

Abstract
TGF‐β controls a variety of cellular functions during development. Abnormal TGF‐β responses are commonly found in human diseases such as cancer, suggesting that TGF‐β signaling must be tightly regulated. Here, we report that protein tyrosine phosphatase non‐receptor 3 (PTPN3) profoundly potentiates TGF‐β signaling independent of its phosphatase activity. PTPN3 stabilizes TGF‐β type I receptor (TβRI) through attenuating the interaction between Smurf2 and TβRI. Consequently, PTPN3 facilitates TGF‐β‐induced R‐Smad phosphorylation, transcriptional responses, and subsequent physiological responses. Importantly, the leucine‐to‐arginine substitution at amino acid residue 232 (L232R) of PTPN3, a frequent mutation found in intrahepatic cholangiocarcinoma (ICC), disables its role in enhancing TGF‐β signaling and abolishes its tumor‐suppressive function. Our findings have revealed a vital role of PTPN3 in regulating TGF‐β signaling during normal physiology and pathogenesis.
Keywords: cholangiocarcinoma, phosphatase, Smurf2, TGF‐β signaling, TβRI
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
As the second leading cause of cancer‐related death worldwide, liver cancer has high incidence and mortality rate but poor prognosis and limited treatment options. Among all primary liver cancers, hepatocellular carcinoma (HCC) occupies about 80%, while intrahepatic cholangiocarcinoma (ICC) occupies an estimated 10–15% (Torre et al, 2015; Llovet et al, 2016; Sia et al, 2017). Extensive studies have established the link between functions of the transforming growth factor‐β (TGF‐β) and liver fibrosis, cirrhosis, and subsequent progression to HCC (Bissell et al, 2001; Dooley & ten Dijke, 2012; Meng et al, 2016). Loss of type II TGF‐β receptor (TβRII) or Smad4 is common in ICC (Ong et al, 2012; Jiao et al, 2013). Still, better understanding of liver tumor biology is urgently needed to improve prognosis and treatments.
TGF‐β and related growth factors regulate diverse cellular responses such as cell proliferation, differentiation, apoptosis, and embryonic development in metazoan (Feng & Derynck, 2005; Derynck & Akhurst, 2007; Wu & Hill, 2009). Dysregulation of TGF‐β signaling leads to abnormal development and severe diseases (Gordon & Blobe, 2008; Massague, 2008; Majumdar et al, 2012; Karimi‐Googheri et al, 2014). On the cell surface, the ligand binds to the TβRII, which phosphorylates the type I TGF‐β receptor (TβRI, also known as ALK5). In canonical signaling, the activated TβRI phosphorylates Smad2/3 in their C‐terminal SXS motif. Phosphorylated Smad2/3 form a complex with Smad4, which leads to Smad accumulation in the nucleus that in turn recruits transcription co‐factors to regulate gene transcription in a context‐dependent manner (Feng & Derynck, 2005; Massague, 2012). Moreover, TGF‐β signaling is also regulated by inhibitory Smads (I‐Smads, i.e., Smad6/7). Smad7 acts as a scaffold protein to recruit members of the Smurf E3 ubiquitin ligase family that ubiquitinates TβRI and leads to proteasome‐mediated TβRI degradation (Afrakhte et al, 1998; Kavsak et al, 2000; Yan et al, 2009; Miyazawa & Miyazono, 2017). Smurf proteins also directly target R‐Smads for proteasomal degradation (Xu et al, 2016).
PTPN3 (also known as PTPH1), belonging to the non‐transmembrane protein tyrosine phosphatase (PTP) family (Yang & Tonks, 1991), possesses three major structural and functional domains. The N‐terminal FERM (band 4.1, ezrin, radixin, moesin) domain of PTPN3 is responsible for targeting its interacting proteins to the cytoskeleton–membrane interface or interaction with transmembrane proteins. The central PDZ (PSD‐95, Dlg, ZO‐1) domain of PTPN3 mediates the interaction with other proteins. The C‐terminal PTP domain harbors the catalytic function of PTPN3 (Yang & Tonks, 1991; Arpin et al, 1994; Ponting et al, 1997; Zhang et al, 1999). As a tyrosine phosphatase, PTPN3 is capable of dephosphorylating several substrates (Chen et al, 2015; Parker, 2015). For instance, PTPN3 interacts with and dephosphorylates TCRζ (Sozio et al, 2004). Accumulating evidence suggests that PTPN3 plays a critical role in the progression of variety of human cancers. PTPN3 directly catalyzes Y1173 dephosphorylation of EGFR and cooperates with tyrosine kinase inhibitor (TKI) to inhibit breast cancer progression (Ma et al, 2015). In lung cancer, PTPN3 dephosphorylates Eps15 to promote EGFR degradation, thereby inhibiting lung cancer cell proliferation and migration (Li et al, 2015). Somatic mutations in PTPN3 are found in human colon cancers, suggesting a loss of its tumor suppressor activity (Wang et al, 2004). More strikingly, about 41% of ICC have been reported to carry mutations in PTPN3 (Gao et al, 2014). For instance, the leucine‐to‐arginine substitution at amino acid 232 (L232R) in PTPN3, a frequent mutation found in ICC, has been shown to promote cholangiocarcinoma cell proliferation and migration and associate with increased risk of tumor recurrence in patients (Gao et al, 2014). In addition, expression of PTPN3 in human hepatoma cells significantly reduces the level of human hepatitis B viral (HBV) replication, whereas deletion of the FERM domain of PTPN3 impairs this effect (Hsu et al, 2007). Therefore, PTPN3 acts as a tumor suppressor in ICC.
Despite its critical role in cancer progression, the underlying mechanisms of how PTPN3 regulates tumor progression especially in liver cancer remain elusive. Here, we report that PTPN3 suppresses liver cancer progression through positively regulating TGF‐β signaling. PTPN3 interacts with TβRI and promotes TβRI stability, thereby enhancing TGF‐β‐mediated tumor‐suppressive functions. Mechanistically, PTPN3 blocks Smurf2‐mediated TβRI ubiquitination in a phosphatase‐independent manner. Furthermore, the recurrent L232R mutation in liver cancer disables the function of PTPN3 on enhancing TGF‐β signaling. Our study suggests that PTPN3 achieves its tumor suppressor functions through the TGF‐β signaling pathway and thus provides an unprecedented mechanism of liver cancer pathogenesis.
Results
PTPN3 enhances TGF‐β‐induced transcriptional responses
During our recent search for protein phosphatases that regulate TGF‐β signaling, we found that PTPN3 promoted TGF‐β‐induced transcriptional responses. We first evaluated the function of PTPN3 using luciferase reporter assays in TGF‐β‐responsive cells. CAGA‐luc, a synthetic TGF‐β‐induced luciferase reporter driven by the promoter containing twelve copies of Smad‐binding element (SBE), was used (Dennler et al, 1998). Transient expression of PTPN3 markedly enhanced the TGF‐β‐induced CAGA‐luc response in a variety of cell lines, including immortalized human keratinocyte HaCaT cells (Fig 1A), adenocarcinoma human alveolar basal epithelial A549 cells (Fig EV1A), HCC Huh7 (Fig EV1B), and SNU449 cells (Fig EV1C). Furthermore, efficient knockdown of PTPN3 by siRNA (Fig EV1D) decreased TGF‐β‐induced reporter gene activity (Figs 1B and EV1E, F and G). The siRNA‐resistant PTPN3 variant (Fig EV1H) completely rescued the effect of siPTPN3 on TGF‐β‐induced reporter gene activities (Fig EV1E and G), demonstrating the specific on‐target effect of siPTPN3. These data suggest that PTPN3 enhances Smad‐mediated TGF‐β transcriptional responses.
Figure 1. PTPN3 enhances TGF‐β‐induced transcriptional responses.
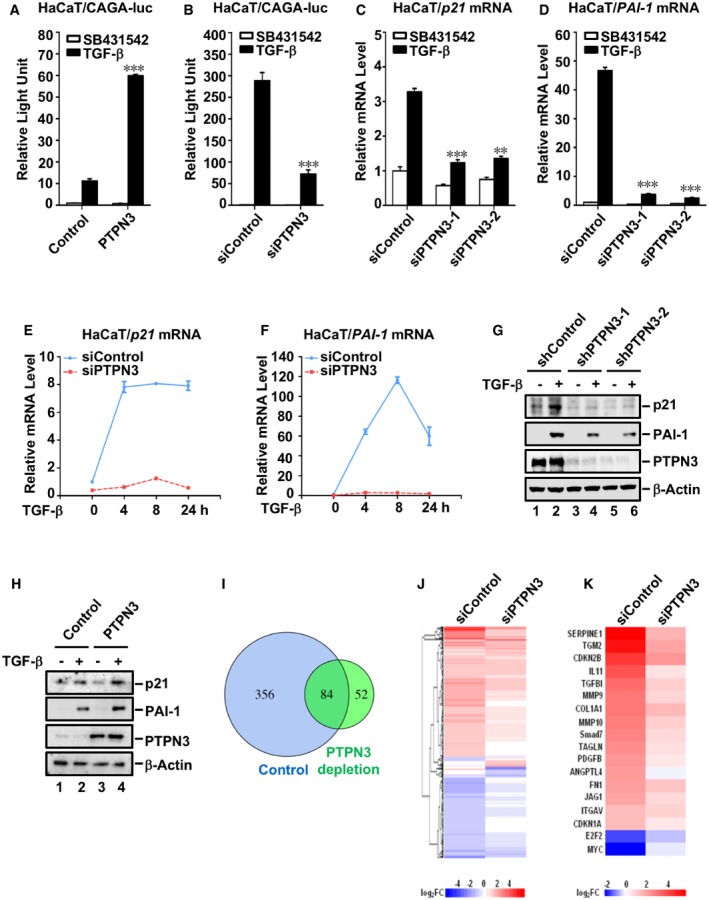
- PTPN3 promotes TGF‐β‐induced CAGA‐luc reporter gene activity. HaCaT cells were transfected with expression plasmids for CAGA‐luc reporter gene, Renilla‐luc reporter gene, and PTPN3 as indicated and treated with TGF‐β (2 ng/ml, 8 h). Relative luciferase activity was measured as described in the text. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Knockdown of PTPN3 decreases TGF‐β‐induced CAGA‐luc reporter gene activity. HaCaT cells were transfected with siRNA against PTPN3 or control siRNA. Twenty‐four hours post‐transfection, expression plasmids for CAGA‐luc reporter and Renilla‐luc reporter were transfected as indicated. Cells were harvested for relative luciferase assay after another 24 h with TGF‐β treatment (2 ng/ml, 8 h). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Knockdown of PTPN3 blocks TGF‐β‐induced p21 transcription. HaCaT cells were transfected with two independent siRNAs against PTPN3 and treated with TGF‐β (2 ng/ml) for 8 h before harvested. Total mRNA was analyzed by qRT–PCR using primers specific to p21. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. **P < 0.01, ***P < 0.001.
