TP53 has been reported to act as a barrier to reprogramming to pluripotent cells (Liu et al., 2015). We noted that BRAFV600E mutant melanoma cell lines MRA5 and MRA6 showed low efficiency or no reprogramming to iPSCs (Figure 1). Therefore, we sought to evaluate the effect of expression of oncogenic BRAFV600E on melanocyte plasticity. We transduced normal human melanocytes with BRAFV600E-GFP, empty vector-GFP, or NOTCH intracellular active domain (NICD-GFP) lentiviruses followed (after 1–2 weeks) by lentiviruses for reprogramming. As shown in Figure 3, while empty vector-transduced melanocytes efficiently generated iPSC colonies, BRAFV600E-transduced melanocytes did not produce iPSC colonies even in the presence of chemical inducers. NICD-expressing melanocytes formed iPSC colonies, suggesting that the effect of BRAFV600E on inhibition of reprogramming is highly specific and not due to a nonspecific overexpression of a signaling protein.
Figure 3.

Effect of BRAFV600E Expression on Reprogramming
(A) Top panels (day 0), from left to right, show melanocytes transduced with empty vector-GFP (control), BRAFV600-GFP, or NICD-GFP lentiviruses. Middle panels (3 weeks), show colony formation in empty vector-GFP and NICD-GFP cells. Bottom panels (passage 1), show miPSC colony maturation of empty vector-GFP transduced and NCID-GFP-expressing cells. BRAFV600-GFP-expressing cells did not exhibit reprogramming.
(B) Representative images of colonies (passage 1) growing for 2 weeks in six-well plates (n = 3 replicate wells) from one experiment are shown.
Expression of Stem Cell Markers in miPSCs
To characterize the iPSC-like cells generated from melanoma cells (miPSCs), we verified the co-expression of reprogramming factors OCT4 and SOX2 with stem cell surface markers SSEA4 (stage-specific embryonic antigen-4) and CDH1 (E-cadherin) (Figures S3A–S3C). Double staining of OCT4 and SOX2 showed that expression of these factors was consistent with live-cell AP staining (Figures 1B and 1C) and expression of reprogramming factors determined by western blots (Figure 1G). An exception to this observation was WM115, which showed weak expression of OCT4 and SOX2, and also weak AP activity (Figure S1B). E-Cadherin expression was higher in WM1862- and MRA2-derived miPSCs than most cells. Primary melanoma WM115, WM1862, and metastatic MRA2-derived miPSCs had higher expression of surface marker SSE4 than miPSCs derived from other cells.
Embryoid Body Formation by miPSCs
To further characterize the stem cell features of miPSCs, we performed embryoid body (EB) formation assays by the hanging drop method for up to 3 weeks (Figure S3D). WM115-, WM1862-, and MRA2-derived miPSCs, but not WM1552C-, WM1361A-, and MRA5-derived miPSCs formed EBs. The ability to form EBs appeared to correlate with strong expression of stem cell surface markers SSEA4 and E-cadherin, which are stem cell surface markers involved in cell-cell contacts necessary for efficient EB formation (Choi et al., 2014). Accordingly, EB-forming WM1862 and MRA2 miPSCs showed strong expression of both SSEA4 and E-cadherin, similar to fibroblast-derived iPSCs, whereas WM1552C miPSCs showed weak expression of E-cadherin and SSEA4; MRA5 miPSCs, on the other hand, expressed E-cadherin but not SSEA4. An exception to this pattern was WM115 miPSCs, which, despite showing low expression of AP, expressed SSEA4 but not E-cadherin and produced small EBs. Importantly, however, the ability of melanoma-iPSCs to form EBs does not seem to be the limiting factor for their differentiation into three germ layers (Figure 4), melanocytes (Figure 5C) and neural cells (Figure S6).
Figure 4.

Differentiation of Fibroblast-iPSCs and miPSCs to Precursors of Three Germ Layers
(A) Ectoderm differentiation and expression of ectoderm marker OTX2.
(B) Mesoderm differentiation and expression of mesoderm marker BRACHYURY.
(C) Endoderm differentiation and expression of endoderm marker SOX17.
Figure 5.

Melanocyte Differentiation of Fibroblast-iPSCs, Melanocyte-iPSCs, and Melanoma-iPSCs
(A) Morphology of parental and iPSCs induced to differentiate in melanocyte medium. Top–bottom: cells differentiated from fibroblast-iPSCs, melanocyte-iPSCs, and primary melanoma-iPSCs: WM115, WM1552C, WM1361A, and WM1862; and metastatic iPSCs: MRA2 and MRA5.
(B) Cell pellets from parental melanoma and miPSC-differentiated cells.
(C) Reprogramming of human melanocytes expressing NICD-GFP, mouse fibroblasts, and BRAFV600/Pten−/– mouse tumor cells, EB formation, and melanocyte differentiation.
(D) Immunofluorescence staining of cells differentiated from fibroblast-iPSCs in melanocyte differentiation medium (top panels) and neuronal differentiation medium (bottom panels) with melanocytic markers SOX10 and MITF, and neuronal markers, TUJ1 and SYN1.