- Knockdown of PTPN3 blocks TGF‐β‐induced PAI‐1 gene transcription. Experiment was done as in (C) with qRT–PCR primers specific to PAI‐1. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Knockdown of PTPN3 abolishes TGF‐β‐induced p21 transcription. HaCaT cells were transfected with siRNA and then treated with TGF‐β (2 ng/ml) for 0, 4, 8, or 24 h as indicated. qRT–PCR was carried out as in (C). Data are shown as mean ± SEM; n = 3.
- Knockdown of PTPN3 abolishes TGF‐β‐induced transcription of PAI‐1. Experiment was done as in (E). Data are shown as mean ± SEM; n = 3.
- Depletion of PTPN3 abolishes TGF‐β‐induced target protein expression. HaCaT cells with stable knockdown of PTPN3 were treated with TGF‐β (2 ng/ml) for 6 h before harvested. Cells were harvested for Western blotting with anti‐p21, anti‐PAI‐1, anti‐PTPN3, and anti‐β‐actin antibodies.
- PTPN3 enhances TGF‐β‐induced target protein expression. HaCaT cells stably expressing HA‐PTPN3 were stimulated with 2 ng/ml of TGF‐β for 6 h and then harvested for cell lysates. Protein levels were examined by Western blotting with appropriate antibodies as indicated.
- Depletion of PTPN3 attenuates TGF‐β global gene responses. Venn diagram shows the number of differential expression genes (DEGs) with TGF‐β treatment in control or PTPN3‐depleted HaCaT cells by RNA‐Seq analysis. The overlap represents TGF‐β target genes that are commonly regulated in both cell lines. HaCaT cells transfected with siControl or siPTPN3 were harvested for RNA‐Seq after treated with or without TGF‐β (2 ng/ml, 8 h).
- Loss of PTPN3 markedly decreases TGF‐β‐dependent upregulation or downregulation of target genes. Heat map of DEGs in control HaCaT cells (siControl) or HaCaT cells depleted of PTPN3 (siPTPN3). Log2FC data represent fold change of TGF‐β (2 ng/ml) treatment over vehicle for 8 h.
- Heat map of Log2FC of select TGF‐β target genes.
Figure EV1. PTPN3 enhances TGF‐β‐induced transcriptional responses.
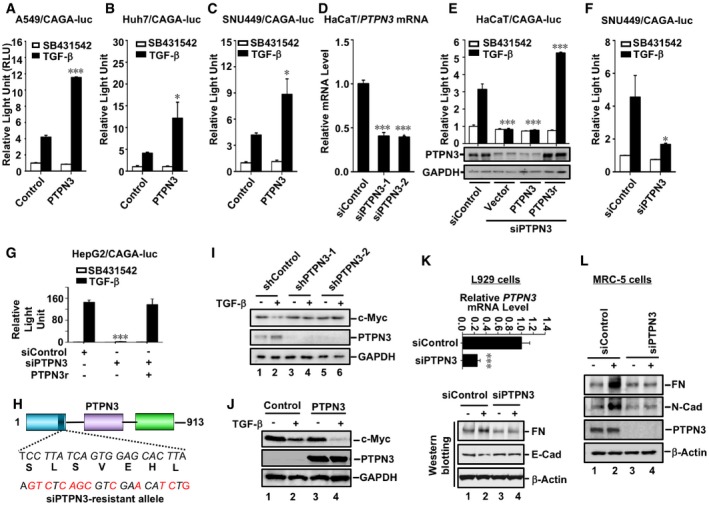
- PTPN3 promotes TGF‐β‐induced CAGA‐luc reporter activity. A549 cells were transfected with expression plasmids for PTPN3, CAGA‐luc reporter, and Renilla‐luc reporter as indicated and treated with TGF‐β (2 ng/ml, 8 h). Relative luciferase activity was measured as described in the text. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- PTPN3 augments TGF‐β‐induced CAGA‐luc reporter activity in Huh7 cells. Cell transfection, TGF‐β treatment, and luciferase assays were similarly done as in (A). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05.
- PTPN3 facilitates TGF‐β‐induced CAGA‐luc reporter activity in SNU449 cells. Cell transfection, TGF‐β treatment, and luciferase assays were similarly done as in (A). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05.
- PTPN3 is efficiently knocked down by siRNA. Two independent siRNAs against PTPN3 were transfected into HaCaT cells. PTPN3 mRNA level was detected by using qRT–PCR. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Knockdown of PTPN3 decreases TGF‐β‐induced CAGA‐luc reporter activity, and siRNA‐resistant variant of PTPN3 rescues the siPTPN3 effect. HaCaT cells were transfected with siPTPN3, and expression plasmids for PTPN3 or RNAi‐resistant variant of PTPN3 (PTPN3r), CAGA‐luc reporter, and Renilla‐luc reporter as indicated and treated with TGF‐β (2 ng/ml, 8 h). Twenty‐four hours later, cells were harvested for luciferase analysis. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Knockdown of PTPN3 decreases TGF‐β‐induced CAGA‐luc reporter activity in SNU449 cells. Cells were transfected with siRNA against PTPN3 or control siRNA; 24 h post‐transfection, expression plasmids for CAGA‐luc reporter and Renilla‐luc reporter were transfected as indicated. Cells were harvested for relative luciferase assay after another 24 h with TGF‐β treatment (2 ng/ml, 8 h). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05.
- Knockdown of PTPN3 decreases TGF‐β‐induced CAGA‐luc reporter activity, and siRNA‐resistant variant of PTPN3 rescues the siPTPN3 effect in HepG2 cells. Cells were transfected with siControl, siPTPN3, expression plasmids for RNAi‐resistant variant of PTPN3 (PTPN3r), CAGA‐luc reporter, and Renilla‐luc reporter as indicated and treated with TGF‐β (2 ng/ml, 8 h). Twenty‐four hours later, cells were harvested for luciferase analysis. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Schematic representation of the RNAi‐resistant PTPN3 variant. The siPTPN3 target sequence (corresponding to amino acids 214–220) is indicated. Silent mutations introduced are shown by italic/red letters.
- Depletion of PTPN3 abolishes TGF‐β‐induced target protein expression. HaCaT cells with stable knockdown of PTPN3 were treated with TGF‐β (2 ng/ml) for 6 h before harvested. Cells were harvested for Western blotting with appropriate antibodies as indicated.
- PTPN3 facilitates TGF‐β‐induced target protein expression. HaCaT cells stably expressing HA‐PTPN3 were stimulated with 2 ng/ml of TGF‐β for 6 h and then harvested for cell lysates. Protein levels were examined by Western blotting with anti‐c‐myc, anti‐PTPN3, and anti‐GAPDH antibodies.
- Knocking down efficiency of siPTPN3 in L929 cells. L929 cells were transfected with siControl or siPTPN3 with RNAiMax. Forty‐eight hours after transfection, cells were harvested for qRT–PCR analysis as indicated. Data in the upper panel are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001. Depletion of PTPN3 attenuates TGF‐β‐induced EMT. L929 cells were transfected with siControl or siPTPN3 with RNAiMax. Forty‐eight hours later, cells were harvested for Western blotting analysis with antibodies as indicated.
- Depletion of PTPN3 attenuates TGF‐β‐induced EMT. MRC‐5 cells were transfected with siControl or siPTPN3 with RNAiMax. Forty‐eight hours later, cells were harvested for Western blotting analysis with antibodies as indicated.
To further investigate the physiological function of PTPN3 in TGF‐β signaling, we assessed the impact of PTPN3 knockdown on TGF‐β‐induced transcription of PAI‐1, p21, and c‐Myc, which are the direct target genes of TGF‐β (Datto et al, 1995; Reynisdottir et al, 1995; Dennler et al, 1998; Warner et al, 1999; Datta et al, 2000). TGF‐β stimulation induces expression of PAI‐1 and p21 and represses expression of c‐Myc. qRT–PCR analysis showed that efficient knockdown of PTPN3 using two independent siRNAs abolished TGF‐β‐induced expression of endogenous p21 and PAI‐1 mRNA in HaCaT cells (Fig 1C and D). While TGF‐β increased PAI‐1 and p21 mRNAs in a time‐dependent manner, PTPN3‐deficient cells were irresponsive or less responsive to TGF‐β (Fig 1E and F). Western blotting analysis also demonstrated that efficient stable knockdown of PTPN3 completely blocked TGF‐β‐induced upregulation of PAI‐1 and p21 proteins as well as downregulation of c‐Myc protein in HaCaT cells (Figs 1G and EV1I). Conversely, stable expression of PTPN3 in HaCaT cells substantially enhanced TGF‐β responses evidenced by upregulation of PAI‐1 and p21 proteins (Fig 1H) and downregulation of c‐Myc protein (Fig EV1J). It is notable that depletion of PTPN3 could also attenuate TGF‐β‐mediated upregulation of fibronectin (FN) and N‐cadherin and downregulation of E‐cadherin in L929 and MRC‐5 fibroblasts (Fig EV1K and L).
To examine whether PTPN3 regulates global TGF‐β gene responses, we carried out RNA‐seq experiments in parental and PTPN3‐depleted HaCaT cells. RNA‐seq analyses showed that 440 genes were up‐ or downregulated (fold change > 2) by TGF‐β in parental HaCaT cells, whereas only 84 of them were responsive to TGF‐β in PTPN3‐deficient cells (Fig 1I and J), suggesting that PTPN3 deficiency disables TGF‐β responsiveness. Next, we selectively examined a group of known TGF‐β target genes, including SERPINE1, TGM2, CDKN2B, IL11, TGFBI, MMP9, COL1A1, MMP10, SMAD7, TAGLN, PDGFB, ANGTL4, FN, JAG1, ITGAV, CDKN1A, E2F2, and MYC. As shown in Fig 1K, PTPN3 deficiency significantly compromised the regulation of these target genes by TGF‐β. Together, our genome‐wide transcriptional analyses support the conclusion that PTPN3 is required for robust TGF‐β‐induced transcriptional responses.
PTPN3 promotes TGF‐β signaling independent of its phosphatase activity
Since PTPN3 is a protein tyrosine phosphatase, we sought to determine whether the phosphatase activity of PTPN3 is required for its regulation of TGF‐β‐induced transcriptional responses. We first generated catalytically inactive mutants of PTPN3, one with aspartic acid‐to‐alanine substitution at amino acid residue 811 (D811A) and the other with cysteine‐to‐serine substitution at amino acid residue 842 (C842S) of PTPN3 (Zhang et al, 1999; Fig 2A). In agreement with a previous report (Ma et al, 2015), PTPN3 dephosphorylated EGF‐induced EGFR pY1173, whereas these two catalytically inactive mutants failed to dephosphorylate EGFR pY1173 (Fig 2B), confirming the loss of phosphatase activity in these two mutants. Unexpectedly, like PTPN3, PTPN3 (D811A) potently augmented TGF‐β‐induced upregulation of p21 and downregulation of c‐Myc in HaCaT cells (Fig 2C). Consistently, PTPN3 (D811A) and PTPN3 (C842S) increased TGF‐β‐induced CAGA‐luc activity when expressed at a level comparable to wild‐type PTPN3 (Fig 2D). These results suggest that the phosphatase activity of PTPN3 is dispensable for its regulation of TGF‐β signaling.
Figure 2. PTPN3 promotes TGF‐β signaling independent of its phosphatase activity.
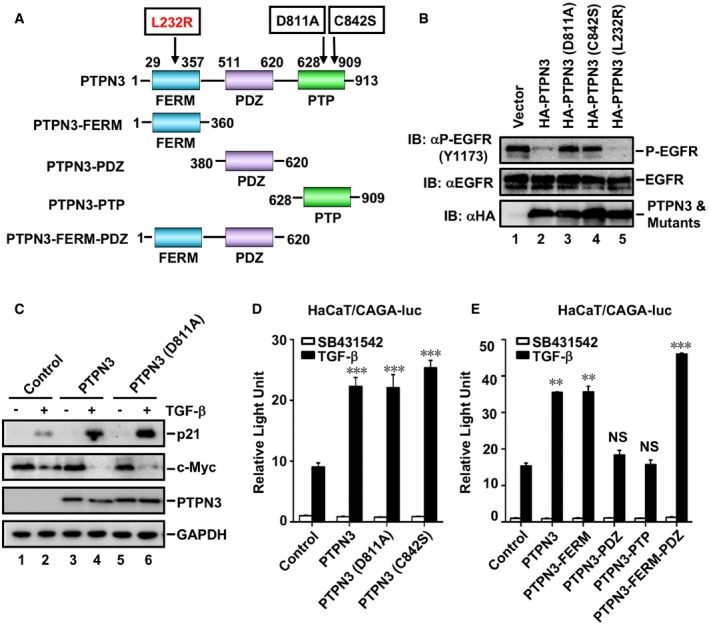
- Schematic diagram of PTPN3 domains. Deletion and point mutation of PTPN3 were shown.
- Both PTPN3 and PTPN3 (L232R), but not the phosphatase‐dead mutants PTPN3 (D811A) and PTPN3 (C842S), dephosphorylate P‐EGFR (Y1173). Wild‐type PTPN3 or its mutants were co‐transfected with EGFR into HEK293T cells as indicated. Twenty‐four hours later, cells were harvested for Western blotting. Levels of P‐EGFR (Y1173), EGFR, PTPN3, and its mutants were examined by appropriate antibodies.
- PTPN3 and PTPN3 (D811A) enhance TGF‐β‐induced target protein expression. HaCaT cells stably expressing HA‐PTPN3 or HA‐PTPN3 (D811A) were stimulated with 2 ng/ml of TGF‐β for 6 h and then harvested for cell lysates. Protein levels were examined by Western blotting with appropriate antibodies as indicated.
- PTPN3 and its catalytically inactive mutants PTPN3 (D811A) and PTPN3 (C842S) equally enhance TGF‐β‐induced reporter gene activity. HaCaT cells were transfected with expression plasmids for each PTPN3 variant and CAGA‐luc reporter and Renilla‐luc reporter. Cells were harvested for relative luciferase assay after TGF‐β (2 ng/ml, 8 h) treatment. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- PTPN3 FERM domain enhances TGF‐β‐induced reporter gene activity as potently as wild‐type PTPN3. HaCaT cells were transfected with expression plasmids for PTPN3 truncations and CAGA‐luc reporter, and internal control Renilla‐luc reporter. Cells were harvested for relative luciferase assay with TGF‐β treatment (2 ng/ml, 8 h). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. **P < 0.01, ***P < 0.001. NS, non‐significant.
Having found that the phosphatase activity is not required in TGF‐β signaling, we sought to identify the specific domain of PTPN3 that regulates TGF‐β signaling. As shown in Fig 2A, we generated PTPN3 mutants that delete various domains, namely PTPN3‐FERM, PTPN3‐PDZ, PTPN3‐PTP, and PTPN3‐FERM‐PDZ. We found that the presence of FERM domain (as in PTPN3‐FERM and PTPN3‐FERM‐PDZ) was sufficient to increase TGF‐β‐induced CAGA‐luc activity in a comparable level to PTPN3, whereas the deletion of FERM (as in PTPN3‐PDZ and PTPN3‐PTP) abolished the regulation of TGF‐β signaling (Fig 2E). These suggest that the FERM domain of PTPN3 is essential in regulating TGF‐β signaling.
PTPN3 regulates TβRI ubiquitination and stability
How does PTPN3 enhance TGF‐β‐induced transcriptional responses? We found that endogenous TβRI protein level was increased in HaCaT cells stably expressing PTPN3 or PTPN3 (D811A) (Fig 3A, lanes 2 and 3). Conversely, stable knockdown of PTPN3 by shPTPN3 profoundly decreased the protein level of endogenous TβRI (Fig 3B, lanes 2 and 3). siRNA‐mediated transient knockdown of PTPN3 also reduced the protein level of TβRI, which was rescued by RNAi‐resistant variant of PTPN3 (Fig EV2A). Furthermore, the TβRI mRNA level was not altered upon PTPN3 depletion (Fig EV2B), suggesting that PTPN3 regulates the stability of TβRI protein. Since TβRI stability is mainly regulated by the ubiquitin‐proteasome pathway (Kavsak et al, 2000; Ebisawa et al, 2001; Xu et al, 2012), we asked whether PTPN3 suppresses the proteasomal degradation of TβRI. Indeed, proteasome inhibitor MG132 restored the level of TβRI in PTPN3 knockdown cells (Fig 3B, lanes 5 and 6).
Figure 3. PTPN3 attenuates ubiquitination of TβRI to stabilize TβRI.
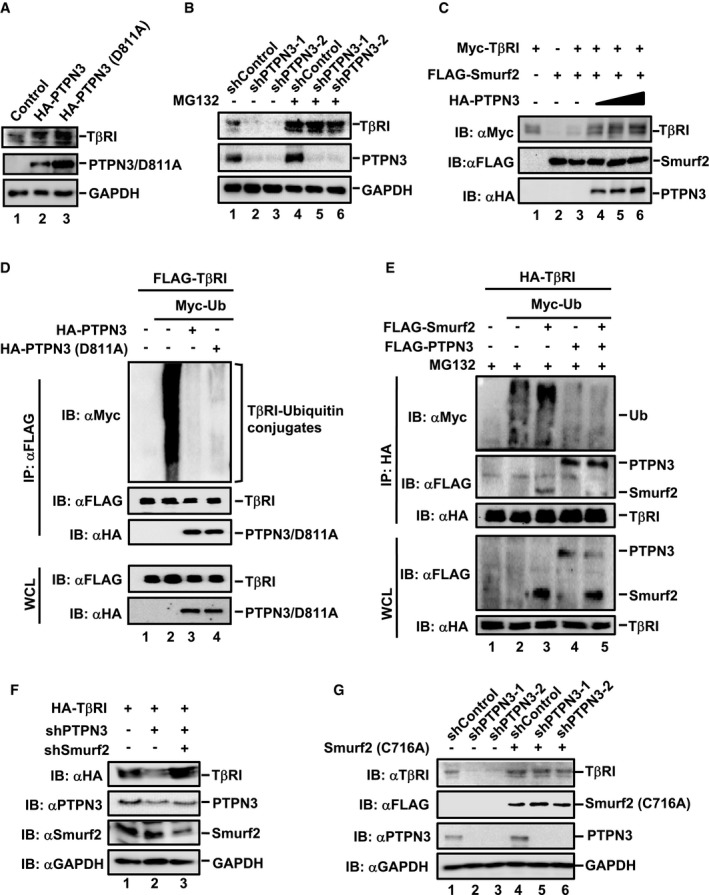
- PTPN3 and PTPN3 (D811A) stabilize TβRI. TβRI and PTPN3 were detected by Western blotting with appropriate antibodies as indicated in HaCaT cells stably expressing PTPN3 or PTPN3 (D811A).