(E and F) Immunofluorescence of cells differentiated from primary WM1862 (E) and metastatic MRA5 (F) melanoma miPSCs in melanocyte differentiation medium with melanocytic marker MITF and neural markers GFAP and MAP2.
miPSCs Differentiate into Three Germ Layers
Pluripotency of miPSCs was assessed using in vitro differentiation assays and immunofluorescence staining for early markers specific for the three germ layers ectoderm, mesoderm, and endoderm (Deshpande et al., 2017, Shinozawa et al., 2017). Differentiation of miPSCs to ectoderm was verified by OTX2 expression, to mesoderm by the expression of BRACHYURY, and to endoderm by the expression of SOX17. Data in Figure 4 show that all miPSCs were able to differentiate into precursors of the three germ layers.
miPSCs Do Not Differentiate into Melanocytes but Show Neural-like Dysplasia
We next asked whether miPSCs can differentiate back to melanocyte/melanocyte-like cells. First, we validated the protocol and conditions for melanocyte differentiation using fibroblast- and melanocyte-derived iPSCs. As expected, when cultured in melanocyte differentiation medium, both fibroblast- and melanocyte-derived iPSCs generated pigmented cells (Figures 5A and 5B). Immunofluorescence staining showed that these cells differentiated from fibroblast-derived iPSCs expressed MITF and SOX10, but did not express TUJ1, the neuronal marker neuronal tubulin β-III (TUBB) (Figure 5D).
In contrast, miPSCs cultured in melanocyte differentiation medium, did not generate cells with melanocytic features, but dysplastic cells with neural-like features (Figure 5A), and co-expressed melanocyte marker MITF with neural markers TUBB3, MAP2, and GFAP (Figures 5E and 5F). Unlike fibroblast- and melanocyte-iPSC-differentiated cells in melanocyte differentiation medium, none of melanoma-iPSCs showed either melanocyte morphology or pigmentation. Although the parental metastatic melanoma MRA2 cells were pigmented, they lost pigment after iPSC reprogramming and did not recover pigmentation. Importantly, cells differentiated in melanocyte differentiation medium no longer displayed iPSC morphology or expression of reprogramming factors (Figure S4A).
To further evaluate melanocyte/neural differentiation, we determined the expression of melanocyte markers SOX10 and MITF; and neuronal markers TUBB3 and MAP2 in parental and melanoma-iPSC-differentiated (miPSC differentiated) in melanocyte differentiation medium. Interestingly, most parental melanoma cell lines and miPSC-differentiated cells expressed relatively low levels of melanocyte markers MITF and SOX10 (Figures S4B and S5A). The pigmented MRA2 parental cells showed the strongest expression of melanocyte markers, but differentiated cells exhibited weak expression of these melanocyte markers. In contrast, neural markers in parental and miPSC-differentiated cells exhibited stronger expression than melanocyte markers (Figures S4C and S5B). Cells differentiated from MRA2-miPSCs showed relatively weak expression of TUBB3 and MAP2, but expression of these markers was strong in cells with neural-like dysplastic morphology (Figures S4C and S5B, arrows).
In view of the neural-like dysplasia exhibited by cells differentiated from miPSCs, we sought to also evaluate the ability of these cells to differentiate along neuronal lineage. We performed neuronal differentiation of fibroblast- and melanocyte-derived iPSCs and found that they generated neuronal-like cells that express neuronal markers (Figures 5D and S6). When cultured in neuronal differentiation medium, melanoma-iPSCs were able to differentiate into cells with neuronal-like morphology and expressed terminal neuronal differentiation markers TUBB3, SYN1 (Synapsin 1), and MAP2 (Figure S6), whereas MRA2-derived miPSCs even expressed the glial marker GFAP (Figure S6B). These data show that neuronal-directed differentiation of fibroblast-, melanocyte-, and melanoma-iPSCs generates cells with neural-like features.
We asked if neural-like dysplastic features of melanoma cells are related to their origin from cells of the neural crest/neuro-ectodermal lineage. To address this, we transduced human melanocytes with NICD-GFP and subjected these NICD-expressing cells to iPSC reprogramming. NOTCH is a neural crest marker that plays a crucial role in the development of brain and neuronal cells, and it has been implicated in melanoma and other cancers (Pinnix et al., 2009, Raafat et al., 2004, Zagouras et al., 1995). In addition, expression of NICD has been reported to induce neural crest-like reprogramming in melanocytes (Zabierowski et al., 2011a). Results showed that NICD-expressing melanocytes formed iPSC colonies that express NICD-GFP (Figure 3). NICD-expressing iPSC colonies were then subjected to EB formation and subsequently to melanocyte differentiation. Interestingly, iPSC-expressing NICD-GFP formed melanocyte-like cells (Figure 5C, top panels). These data show that NICD expression and neural crest-like state (induced by NICD) did not block reprogramming of melanocytes or (re)differentiation along melanocytic lineage in melanocyte differentiation medium.