- shRNA PTPN3‐mediated reduction of endogenous TβRI level is restored by MG132. HaCaT cells expressing shRNA PTPN3‐1, shRNA PTPN3‐2, or control shRNA were treated with MG132 (20 μM) for 4 h. Cell lysates were analyzed by Western blotting with anti‐TβRI, anti‐PTPN3, and anti‐GAPDH antibodies.
- PTPN3 rescues the degradation of TβRI induced by Smurf2. HEK293T cells were transfected with the indicated plasmids. Cells were harvested after 24 h post‐transfection. Western blotting was done with the indicated antibodies.
- PTPN3 and PTPN3 (D811A) attenuate ubiquitination of TβRI. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, IP and immunoblotting analysis were performed with the anti‐FLAG antibody. Levels of TβRI ubiquitination and other proteins were examined by Western blotting with the indicated antibodies.
- PTPN3 blocks Smurf2‐mediated ubiquitination of TβRI. HEK293T cells were transfected with the indicated plasmids and treated with MG132 (20 μM, 4 h) before harvest. TβRI IP was performed with the anti‐HA antibody, and TβRI ubiquitination was determined by Western blotting with the anti‐Myc antibody. Protein levels were examined by Western blotting with the indicated antibodies.
- Depletion of Smurf2 reverses the shPTPN3‐induced degradation of TβRI. HEK293T cells were transfected with expression plasmids for shPTPN3, shSmurf2, and HA‐TβRI. Forty‐eight hours post‐transfection, cells were harvested for immunoblotting analysis. TβRI level was determined by Western blotting with the anti‐HA antibody. Levels of other proteins were done by Western blotting with the indicated antibodies.
- Dominant‐negative Smurf2 mutant C716A prevents shPTPN3‐induced TβRI degradation. HaCaT cells stably expressing shPTPN3 were transfected with FLAG‐Smurf2 (C716A). Cells were harvested for Western blotting with anti‐TβRI, anti‐FLAG, anti‐PTPN3, and anti‐GAPDH antibodies.
Figure EV2. Assessment of PTPN3 mutants and RNAi knockdown efficiency.
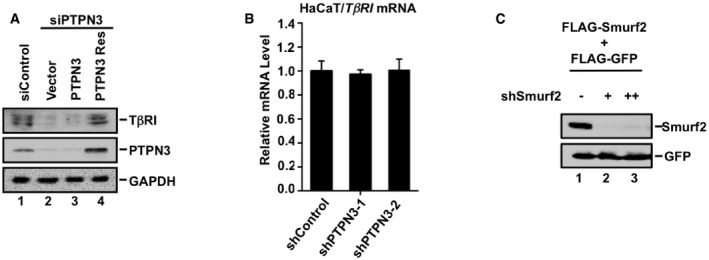
- Knockdown of PTPN3 decreases the TβRI protein level and RNAi‐resistant variant of PTPN3 rescues the siPTPN3 effect. HaCaT cells were transfected with siPTPN3, together with PTPN3 and RNAi‐resistant variant of PTPN3. Twenty‐four hours later, cells were harvested for Western blotting analysis.
- Depletion of PTPN3 has no effect on the mRNA level of TβRI. qRT–PCR was used to examine the mRNA level of TβRI in HaCaT cells expressing shRNA Control, shPTPN3‐1, and shPTPN3‐2. Data are shown as mean ± SEM; n = 3.
- shSmurf2 efficiently knocks down Smurf2. HEK293T cells were co‐transfected with FLAG‐Smurf2, FLAG‐GFP, and increasing amounts of pSRGshSmurf2 as indicated. Cells were harvested for Western blotting analysis with anti‐FLAG antibodies.
Smurf2 is a primary E3 ligase for ubiquitin‐proteasomal degradation of TβRI (Kavsak et al, 2000). We asked whether PTPN3 stabilized TβRI through regulating Smurf2 function. In HEK293T cells, increasing amounts of PTPN3 gradually increased the level of TβRI even in the presence of Smurf2 (Fig 3C, compare lanes 4–6 to lane 3), suggesting that PTPN3 blocked the degradation of TβRI by Smurf2. To examine the effect of PTPN3 on TβRI ubiquitination, FLAG‐TβRI and Myc‐ubiquitin, together with HA‐PTPN3 or HA‐PTPN3 (D811A), were co‐transfected in HEK293T cells. Anti‐FLAG immunoprecipitation (IP) was carried out to retrieve TβRI, which was followed by anti‐Myc Western blotting analysis to detect the ubiquitination of TβRI. As shown in Fig 3D, PTPN3 and PTPN3 (D811A) substantially reduced the ubiquitination of TβRI. Moreover, the impact of PTPN3 on the stabilization of TβRI was reflected by its blocking effect on Smurf2‐mediated ubiquitination of TβRI (Fig 3E).
To further prove that PTPN3‐regulated TβRI stability is dependent on Smurf2, we tested whether depletion of Smurf2 or inhibition of Smurf2 activity could reverse TβRI degradation induced by PTPN3 knockdown. We used an shSmurf2 to effectively knock down Smurf2 (Wrighton et al, 2008) (Fig EV2C) or Smurf2 (C716A), a catalytically dead and dominant‐negative mutant of Smurf2, to block Smurf2 activity (Kavsak et al, 2000; Lin et al, 2000). In HEK293T cells, the level of TβRI was clearly reduced by shPTPN3 (Fig 3F, lane 2). Depletion of Smurf2 rescued the level of TβRI (Fig 3F, lane 3). Likewise, Smurf2 (C716A) also restored the level of TβRI in the shPTPN3 cells (Fig 3G, lanes 5 and 6). These results demonstrate that PTPN3 plays a critical role in the regulation of TβRI stability by antagonizing Smurf2.
PTPN3 attenuates the interaction of TβRI with the Smad7‐Smurf2 E3 ligase
One obvious question is that whether and how PTPN3 affects Smurf2‐mediated TβRI degradation. To this end, we first performed co‐immunoprecipitation (co‐IP) to assess the interaction between PTPN3 and TβRI. Under the physiological condition, IP of endogenous PTPN3 using an anti‐PTPN3 antibody pulled down the co‐precipitated endogenous TβRI in HaCaT cells (Fig 4A, lanes 3 and 4). TGF‐β had little or no effect on the TβRI‐PTPN3 interaction (Fig 4A, lane 4). Furthermore, the catalytically inactive mutant PTPN3 (D811A) bound to TβRI to the same extent as wild‐type PTPN3 (Fig 4B, lanes 3 and 4).
Figure 4. PTPN3 attenuates the binding the Smad7‐Smurf2 E3 ligase to TβRI.
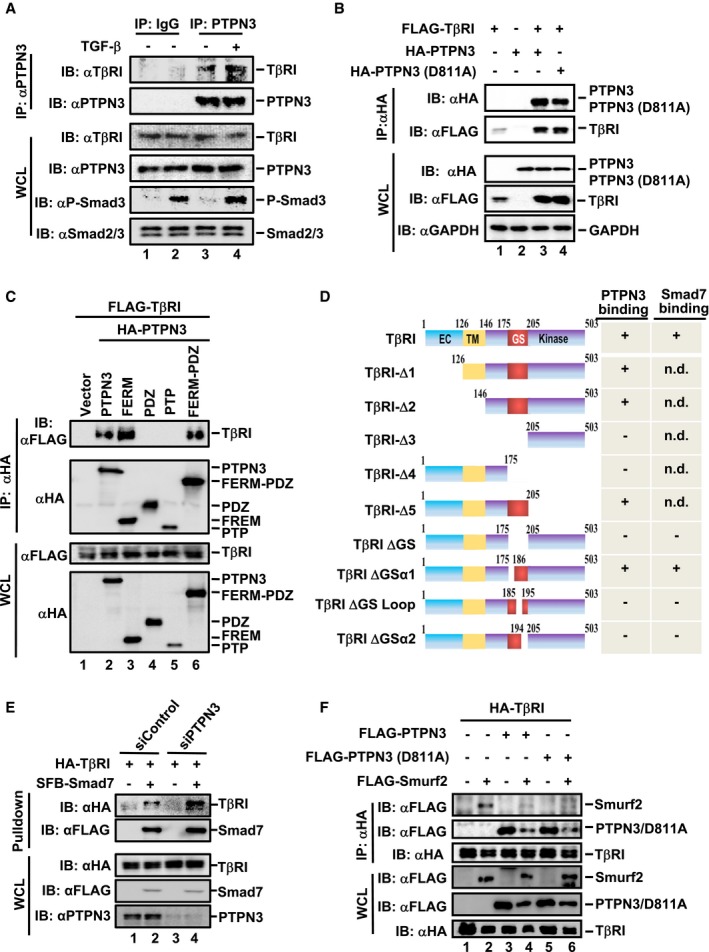
- PTPN3 interacts with TβRI at endogenous levels. HaCaT cells were harvested after TGF‐β treatment (2 ng/ml, 1 h) and then subjected to IP by PTPN3 antibody or control IgG. PTPN3 and TβRI were detected by Western blotting.