Melanoma cells accumulate multiple mutations that may cause dysplasia and transdifferentiation (Bhat et al., 2006, Maddodi et al., 2010). We asked if neural-like dysplasia of miPSCs might also be related with oncogenic mutations. To address this question we focused on primary melanoma cells harboring BRAFV600E/Pten−/– genotypes. We used cells isolated and cultured from BRAFV600E/Pten−/– mouse melanoma tumors (Dankort et al., 2009) to perform iPSC reprogramming and melanocyte differentiation. As a control, we also reprogrammed mouse fibroblasts into iPSCs. Results showed that both BRAFV600E/Pten−/– primary tumor cells and mouse fibroblasts were able to readily generate iPSC colonies and both cells formed EBs (Figure 5C, middle and bottom panels; Figure S7). EBs were subjected to melanocyte differentiation. Results showed that mouse fibroblast-iPSC produced pigmented cells with mouse melanocyte-like morphology. However, BRAFV600E/Pten−/– mouse tumor cell-derived iPSC-generated amelanotic cells with neural-like dysplastic morphology (Figure 5C, bottom panels). These data suggest that neural-like dysplasia of melanoma-iPSCs is related to malignant transformation and activation of BRAF and loss of Pten is sufficient to block melanocyte differentiation.
miPSC-Differentiated Cells Retain Tumorigenicity and Exhibit Neural-like Dysplasia In Vivo
Melanocytes derived from human iPSCs, when transplanted on to nude mouse skin, produce pigmented melanocytic aggregates and express melanocyte markers (Kawakami et al., 2018). We asked whether miPSC-differentiated cells in melanocyte medium (miPSC differentiated) retain tumorigenicity and whether mouse skin environment in vivo induces melanocytic differentiation. For this, we selected primary WM1862 and metastatic MRA2 cell line-derived miPSCs differentiated in melanocyte medium. Both primary WM1862 and metastatic MRA2 miPSC-differentiated cells formed amelanotic tumors (Figure S5C). Western blot analysis of tumor lysates for melanocyte differentiation markers (MITF, SOX10, TYR, TYRP1, and TYRP2), neural/neural crest markers (NOTCH/NICD, PAX3, TUBB3, MAP2, and GFAP), and melanoma stem cell markers (SOX9, ALDH1, and CD271) showed a pattern of expression consistent with that observed by immunofluorescence analyses. We compared the expression of these markers in parental melanoma cells, miPSC-differentiated cells in vitro and tumors derived in vivo from miPSC-differentiated cells. Parental primary melanoma cell line WM1862 and WM1862-miPSC-differentiated cells showed weak or no expression of melanocyte markers in vitro, but a weak induction of these markers was noted in tumors in vivo (Figure S5D). Parental metastatic melanoma MRA2 cells showed strong expression of melanocyte markers TYR, TYRP1, TYRP2, and MART1 but weak expression of MITF and SOX10. However, the expression of a subset of these markers was extinguished in MRA2-miPSC-differentiated cells both in vitro and in vivo. These data suggest that miPSC-differentiated cells fail to express melanocyte markers, even when present in the cutaneous environment.
Both primary WM1862 and metastatic MRA2 cells showed a highly similar pattern of expression of neural markers. NICD was present in WM1862 parental and WM1862-miPSC-differentiated cells and MRA2-miPSC-differentiated cells. PAX3 was present in tumors produced by both cell lines. Both cell lines retained the neuronal markers TUBB3 and MAP2 under all conditions. In MRA2 cells, TUBB3 was detected in parental pigmented cells and the miPSC-differentiated cells in vivo (tumor) but not in vitro (Figure S5B). GFAP expression was induced in tumors derived from both WM1862 and MRA2 miPSC-differentiated cells.
Skin stem cell marker SOX9 and melanoma stem cell markers ALDH1 and CD271 were present only in vivo (tumors). These data suggest that miPSC-derived cells display mixed dysplastic features of neural crest-, neural-, melanocyte-, and melanoma stem cell-like differentiation. However, while weak expression of melanocyte markers is retained, strong expression of neural crest, neural, and melanoma/stem cell markers are retained throughout the reprogramming and differentiation.
miPSC-Differentiated Cells Exhibit Acquired Resistance to MAPK Inhibition
Next, we asked whether BRAFV600E mutant miPSC-differentiated cells retain sensitivity to BRAFi PLX4032/vemurafenib and MEKi AZD6244/selumetinib. Based on the half maximal inhibitory concentration of the parental metastatic MRA5 and MRA6 melanoma cell lines (Rodríguez et al., 2017), we evaluated whether miPSC-differentiated cells are also sensitive to concentration of MAPKi inhibitors that kill parental cells. As shown in Figure 6, both primary and metastatic melanoma parental cells were sensitive to MEKi AZD6244 and parental cells with BRAFV600E mutation were sensitive to killing by PLX4032. However, miPSC differentiated in melanocyte medium (miPSC differentiated) showed resistance to killing. MRA5 is intrinsically resistant to both drugs and MRA5-miPSC-differentiated cells remain resistant to both inhibitors similar to the parental MRA5 cells.
Figure 6.

Acquired MAPKi Resistance in Melanoma-iPSC Differentiated Cells
Melanoma cells (parental) were reprogrammed into melanoma-iPSCs (miPSCs) and embryoid bodies generated and allowed to differentiate in melanocyte medium (miPSC differentiated). The sensitivity of parental melanoma cells and miPSC-differentiated cells to MAPKi (at previously determined half maximal inhibitory concentrations) was evaluated. Survival of each parental melanoma cell line and miPSC-differentiated cells treated with DMSO (D), PLX4032 (P) (0.5 μM), or AZD6244 (A) (0.5 μM) 72 h was estimated by MTT assay. Data (mean ± SD; n = 6–8 replicate wells/cell line) from one experiment are shown. Employing two-factor linear regression (factors cell line and parental versus induced), we tested contrasts between parental and induced cells for each drug and cell line. ∗∗p < 10−4 for all contrasts, except for MRA5 (ns, not significant).