- Both PTPN3 and PTPN3 (D811A) interact with TβRI. FLAG‐TβRI was co‐transfected with HA‐PTPN3 or HA‐PTPN3 (D811A) into HEK293T cells. Cell lysates were harvested and subjected to IP with FLAG antibody. PTPN3, PTPN3 (D811A), and TβRI were detected by Western blotting with appropriate antibodies.
- The FERM domain of PTPN3 is necessary for its binding to TβRI. FLAG‐TβRI and HA‐PTPN3 (wild‐type or truncations) were co‐transfected into HEK293T cells. Cells were harvested after 24 h and subjected to IP by HA antibody. TβRI, PTPN3, and PTPN3 truncations were detected by Western blotting with appropriate antibodies.
- Schematic representation of TβRI deletion mutants and their interactions with PTPN3 or Smad7. EC, extracellular domain; TM, transmembrane domain; GS, Gly‐Ser‐rich motif; +, positive interaction; −, no interaction.
- Knockdown of endogenous PTPN3 promotes the Smad7‐TβRI association. HEK293T cells were transfected with the indicated plasmids and treated with MG132 (20 μM) for 4 h before harvested. SFB‐Smad7 proteins were retrieved with the streptavidin pulldown. Levels of TβRI and other proteins were examined by Western blotting with the indicated antibodies.
- PTPN3 and PTPN3 (D811A) abolish the interaction between TβRI and Smurf2 in HEK293T cells. HEK293T cells were co‐transfected with plasmids as indicated. Twenty hours later, cells were treated with MG132 (20 μM) for 4 h, followed by IP and Western blotting.
Source data are available online for this figure.
We further determined the domain of PTPN3 that interacted with TβRI. In consistent with the essential role of the FERM domain in PTPN3 functions (Fig 2), all PTPN3 variants with the FERM domain, including wild‐type PTPN3, PTPN3‐FERM, and PTPN3‐FERM‐PDZ, interacted with TβRI (Fig 4C, lanes 2, 3 and 6; for domain diagram, see Fig 2A). However, the PDZ or PTP domain alone failed to associate with TβRI (Fig 4C, lanes 4 and 5). Our results suggest that the FERM domain of PTPN3 is necessary and sufficient for PTPN3 binding to TβRI.
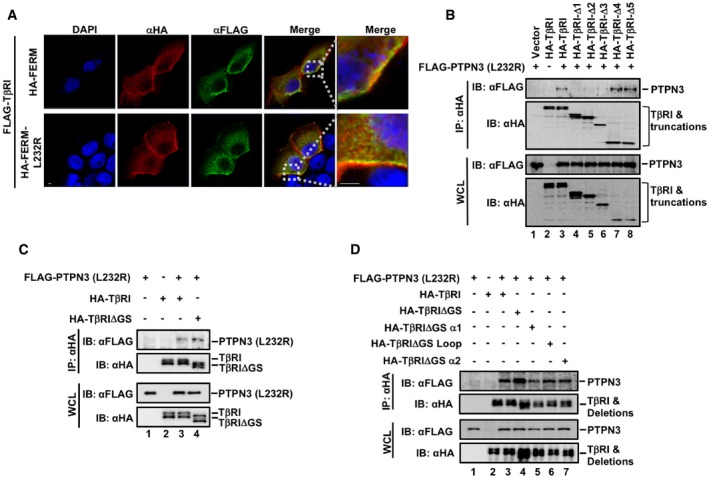
Similarly, we delineated the domain of TβRI for PTPN3 binding to help understand how PTPN3 stabilizes TβRI. We found that PTPN3 interacted with the Gly‐Ser (GS) region of TβRI (Fig 4D). Deletions of the GS motif disable the binding to PTPN3 (Figs 4D and EV3A and B). To further define the critical PTPN3‐binding domain in the GS region, we made deletions of small regions around the GS motif, including the α1 (aa 175–184), the GS loop (aa 185–194), or α2 (aa 195–205), and tested their binding to PTPN3 (Fig 4D). Deletion of the GS loop or α2, but not α1, abolished their interactions with PTPN3 (Figs 4D and EV3C). Importantly, PTPN3 and Smad7 bound to the same GS loop‐α2 region of TβRI (Figs 4D and EV3D), implying that they may compete for binding on TβRI. Indeed, depletion of PTPN3 profoundly enhanced the Smad7‐TβRI association (Fig 4E).
Figure EV3. PTPN3 binds to TβRI and disrupts its interaction with the Smad7‐Smurf2 E3 ligase.
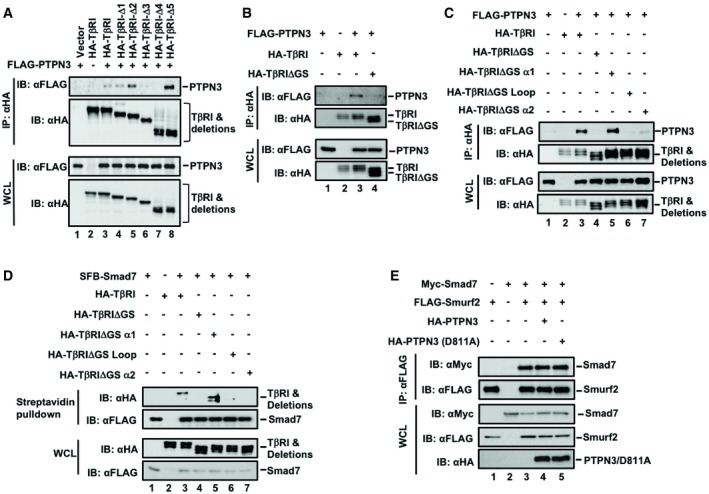
- Deletion of the GS region of TβRI disabled its interaction with PTPN3. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, immunoprecipitation and immunoblotting analysis were performed with the anti‐HA antibody. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
- GS region of TβRI is necessary for its binding to PTPN3. FLAG‐PTPN3, HA‐TβRI, and HA‐TβRI truncations were transfected into HEK293T cells. Cells were harvested after 24 h and immunoprecipitated by HA antibody. PTPN3, TβRI, and TβRI truncations were detected by Western blotting with anti‐Flag and anti‐HA antibodies.
- Deletion of the GS or GS‐loop or GS‐α2 region of TβRI disabled its interaction with PTPN3. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, immunoprecipitation and immunoblotting analyses were performed with the anti‐HA antibody. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
- Deletion of the GS or GS‐loop or GS‐α2 region of TβRI disabled its interaction with Smad7. HEK293T cells were transfected with the indicated plasmids. Cells were treated with MG132 (20 μM, 4 h) before harvested. Then, immunoprecipitation was performed with the streptavidin. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
- PTPN3 wild‐type or the D811A mutant did not affect the Smurf‐Smad7 interaction. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, immunoprecipitation and immunoblotting analyses were performed with the anti‐HA antibody. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
If PTPN3 associates TβRI, does this interaction affect the binding of the E3 ligase Smurf2 to TβRI? We then investigated whether PTPN3 blocks the interaction between Smad7/Smurf2 and TβRI. As shown in Fig 4F, PTPN3 and PTPN3 (D811A) attenuated the interaction between Smurf2 and TβRI (Fig 4F, lanes 4 and 6). Interestingly, we also noticed that PTPN3 was partly degraded in the presence of Smurf2 (Fig 4F, lanes 4 and 6). In addition, PTPN3 exhibited little effect on the Smad7‐Smurf2 association (Fig EV3E). Taken together, these results demonstrate that PTPN3 blocks the binding of the Smad7‐Smurf2 ligase to TβRI, thereby stabilizing the TβRI protein.
PTPN3 (L232R) fails to augment TGF‐β signaling
PTPN3 is frequently mutated in ICC. Since the L232R mutation located in the PTPN3 FERM domain is commonly found in ICC (Gao et al, 2014), we hypothesized that it might alter the PTPN3 functions through its effect on TGF‐β signaling. To test this, we examined whether L232R affects TGF‐β signaling events and downstream gene responses. We noticed that all PTPN3 variants were similarly localized and co‐localized with TβRI at or adjacent to the plasma membrane, indicating that L232R mutation did not alter its cellular localization (Figs 5A and EV4A). We then compared L232R with wild‐type PTPN3 and/or phosphatase‐dead mutant PTPN3 (D811A) in their ability to stabilize TβRI. While stable expression of PTPN3 or PTPN3 (D811A) enhanced the level of endogenous TβRI in HaCaT cells (Fig 5B, lanes 2 and 3), PTPN3 (L232R) was defective in regulating TβRI (Fig 5B, lane 4). Increasing amounts of PTPN3 or PTPN3 (D811A) gradually increased the protein level of TβRI in the presence of Smurf2 (Fig 5C, lanes 3–8). In contrast, PTPN3 (L232R) clearly failed to restore the level of TβRI (Fig 5C, lanes 9–11). Thus, L232R mutation led to the loss of PTPN3 function in regulating TβRI stability.
Figure 5. PTPN3 (L232R) fails to augment TGF‐β signaling.
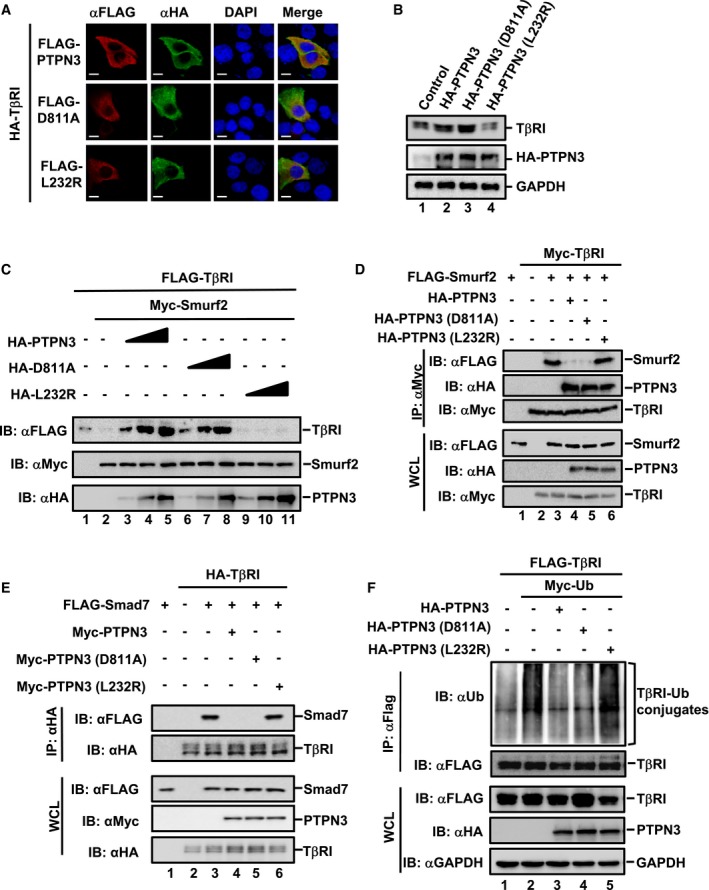
- PTPN3 wild‐type, phosphatase‐dead D811A and ICC‐derived mutant L232R exhibit similar patterns of subcellular localization and co‐localization with TβRI. HaCaT cells were transfected with HA‐TβRI and Flag‐PTPN3 (wild‐type or its mutants). Twenty‐four hours later, cells were harvested for immunofluorescence with the indicated antibodies. Scale bar = 10 μm.
- PTPN3 (L232R) does not increase the TβRI protein level. TβRI and PTPN3 were determined by Western blotting in HaCaT cells stably expressing PTPN3, PTPN3 (D811A), or PTPN3 (L232R).
- PTPN3 (L232R) fails to rescue the Smurf2‐mediated degradation of TβRI. HEK293T cells were transfected with FLAG‐TβRI, Myc‐Smurf2, and the increasing amount of PTPN3 (wild‐type and mutants). Cells were harvested 24 h post‐transfection. Protein levels were examined by Western blotting with the indicated antibodies.
- PTPN3 (L232R) does not block the interaction between TβRI and Smurf2. HEK293T cells were co‐transfected with plasmids as indicated. Twenty hours later, cells were treated with MG132 (20 μM) for 4 h and analyzed by IP/Western blotting.
- Ectopic expression of PTPN3 wild‐type and PTPN3 (D811A), but not PTPN3 (L232R), completely abolishes the Smad7‐TβRI interaction. HEK293T cells were co‐transfected with plasmids as indicated. Twenty hours later, cells were treated with MG132 (20 μM) for 4 h, followed by IP and Western blotting.
- PTPN3 and PTPN3 (D811A), but not PTPN3 (L232R), attenuate ubiquitination of TβRI. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, IP/Western blotting analysis was performed with anti‐Flag antibody IP and followed by Western blotting with the indicated antibodies.
Figure EV4. Subcellular localization and TβRI‐binding feature of PTPN3 L232R.
- PTPN3‐FERM and PTPN3‐FERM (L232R) share similar patterns of subcellular localization and co‐localization with TβRI. HaCaT cells were transfected with FLAG‐TβRI, HA‐PTPN3‐FERM, and HA‐PTPN3‐FERM (L232R). Twenty‐four hours later, cells were harvested for immunofluorescence with the indicated antibodies. Scale bar = 10 μm (except scale bar = 5 μm in the last column).
- The interactions between PTPN3 (L232R) and TβRI truncations. HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, immunoprecipitation and immunoblotting analysis were performed with the anti‐HA antibody. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
- Deletion of the GS region of TβRI had no effect on its interaction with PTPN3 (L232R). HEK293T cells were transfected with the indicated plasmids. Twenty‐four hours after transfection, immunoprecipitation and immunoblotting analyses were performed with the anti‐HA antibody. Levels of indicated proteins were examined by Western blotting with the indicated antibodies.
- Deletion of the GS, GS‐loop, GS‐α1, or GS‐α2 region of TβRI had no effect on its interaction with PTPN3 (L232R). HEK293T cells were transfected with the indicated plasmids. Twenty hours later, cells were treated with MG132 (20 μM) for 4 h, followed by Western blotting with the indicated antibodies.
Since PTPN3 or PTPN3 (D811A) interacted with TβRI to block its binding to Smurf2 (Fig 4F) as the mechanism to stabilize TβRI and, hence, enhance TGF‐β activity, we further assessed the ability of PTPN3 (L232R) in regulating the TβRI‐Smurf2 interaction. PTPN3 (L232R) failed to block the TβRI‐Smurf2 (Fig 5D, lane 6) or TβRI‐Smad7 interactions (Fig 5E, lane 6) as compared to the blocking ability of PTPN3 or PTPN3 (D811A) (Fig 5D and E, lanes 4 and 5). Interestingly, PTPN3 (L232R) bound to the TβRI‐Δ4 (Fig EV4B, lane 7), TβRI‐ΔGS (Fig EV4C, lane 3), and TβRI mutants with smaller deletions in the GS region (Fig EV4D, lanes 5–7), whereas wild‐type PTPN3 did not (Fig 4D), suggesting that they bind to TβRI in different modes. Consistently, unlike PTPN3 or PTPN3 (D811A), PTPN3 (L232R) was unable to effectively attenuate TβRI ubiquitination (Fig 5F, lane 5).
Phosphorylation of Smad2/3 is the indicator of TβRI activity. Next, we examined the effect of PTPN3 (L232R) on TGF‐β‐induced Smad2 and Smad3 phosphorylation in HaCaT cells in comparison with that of PTPN3 and PTPN3 (D811A). As expected, TGF‐β (2 ng/ml, 1 h) induced Smad2 and Smad3 phosphorylation, indicated by increased levels of P‐Smad2 and P‐Smad3, respectively (Fig 6A, lane 2). Both PTPN3 and phosphatase‐dead PTPN3 (D811A) apparently increased levels of P‐Smad2 and P‐Smad3 in HaCaT stable cells (Fig 6A, lanes 4 and 6), suggesting that PTPN3 enhances the phosphorylation of Smad2 and Smad3 in a phosphatase‐independent manner. Notably, PTPN3 (L232R) lost its effect in enhancing the levels of P‐Smad2 and P‐Smad3 in comparison with those of PTPN3 and PTPN3 (D811A) (Fig 6A, compare lane 8 with lanes 4 and 6).
Figure 6. PTPN3 enhances TGF‐β‐induced growth inhibitory responses.
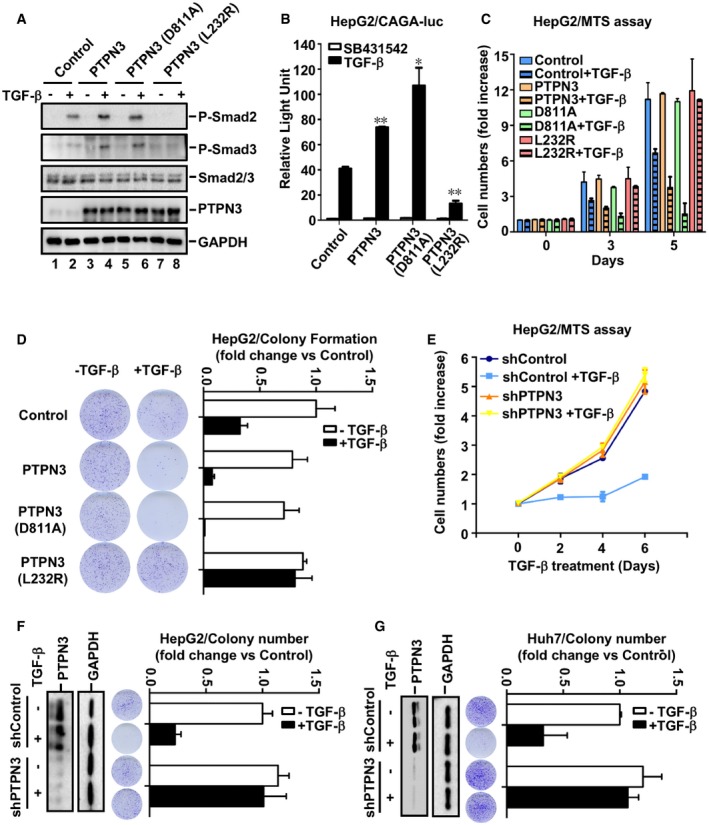
- PTPN3 and PTPN3 (D811A), but not PTPN3 (L232R), enhance Smad2 and Smad3 phosphorylation. HaCaT cells were stimulated with TGF‐β (2 ng/ml) for 1 h. Levels of P‐Smad2, P‐Smad3, Smad2, Smad3, PTPN3, and GAPDH were detected by Western blotting.
- PTPN3 (L232R) inhibits TGF‐β‐induced reporter gene activity. HepG2 cells were transfected with expression plasmids for PTPN3, PTPN3 mutants and CAGA‐luc reporter, and Renilla‐luc reporter. Cells were harvested for luciferase assay after 8 h of TGF‐β (2 ng/ml) treatment. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05, **P < 0.01.