Resistance to MAPK Inhibitors Correlates with Loss of Plasticity for Reprogramming
We investigated whether MAPKi resistance is related to reprogramming to the iPSC-like state. We selected two BRAFV600E melanoma cell lines––MRA5 shows intrinsic resistance to both BRAFi and MEKi, and MRA6 is sensitive to both drugs. We also generated two MAPKi-resistant cell lines from MRA6: MRA6BR and MRA6MR, which are resistant, respectively, to BRAFi and MEKi (Rodríguez et al., 2017). Data showed that addition of BRAFi during reprogramming significantly increased the efficiency of reprogramming of BRAFi-sensitive MRA6 cells to iPSC-like cells (Figures 7A–7C). In contrast, intrinsically MAPKi-resistant MRA5 and MRA6BR cells that acquired resistance to BRAFi did not yield iPSC colonies even in the presence of BRAFi. Although MEKi also enhanced the plasticity of MAR6MR cells into stem cell state, the colonies did not exhibit iPSC-like morphology or express the stem cell marker AP and did not survive beyond passage 2. These results suggest that treatment of BRAFV600E mutant melanoma with BRAFi can enhance their plasticity to activate pluripotent stem cell-like state, whereas BRAFV600E mutant melanomas that are intrinsically resistant or have acquired resistance to BRAFi are refractory to the effects of BRAFi on their plasticity. MRA6-derived miPSC-differentiated cells in melanocyte differentiation medium did not generate melanocytes and showed acquired resistance to both BRAFi and MEKi, similar to MRA6BR and MRA6MR cells, respectively (Figure 7D).
Figure 7.

Resistance to MAPKi Inhibits Reprogramming of BRAFV600E Mutant Cells
(A) Morphology of BRAF mutant cells during reprogramming. Intrinsically MAPKi-resistant MRA5, MAPKi-sensitive MRA6, cells with acquired resistance to BRAFi (MRA6BR), and MEKi (MRA6MR) at 3 weeks post-iPSC induction and at passage 1 in the absence of MAPKi.
(B) Reprogramming in the absence or presence of BRAFi (for MRA5, MRA6, and MRA6BR) and MEKi (for MRA6MR).
(C) Quantitation of iPSC reprogramming shows number of colonies generated. Data (mean ± SD; n = 6 replicate well/cell line) from one experiment are shown.
(D) miPSC-differentiated cells from MAPKi-sensitive MRA6 cells show acquired resistance to BRAFi and MEKi. Survival (MTT assays) of MRA6 (MAPKi sensitive) parental melanoma cells, MRA6-miPSC-differentiated cells in melanocyte medium, MRA6BR and MRA6MR treated with DMSO (D), PLX4032 (P, 0.5 μM), or AZD6244 (A, 0.5 μM) for 72 h. Data (mean ± SD; n = 6–8 replicate wells/cell line) shown are from one experiment. Student's t test for parental versus differentiated (D versus D, ns, not significant; P versus P, ∗∗∗p = 0.0006; A versus A, ∗∗∗p = 0.0010). No significant difference was noted in MRA6-miPSC-differentiated cells compared with MRA6BR and MRA6MR cell lines.
(E and F) RNA sequencing differential expression data for genes in BRAFV600E mutant MAPKi-resistant (MRA5, MRA6BR, and MRA6MR) and MAPKi-sensitive (MRA6) cells. (E) MAPK Cascade (GO: 0000165) 177 genes with highest marginal variance. (F) Stem Cell Proliferation (GO: 0072089) top 24 genes with highest marginal variance.
Acquired resistance to MAPKi and recurrence of highly aggressive melanoma are known to be associated with stem cell pathways (Roesch et al., 2010). We asked whether the difference in plasticity (to reprogramming to the iPSC-like state) between MAPKi-sensitive and -resistant cells could be related to differences in the elaboration of oncogenic MAPK and stem cell pathway gene expression programs. Whole transcriptome RNA sequencing analysis showed that gene expression profiles for MAPKi-resistant and MAPKi-sensitive cells were distinct. Figures 7E and 7F show that stem cell proliferation and MAPK cascade genes are the most significantly differentially expressed genes. MAPKi-sensitive MRA6 cells show higher expression levels of stem cell proliferation genes including SOX10, HHIP, ID4, and LEF1, genes related to growth and development including TGFA, GFRA1, and FGF13 compared with MAPKi-resistant cells (Figures 7E and 7F). These data suggest that oncogenic MAPK signaling restricts the plasticity of melanoma cells (for iPSC-like reprogramming) and that this inhibitory effect can be overcome by inhibition of the MAPK pathway.
Discussion
In this study we investigated the plasticity of melanocytes and melanoma cells for reprogramming to iPSCs and differentiation into melanocytes. We show that expression of oncogenic BRAFV600E inhibits the plasticity of melanocytes and that pharmacological inhibition of BRAFV600E promotes the reprogramming of melanoma cells. These observations are consistent with previous reports that showed that oncogene c-Jun inhibited reprogramming of somatic cells (Liu et al., 2015).