- PTPN3 and PTPN3 (D811A), but not PTPN3 (L232R), enhance TGF‐β‐induced growth inhibitory effect. HepG2 cells stably expressing PTPN3, PTPN3 (D811A), or PTPN3 (L232R) were treated with or without TGF‐β (1 ng/ml) for indicated days. Cells were analyzed for cell proliferation by MTS assay. Data are shown as mean ± SEM; n = 3.
- Depletion of PTPN3 attenuates TGF‐β‐induced growth inhibitory effect. HepG2 cells stably expressing shPTPN3 were treated with or without TGF‐β (1 ng/ml) for indicated days. Cells were analyzed for cell proliferation by using MTS assay. Data are shown as mean ± SEM; n = 3.
- PTPN3 (L232R) attenuates TGF‐β‐induced growth inhibition on colony formation in HepG2 cells. Cells were treated with or without TGF‐β (1 ng/ml) treatment for 10 days. Clonogenic assay is described in Materials and Methods. Quantification of the results in clonogenic assays was analyzed by ImageJ. Data are shown as mean ± SEM; n = 3.
- Depletion of PTPN3 attenuates TGF‐β‐induced growth inhibitory effect in HepG2 cells. Cells with stable knockdown of PTPN3 were treated with or without TGF‐β (1 ng/ml) for 10 days. Clonogenic assay is described in the main text. Quantification of the results in clonogenic assays was analyzed by ImageJ. Knockdown efficiency of PTPN3 in HepG2 cells was analyzed by Western blotting with the anti‐PTPN3 antibody. Data are shown as mean ± SEM; n = 3.
- Depletion of PTPN3 attenuates TGF‐β‐induced growth inhibitory effect in Huh7 cells. Experiments and analysis were similarly done as in (F). Data are shown as mean ± SEM; n = 3.
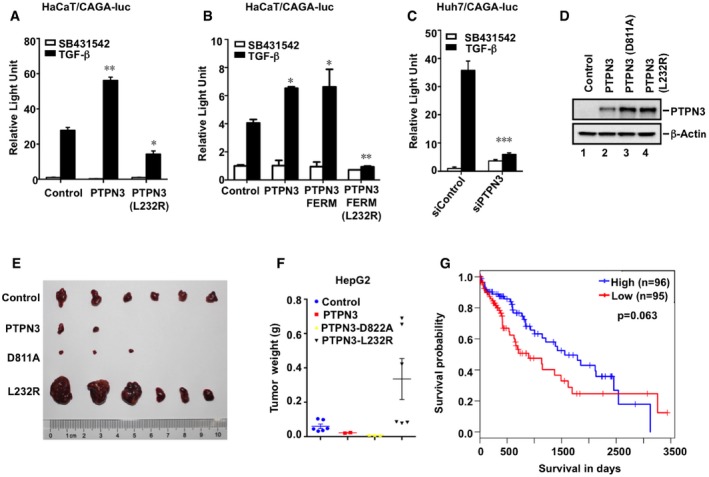
To analyze the functional outcome of the L232R mutation, we sought to determine the effect of PTPN3 (L232R) on TGF‐β gene responses. In consistent with the result in Figs 1 and 2, PTPN3 and PTPN3 (D811A) further potentiated TGF‐β‐induced transcription. In contrast, PTPN3 (L232R) significantly attenuated such responses in HepG2 (Fig 6B) or HaCaT cells (Fig EV5A). Since the L232R mutation is located in the FERM domain, HaCaT cells expressing PTPN3‐FERM (L232R) lost TGF‐β responsiveness (Fig EV5B). These results demonstrate that PTPN3 (L232R) completely lost its ability to potentiate TGF‐β signaling.
Figure EV5. Role of PTPN3 in tumorigenesis.
- PTPN3 (L232R) inhibits TGF‐β‐induced reporter gene activity. Transfection of HaCaT cells, TGF‐β treatment, and relative luciferase assays were similarly done as in Fig EV1A. Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05, **P < 0.01.
- PTPN3‐FERM (L232R) inhibits TGF‐β‐induced reporter gene activity. Experiments were done as in (A). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. *P < 0.05, **P < 0.01.
- Knockdown of PTPN3 decreases TGF‐β‐induced CAGA‐luc reporter activity in Huh7 cells. Transfection of Huh7 cells, TGF‐β treatment, and luciferase assays were similarly done as in (A). Data are shown as mean ± SEM; n = 3. Statistical analysis was performed using two‐tailed Student's t‐test. ***P < 0.001.
- Expression levels of PTPN3, PTPN3 (D811A), and PTPN3 (L232R) of HepG2 stable cells were analyzed by Western blotting.
- PTPN3 attenuates tumorigenesis in mouse models, while PTPN3 (L232R) exhibited the blocking effect. HepG2 cells stably expressing PTPN3, PTPN3 (D811A), and PTPN3 (L232R) were subcutaneously injected into nude mice. Fifty days after cell implantation, tumors were photographed.
- Weight from all tumors in (E) was recorded from Control, PTPN3, PTPN3 (D811A), and PTPN3 (L232R) groups. Data are shown as mean ± SEM; n = 6 tumors for each group.
- Higher PTPN3 expression is corrected with better survival in LIHC patient. A total of 191 samples from the TCGA LIHC database were analyzed. The log‐rank test was used to test whether gene levels were significantly associated with overall patient survival. Kaplan–Meier curve shows that LIHC patients with lower PTPN3 expression had a significantly high risk of recurrence than those with higher PTPN3 expression.
PTPN3 enhances TGF‐β‐induced growth inhibitory responses
After establishing the positive role of PTPN3 in TGF‐β transcriptional responses, we sought to investigate the role of PTPN3 in TGF‐β‐induced physiological responses such as cell proliferation. PTPN3 or its variant (D811A or L232R) was stably expressed in HepG2 cells, and the proliferation of these stable cells was examined by using MTS and the clonogenic assays. As expected, PTPN3 enhanced the TGF‐β‐induced growth inhibition (Fig 6C) and inhibition in colony formation (Fig 6D). The phosphatase‐dead mutant PTPN3 (D811A) behaved similarly as wild‐type PTPN3 in enhancing the TGF‐β effect on cell growth and colony formation (Fig 6C and D), whereas PTPN3 (L232R) lost its function on enhancing TGF‐β‐mediated growth responses (Fig 6C and D). Consistently, knockdown of PTPN3 significantly inhibited TGF‐β‐induced arrest on cell growth and colony formation in HepG2 (Fig 6E and F). The promoting effect of PTPN3 on TGF‐β responses was also observed in another HCC cell line Huh7, in which depletion of PTPN3 markedly attenuated TGF‐β‐mediated CAGA‐luc expression (Fig EV5C) and growth inhibitory responses (Fig 6G).
To support our findings from in vitro cell‐based assays, we investigated the function of PTPN3 in tumorigenesis. The tumorigenicity of Huh7 cells stably expressing PTPN3, PTPN3 (D811A), and PTPN3 (L232R) was examined (Fig 7A). Huh7 parental cells developed tumors by day 20 after injection of cells in mice as detected by luciferase‐induced bioluminescence (Fig 7B). Expression of PTPN3 or PTPN3 (D811A) significantly blocked tumor formation, whereas PTPN3 (L232R) failed to do so (Fig 7B and C). Using HepG2 cells, which required a long time to induce tumors, similar results were obtained as PTPN3 or PTPN3 (D811A) inhibited the tumor formation, whereas PTPN3 (L232R) lost this function (Fig EV5D–F). Furthermore, depletion of PTPN3 in HepG2 cells accelerated tumor appearance (Fig 7D–F). By examining the molecular events in the TGF‐β pathway, we found that tumors from PTPN3‐depleted cells had decreased TβRI level, reduced Smad2/3 phosphorylation, decreased expression of p15, and increased levels of c‐Myc (Fig 7G). These results demonstrate that knockdown of PTPN3 attenuated TGF‐β growth inhibitory and tumor‐suppressive responses.
Figure 7. PTPN3 enhances TGF‐β‐induced growth inhibitory responses.
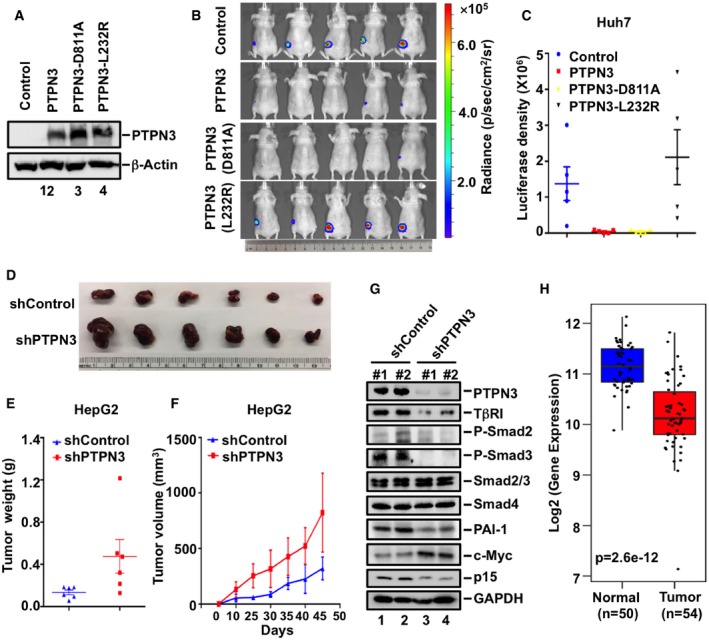
- PTPN3, PTPN3 (D811A), and PTPN3 (L232R) are similarly expressed in Huh7 stable cell lines. Expression levels of indicated proteins were detected with appropriate antibodies in Western blotting.