Limited plasticity of melanoma cells to iPSC reprogramming appears to correlate with tumor progression. Reprogramming of genetically matched primary and metastatic melanoma cells from the same patient showed that metastatic cells are more resistant to reprogramming, suggesting that accumulated mutations in melanoma cells during tumor progression inhibit plasticity (Bozic et al., 2010). We noted that metastatic cells refractory to reprogramming also exhibited high senescence and limited survival and proliferation on iPSC reprogramming, consistent with previous reports that showed that senescence and proliferation affected reprogramming in vitro an in vivo (Banito et al., 2009, Mosteiro et al., 2016). A limitation of these studies is that these cell lines used were propagated in culture. However, freshly isolated cells from primary melanoma tumors are often difficult to acquire due to the size of the excised lesions and medico-legal requirements of diagnostic pathology.
miPSCs exhibited stem cell-like features including expression of stem cell markers and ability to differentiate into three germ layers. More interestingly, miPSCs were more amenable to differentiate along neural dysplastic lineages than melanocytic lineage both in vitro and in vivo. Dedifferentiation is thought to be a hallmark of melanocytic neoplasms, which often resemble various stages of their embryonic development. Dedifferentiation also appears to influence the sensitivity of melanocytic neoplasms to drugs (Tsoi et al., 2018). Cutaneous melanocytic neoplasms are also known to acquire variable characteristics of neural and other neural crest derivatives (Fang et al., 2001, Iyengar and Singh, 2010). Skin stem cells also express neural crest markers and exhibit similarities with melanoma cells including self-renewal and differentiation into multiple neural crest lineages (Zabierowski et al., 2011b).
Expression of active NOTCH (NICD) was shown to be sufficient to reprogram melanocytes into neural crest-like state (Zabierowski et al., 2011a). Our data show that expression of NICD did not block iPSC reprogramming of melanocytes and their ability to redifferentiate to melanocytes. These data suggest that lack of melanocyte differentiation and patterns of neural crest- and neural-like dysplasia of melanoma-iPSCs could not be explained by their origin from neural crest lineage but may be related to de-differentiation/transdifferentiation of melanocytes during malignant transformation. Resetting of melanoma cells by iPSC reprogramming, however, does not restore the melanocytic program disrupted by oncogenic mutations but generates neural-like dysplasia. Consistent with this notion, we previously reported that expression of oncogenic BRAFV600E induces the expression of neuronal marker MAP2 in melanoma cells (Bhat et al., 2006, Maddodi et al., 2010). Another possibility, however, is that different oncogenic mutations might induce different patterns of plasticity such as neuronal, glial, or neural crest lineages. For example, neuronal differentiation from MRA2-miPSCs, which harbor wild-type BRAF but loss of PTEN showed neural-like dysplasia with strong expression of glial cell marker GFAP in neural-directed differentiation. Intriguingly, glial cell precursors from cutaneous innervation were reported to serve as a source of melanocytes in the skin (Adameyko et al., 2009).
Furthermore, the different patterns of lineage marker expression of miPSC-differentiated cells could also be related to clinical and histological subtypes of cutaneous melanoma. Detailed clinical and histopathological studies have grouped melanocytic neoplasms into distinct categories based on association with chronic sun damage and their anatomical location; however, it is not known whether these histologically distinct lesions all arise from melanocytes at the same stage in the melanocytic lineage differentiation, i.e., skin-resident precursors with neural crest-like features, melanocyte stem cells, melanoblasts, or terminally differentiated melanocyte (Grichnik et al., 2014, Kulesa et al., 2006, Yu et al., 2010). Recent studies employing BRAFV600E models of mouse melanoma showed that UVB-induced melanocytic neoplasms can originate in the hair-bearing skin from melanocyte stem cells (Moon et al., 2017), whereas non-UV-induced lesions arise in the interfollicular tail from mature differentiated pigment producing melanocytes, but not from dormant amelanotic melanocytes or melanocyte stem cells from the hair follicle bulge (Köhler et al., 2017). These in vivo mouse models, although elegant and powerful, have limitations when extrapolated to human melanocytic neoplasms. For example, they are limited to BRAFV600E-driven melanomas and do not fully recapitulate the characteristic subtypes and spectrum of histological presentations in human melanomas. In this context, our observations on the differences in plasticity of primary and metastatic human melanoma cells suggest iPSC reprogramming of melanoma cells is a useful model to understand the origin and progression of melanocytic neoplasms. It is of interest to investigate whether the plasticity of miPSCs to differentiate to neural-like dysplastic cells reflects the cell of origin, i.e., differentiated melanocytes and melanocytic stem cells versus precursors of neural crest-derived melanocytic precursors (Yu et al., 2010).
Resistance to BRAFi and MEKi appears to limit the plasticity of melanoma cells into stem cell state. Although cells sensitive to BRAFi expressed higher levels of stem cell proliferation and growth/development-related genes than MAPKi-resistant cells, constitutive activation of the MAPK pathway appears to block reprogramming of MAPKi-sensitive cells, whereas plasticity for reprogramming is unlocked on inhibition of oncogenic BRAF. However, once cells acquire MAPKi resistance, expression of stem cell marker expression is diminished and the cells are less susceptible to reprogramming. BRAFi did not restore plasticity in the MAPKi-resistant cells. In addition, cells differentiated from melanoma-iPSCs in melanocyte differentiation medium exhibited decreased sensitivity to BRAFi and MEKi. Our results are consistent with published data (Bernhardt et al., 2017) and suggest that miPSC-differentiated cells are less dependent on MAPK signaling for survival and proliferation.
Reprogramming of melanoma cells into a stem cell state facilitated by BRAFi (PLX4032) in cells sensitive to BRAFi could serve as a novel strategy to uncover mechanisms of acquired resistance related to cellular plasticity. After reprogramming and differentiation in melanocyte differentiation medium, sensitive cells become resistance to BRAFi. BRAFi-sensitive cells exhibit a stem cell expression signature with oncogenic MAPK cascade, but are not able to reprogram unless BRAFi is added during reprogramming. We suggest that, in patients with melanoma, BRAFi might modulate the expression of stem cell genes involved in melanoma plasticity, transdifferentiation, and acquired resistance through stem cell pathways, and such genes might be targets for overcoming therapy resistance.
Experimental Procedures
Cell Culture
Primary fibroblasts and melanocytes were isolated from human neonatal foreskins in the UW-Skin Disease Research Center. All primary and metastatic cells of the WM series were obtained from Rockland Immunochemicals (Limerick, PA). MRA series of metastatic melanoma cell lines were established and genotyped for BRAF and RAs mutations at UW-Madison by Dr. Mark Albertini. Human primary fibroblasts, all melanoma cells, and mouse tumor cells were cultured in DMEM, 10% fetal bovine serum (FBS), and 1% penicillin and streptomycin (PenStrep). Human melanocytes were cultured in 254 basal media containing HMGS2. MEFs were obtained from WiCell Research Institute and plated following their protocol recommendations. All cells were cultured in a humid incubator at 37°C with 5% CO2 and regularly tested for mycoplasma. Isolation and use of human cells was approved by appropriate institutional review committees.
Reprogramming of iPSCs and AP Live Staining
Five days after… transduction with three reprogramming lentiviruses, cells were plated on six-well plates with MEF feeders (WiCell Research Institute) with stem cell reprogramming medium (KO DMEM, 20% KOSR, 1% GlutaMAX, 1% NEAA, 1% PenStrep, 10 ng/mL basic fibroblast growth factor [bFGF], 2 × 10−4 M of 2-mercaptoethanol) at a density of 2 × 104 cells per well of six-well plates. A cocktail of up to five chemicals was used for reprogramming melanoma cells including FSK (10 μM), VPA (500 μM), CHIR99021 (10 μM), RepSox (5 μM), and TCP (5 μM). Medium was replaced every 3–4 days for 3 weeks. After 3 weeks, colonies were passaged using Versene on fresh MEFs every 2 weeks with maintenance stem cell medium, which is the same as reprogramming but supplemented with 4 ng/mL bFGF, CHIR99021 (3 μM), and PD0325901 (1 μM). AP live staining (Thermo Fisher Scientific, no. A14353) was performed after 3 weeks of reprogramming and after 2 weeks in passage 1 according to the manufacturer's protocol.
EB Formation, and Melanocyte and Neuronal Differentiation
EBs and melanocyte differentiation were performed as reported (Ohta et al., 2011, Yang et al., 2011) with modifications. Neuronal differentiation was performed essentially as reported (Bernhardt et al., 2017) (Supplemental Experimental Procedures).
MTT Assays
miPSC-differentiated cells were first expanded in DMEM, 10% FBS, and 1% PenStrep for 2 weeks and passed at least 5 times. For MTT assays, 4,000 cells/well were plated on 96-well plates with DMEM, 10% FBS, and 1% PenStrep and incubated at 37°C overnight, and, the next day, medium was replaced with medium containing 0.5 μM of PLX4032 or AZD6244 inhibitors for 72 h. The absorbance was read at 540 nm after adding 20 μL of a 5-mg/mL MTT dye solution at 37°C for 45 min.
RNA Sequencing and Transcriptome Analysis
Cells were plated on six-well plates and, after 24 h, RNA samples were collected and purified using the miRNeasy Mini Kit (no. 217004, QIAGEN). Samples were then sent for sequencing at Sanford Burnham Prebys Medical Discovery Institute, Orlando, FL. Transcriptome data were deposited at NCBI (accession no. GEO: GSE110179). Data were analyzed as described in Supplemental Experimental Procedures using RSEM (Li et al., 2010) for abundance estimation and EBSeq (Leng et al., 2013) for differential expression analysis.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 7 and R. Student's unpaired t test was performed for significance studies with a 95% confidence interval (p < 0.05). For the data on drug resistance of miPSCs, we employed two-factor linear regression (factors cell line and parental versus induced) and we tested contrasts between parental and induced cells for each drug and cell line.
Author Contributions
V.S. and E.C.-P. designed the study. E.C.-P performed all the experiments. C.I.R. characterized the MRA cell lines and generated MAPKi-resistant cells. D.M. produced BRAF(V600E) and NICD lentiviruses and contributed to mouse melanoma cell line experiments. S.S. and A.M. assisted E.C.-P. with cell culture experiments, microscopy, and ImageJ. M.A.N. performed statistical analyses. V.S. and E.C.-P. wrote the manuscript with input from M.A.N.
Acknowledgments
This work was supported in part by Department of Defense grant W81XWH-15-1-0529 and VA Merit Award 1 I01 BX002623 and NIH/NIAMS grant P30AR066524 to V.S. M.A.N. was supported in part by the Biostatistics Shared Resource of the UWCCC, P30CA014520-41. We thank Dr. Igor Slukvin for critical reading of the manuscript and comments and suggestions.
Published: June 20, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.05.018.
Supplemental Information
References
- Adameyko I., Lallemend F., Aquino J.B., Pereira J.A., Topilko P., Müller T., Fritz N., Beljajeva A., Mochii M., Liste I. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139:366–379. doi: 10.1016/j.cell.2009.07.049. [DOI] [PubMed] [Google Scholar]
- Banito A., Rashid S.T., Acosta J.C., Li S., Pereira C.F., Geti I., Pinho S., Silva J.C., Azuara V., Walsh M. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–2139. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt M., Novak D., Assenov Y., Orouji E., Knappe N., Weina K., Reith M., Larribere L., Gebhardt C., Plass C. Melanoma-derived iPCCs show differential tumorigenicity and therapy response. Stem Cell Reports. 2017;8:1379–1391. doi: 10.1016/j.stemcr.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K.M., Maddodi N., Shashikant C., Setaluri V. Transcriptional regulation of human MAP2 gene in melanoma: role of neuronal bHLH factors and Notch1 signaling. Nucleic Acids Res. 2006;34:3819–3832. doi: 10.1093/nar/gkl476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic I., Antal T., Ohtsuki H., Carter H., Kim D., Chen S., Karchin R., Kinzler K.W., Vogelstein B., Nowak M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. U S A. 2010;107:18545–18550. doi: 10.1073/pnas.1010978107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M.P., Gentles A.J., Chatterjee S., Lan F., Reinisch A., Corces M.R., Xavy S., Shen J., Haag D., Chanda S. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell. 2017;20:329–344.e7. doi: 10.1016/j.stem.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.S., Kim W.T., Ryu C.J. Antibody approaches to prepare clinically transplantable cells from human embryonic stem cells: identification of human embryonic stem cell surface markers by monoclonal antibodies. Biotechnol. J. 2014;9:915–920. doi: 10.1002/biot.201300495. [DOI] [PubMed] [Google Scholar]
- Dankort D., Curley D.P., Cartlidge R.A., Nelson B., Karnezis A.N., Damsky W.E., You M.J., DePinho R.A., McMahon M., Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A., Yadav S., Dao D.Q., Wu Z.Y., Hokanson K.C., Cahill M.K., Wiita A.P., Jan Y.N., Ullian E.M., Weiss L.A. Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep. 2017;21:2678–2687. doi: 10.1016/j.celrep.2017.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen N., Reis-Filho J.S., da Rocha Dias S., Hayward R., Savage K., Delmas V., Larue L., Pritchard C., Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Fang D., Hallman J., Sangha N., Kute T.E., Hammarback J.A., White W.L., Setaluri V. Expression of microtubule-associated protein 2 in benign and malignant melanocytes: implications for differentiation and progression of cutaneous melanoma. Am. J. Pathol. 2001;158:2107–2115. doi: 10.1016/S0002-9440(10)64682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grichnik J.M., Ross A.L., Schneider S.L., Sanchez M.I., Eller M.S., Hatzistergos K.E. How, and from which cell sources, do nevi really develop? Exp. Dermatol. 2014;23:310–313. doi: 10.1111/exd.12363. [DOI] [PubMed] [Google Scholar]
- Herlyn M., Balaban G., Bennicelli J., Guerry D., Halaban R., Herlyn D., Elder D.E., Maul G.G., Steplewski Z., Nowell P.C. Primary melanoma cells of the vertical growth phase: similarities to metastatic cells. J. Natl. Cancer Inst. 1985;74:283–289. [PubMed] [Google Scholar]
- Hodis E., Watson I.R., Kryukov G.V., Arold S.T., Imielinski M., Theurillat J.P., Nickerson E., Auclair D., Li L., Place C. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P., Li Y., Zhang X., Liu C., Guan J., Li H., Zhao T., Ye J., Yang W., Liu K. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- Iyengar B., Singh A.V. Patterns of neural differentiation in melanomas. J. Biomed. Sci. 2010;17:87. doi: 10.1186/1423-0127-17-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T., Okano T., Takeuchi S., Osumi K., Soma Y., Itoh M., Hirobe T., Jimbow K. Approach for the derivation of melanocytes from induced pluripotent stem cells. J. Invest. Dermatol. 2018;138:150–158. doi: 10.1016/j.jid.2017.07.849. [DOI] [PubMed] [Google Scholar]
- Kemper K., de Goeje P.L., Peeper D.S., van Amerongen R. Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. 2014;74:5937–5941. doi: 10.1158/0008-5472.CAN-14-1174. [DOI] [PubMed] [Google Scholar]
- Köhler C., Nittner D., Rambow F., Radaelli E., Stanchi F., Vandamme N., Baggiolini A., Sommer L., Berx G., van den Oord J.J. Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell. 2017;21:679–693.e6. doi: 10.1016/j.stem.2017.08.003. [DOI] [PubMed] [Google Scholar]
- Kulesa P.M., Kasemeier-Kulesa J.C., Teddy J.M., Margaryan N.V., Seftor E.A., Seftor R.E., Hendrix M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. U S A. 2006;103:3752–3757. doi: 10.1073/pnas.0506977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M., Haag J.D., Gould M.N., Stewart R.M., Kendziorski C. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Ruotti V., Stewart R.M., Thomson J.A., Dewey C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Han Q., Peng T., Peng M., Wei B., Li D., Wang X., Yu S., Yang J., Cao S. The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol. 2015;17:856–867. doi: 10.1038/ncb3193. [DOI] [PubMed] [Google Scholar]
- Maddodi N., Bhat K.M., Devi S., Zhang S.C., Setaluri V. Oncogenic BRAFV600E induces expression of neuronal differentiation marker MAP2 in melanoma cells by promoter demethylation and down-regulation of transcription repressor HES1. J. Biol. Chem. 2010;285:242–254. doi: 10.1074/jbc.M109.068668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C., Vredeveld L.C., Soengas M.S., Denoyelle C., Kuilman T., van der Horst C.M., Majoor D.M., Shay J.W., Mooi W.J., Peeper D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Moon H., Donahue L.R., Choi E., Scumpia P.O., Lowry W.E., Grenier J.K., Zhu J., White A.C. Melanocyte stem cell activation and translocation initiate cutaneous melanoma in response to UV exposure. Cell Stem Cell. 2017;21:665–678.e6. doi: 10.1016/j.stem.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosteiro L., Pantoja C., Alcazar N., Marión R.M., Chondronasiou D., Rovira M., Fernandez-Marcos P.J., Muñoz-Martin M., Blanco-Aparicio C., Pastor J. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354 doi: 10.1126/science.aaf4445. [DOI] [PubMed] [Google Scholar]
- Ohta S., Imaizumi Y., Okada Y., Akamatsu W., Kuwahara R., Ohyama M., Amagai M., Matsuzaki Y., Yamanaka S., Okano H. Generation of human melanocytes from induced pluripotent stem cells. PLoS One. 2011;6:e16182. doi: 10.1371/journal.pone.0016182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnix C.C., Lee J.T., Liu Z.J., McDaid R., Balint K., Beverly L.J., Brafford P.A., Xiao M., Himes B., Zabierowski S.E. Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res. 2009;69:5312–5320. doi: 10.1158/0008-5472.CAN-08-3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raafat A., Bargo S., Anver M.R., Callahan R. Mammary development and tumorigenesis in mice expressing a truncated human Notch4/Int3 intracellular domain (h-Int3sh) Oncogene. 2004;23:9401–9407. doi: 10.1038/sj.onc.1208187. [DOI] [PubMed] [Google Scholar]
- Rodríguez C.I., Castro-Pérez E., Prabhakar K., Block L., Longley B.J., Wisinski J.A., Kimple M.E., Setaluri V. EPAC-RAP1 axis-mediated switch in the response of primary and metastatic melanoma to cyclic AMP. Mol. Cancer Res. 2017;15:1792–1802. doi: 10.1158/1541-7786.MCR-17-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A., Fukunaga-Kalabis M., Schmidt E.C., Zabierowski S.E., Brafford P.A., Vultur A., Basu D., Gimotty P., Vogt T., Herlyn M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A., Paschen A., Landsberg J., Helfrich I., Becker J.C., Schadendorf D. Phenotypic tumour cell plasticity as a resistance mechanism and therapeutic target in melanoma. Eur. J. Cancer. 2016;59:109–112. doi: 10.1016/j.ejca.2016.02.023. [DOI] [PubMed] [Google Scholar]
- Shinozawa T., Nakamura K., Shoji M., Morita M., Kimura M., Furukawa H., Ueda H., Shiramoto M., Matsuguma K., Kaji Y. Recapitulation of clinical individual susceptibility to drug-induced QT prolongation in healthy subjects using iPSC-derived cardiomyocytes. Stem Cell Reports. 2017;8:226–234. doi: 10.1016/j.stemcr.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suknuntha K., Ishii Y., Tao L., Hu K., McIntosh B.E., Yang D., Swanson S., Stewart R., Wang J.Y., Thomson J. Discovery of survival factor for primitive chronic myeloid leukemia cells using induced pluripotent stem cells. Stem Cell Res. 2015;15:678–693. doi: 10.1016/j.scr.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J., Robert L., Paraiso K., Galvan C., Sheu K.M., Lay J., Wong D.J.L., Atefi M., Shirazi R., Wang X. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33:890–904.e5. doi: 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R., Jiang M., Kumar S.M., Xu T., Wang F., Xiang L., Xu X. Generation of melanocytes from induced pluripotent stem cells. J. Invest. Dermatol. 2011;131:2458–2466. doi: 10.1038/jid.2011.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Kumar S.M., Kossenkov A.V., Showe L., Xu X. Stem cells with neural crest characteristics derived from the bulge region of cultured human hair follicles. J. Invest. Dermatol. 2010;130:1227–1236. doi: 10.1038/jid.2009.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabierowski S.E., Baubet V., Himes B., Li L., Fukunaga-Kalabis M., Patel S., McDaid R., Guerra M., Gimotty P., Dahmane N. Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells. 2011;29:1752–1762. doi: 10.1002/stem.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabierowski S.E., Fukunaga-Kalabis M., Li L., Herlyn M. Dermis-derived stem cells: a source of epidermal melanocytes and melanoma? Pigment Cell Melanoma Res. 2011;24:422–429. doi: 10.1111/j.1755-148X.2011.00847.x. [DOI] [PubMed] [Google Scholar]
- Zagouras P., Stifani S., Blaumueller C.M., Carcangiu M.L., Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc. Natl. Acad. Sci. U S A. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.