- PTPN3 and PTPN3 (D811A), but not PTPN3 (L232R), attenuate tumorigenesis. Luciferase‐harboring Huh7 tumor cells (2 × 106 cells/mouse) expressing PTPN3, PTPN3 (D811A) or PTPN3 (L232R), or empty vector (served as negative control) were subcutaneously injected into 5‐week‐old nude mice. Twenty days after injection, mice were analyzed by bioluminescence using Xenogen IVIS imaging system. Tumors expressing luciferase are indicated by radiance bar (p/sec/cm2/sr).
- Quantitation of bioluminescence with data from (B). Data are shown as mean ± SEM; n = 5.
- Depletion of PTPN3 accelerates tumorigenesis. HepG2 cells stably expressing shPTPN3 or shControl were subcutaneously injected into nude mice. Fifty days after cell implantation, tumors were dissected and photographed.
- Depletion of PTPN3 increased tumor sizes. Tumor volume was measured at indicated days post‐implantation. Data are shown as mean ± SEM; n = 6 for each group.
- Depletion of PTPN3 increased tumor weight. Weight from all tumors was recorded from both control and shPTPN3 groups. Data are shown as mean ± SEM; n = 6 for each group.
- PTPN3 depletion impairs TGF‐β signaling in tumors. Levels of PTPN3, GAPDH, and TGF‐β signaling‐associated proteins, such as TβRI, P‐Smad2, P‐Smad3, Smad2/3, Smad4, PAI‐1, c‐Myc, and p15 in tumors, were analyzed by Western blotting analysis.
- Clinical significance of PTPN3 expression in LIHC based on TCGA database. In the boxplots, y‐axis represents the expression level of PTPN3 in normal and tumor samples. The boxes show the median (horizontal line in the box) ± 1 quartile; the upper and lower box limits represent the upper and lower quartiles, respectively. The whiskers (vertical lines) extend from the hinge to the smallest or largest value within 1.5 interquartile range from the box boundaries.
Our results above indicated that PTPN3 plays an important role in facilitating TGF‐β anti‐growth response. We thus speculated that the expression of PTPN3 may be associated with tumorigenesis. We therefore analyzed the gene expression data in the TCGA database. We found that the mRNA level of PTPN3 was significantly downregulated in liver cancer clinical samples compared with normal samples (Fig 7H). Kaplan–Meier survival analyses of TCGA data also show that low expression of PTPN3 predicted poorer survival of liver cancer patients (Fig EV5G). These observations support the notion that PTPN3 acts as a tumor suppressor in liver cancer.
Discussion
Numerous lines of evidence support the importance of maintaining proper strength and duration of TGF‐β signaling in physiology and pathology. Association of the TGF‐β receptors with multiple complexes fine‐tunes the steady‐state levels and activities of TGF‐β receptors. One well‐studied example is the negative feedback product Smad7, which can recruit ubiquitin E3 ligase complexes for TβRI degradation, thereby dampening TGF‐β signaling events (Afrakhte et al, 1998; Kavsak et al, 2000; Yan et al, 2009; Miyazawa & Miyazono, 2017). Several ubiquitin E3 ligases such as Smurf1 and Smurf2 have been shown to target TβRI for ubiquitin‐mediated proteasomal degradation (Kavsak et al, 2000; Ebisawa et al, 2001). However, it is not known whether the negative function of Smad7 can be directly antagonized by yet‐unidentified signaling proteins. Here, we report that PTPN3 serves as such a gatekeeper to protect TβRI from Smad7‐/Smurf2‐mediated degradation. Our studies not only reveal a novel function of PTPN3 in fine‐tuning TGF‐β signaling, but also elucidate an underlying basis for PTPN3 as a suppressor in tumorigenesis.
PTPN3 is a critical regulator of TGF‐β signaling
Reversible phosphorylation of proteins is an important mechanism of signal transduction to maintain cellular homeostasis as well as to adapt to the external environment. The tight temporospatial control of protein tyrosine phosphorylation by protein tyrosine kinases (PTKs) and PTPs are essential in precisely regulating diverse cellular processes. Among PTPs, PTPN3 is an understudied protein tyrosine phosphatase with very limited implications on its functions. Most studies focus on identification and characterization of PTPN3 substrates such as EGFR, Egs15, and TCRζ (Sozio et al, 2004; Bauler et al, 2008; Chen et al, 2015; Parker, 2015). PTPN3 has also been reported to interact with vitamin D receptor (VDR), stabilizes VDR in the cytoplasm, and consequently promotes breast cancer growth (Zhi et al, 2011). What is the molecular basis for how PTPN3 functions in promoting TGF‐β signaling? Strikingly, PTPN3 functions to stabilize TβRI through antagonizing the negative function of Smad7. Smad7, which is induced by TGF‐β at the transcriptional level, can bind directly to TβRI in a feedback loop. It is generally thought that Smad7 targets TβRI for Smurf2‐mediated ubiquitination and proteasomal degradation (Kavsak et al, 2000) and protein phosphatase 1‐mediated dephosphorylation (Shi et al, 2004), thereby resulting in blockade of R‐Smad (e.g., Smad2/3) activation.
Like Smad7, PTPN3 directly interacts with TβRI. A series of biochemical assays demonstrate that the binding of PTPN3 and Smad7 to TβRI is overlapping as both PTPN3 and Smad7 bind to the same GS loop‐α2 region on TβRI (Fig 4D). We reason that through its direct binding to TβRI, PTPN3 blocks the binding of Smurf2 E3 ligase to TβRI, thereby ensuring the stabilization of TβRI. Indeed, ectopic expression of PTPN3 can override the negative action of Smurf2 to maintain the steady‐state level of TβRI (Fig 3C–E), whereas knockdown of PTPN3 enhances the Smurf2 association with TβRI. The L232R substitution in the FERM domain also fails to augment TβRI stability, which is consistent with the inability of the L232R mutant to replace Smad7 on TβRI. The L232R mutant binds to TβRI independent of the GS region, which is apparently in a different binding mode from wild‐type PTPN3. Furthermore, the destructive effect of PTPN3 depletion on TβRI stability can be counterbalanced by simultaneous knockdown of Smurf2 (Fig 3F) or co‐expression of dominant‐negative mutant of Smurf2 (Fig 3G).
Our biochemical experiments demonstrate that the PTPN3‐TβRI interaction requires the FERM domain of PTPN3 and the GS motif of TβRI. Since FERM domain is one of common domains associated with membrane signaling and the GS motif is a signature motif of all type I receptor of members of the TGF‐β superfamily, this raises two interesting questions: First, does PTPN3 or a related protein bind to other members of the TGF‐β receptor superfamily and protect the receptor from proteasomal degradation? Second, do other FERM domain‐containing proteins regulate TGF‐β receptor superfamily signaling? Extended investigation is needed to answer these questions, which should help understand the fine‐tuning regulation of TGF‐β superfamily signaling in physiological and pathological processes.
Collectively, our findings illustrate that PTPN3 disrupts the Smad7/Smurf2 negative loop and ensures robust TGF‐β signaling responses. This represents an important regulatory mechanism in TGF‐β signaling. It would also be interesting to investigate whether PTPN3‐mediated stabilization of VDR utilizes a similar mechanism to that of TβRI described in this study.
PTPN3 acts as a tumor suppressor
TGF‐β is a potent inhibitor of cell proliferation in a wide variety of cell types, particularly epithelial and endothelial cells. Loss of TGF‐β tumor‐suppressive signaling is a hallmark in cancer. Deletions or mutations of the genes encoding TGF‐β receptors or Smads are frequent, yet largely restricted in a few cancer types, including pancreatic and colon cancers. Somatic mutation or depletion of Smad4 is also common in cholangiocarcinoma and loss of TGF‐β receptor II has been found in ICC (Ong et al, 2012; Jiao et al, 2013). TGF‐β signaling inhibits cholangiocyte proliferation at later stages so that it attenuates the development of cholangiocarcinoma arising from hepatocytes and cholangiocytes (Mu et al, 2016). Our observations that the PTPN3 promotes TGF‐β signaling provide compelling evidence that PTPN3 fits well as a tumor suppressor as other components in the TGF‐β pathway.
Tyrosine phosphorylation plays a vital role in malignant transformation and tumor progression. Numerous studies have shown that PTPs suppress tumorigenesis (Tonks, 2013). However, there are two features that distinguish the tumor suppressor role of PTPN3 from other PTPs. First, while most PTPs negatively regulate the growth‐promoting functions of PTKs (Tonks, 2013), PTPN3 positively regulates the growth inhibitory functions of TGF‐β signaling (Figs 1 and 7). Second, most PTPs function through their tyrosine phosphatase activities toward growth signal‐induced PTK activation, whereas PTPN3 enhances TGF‐β signaling completely independent of its catalytic activity (Fig 2). We found both PTPN3 and its catalytically dead mutant equally stabilize TβRI, facilitate R‐Smad phosphorylation, and promote TGF‐β physiological responses (Figs 3, 5 and 6). Knockdown of PTPN3 profoundly inhibits TGF‐β growth inhibitory and gene responses (Figs 1 and 6). Thus, PTPN3 enables TGF‐β to induce higher amplitude of responses, which means more potent growth suppression in epithelial cells. This is not totally surprising since growth‐promoting tyrosine phosphorylation unlikely exists on TGF‐β receptors or Smads, even though the type II TGF‐β receptor is auto‐phosphorylated on tyrosine (Lawler et al, 1997) and Smad4 is tyrosine‐phosphorylated in cancer (Zhang et al, 2019). Therefore, our findings suggest that PTPN3 acts as a growth inhibitor likely through at least two mechanisms: dephosphorylation‐mediated inactivation of oncogenic proteins (previously reported) and dephosphorylation‐independent stabilization of tumor suppressor TβRI (this study).
Given its role in promoting TGF‐β signaling, the level or activity of PTPN3 should be tightly controlled and loss of its function may contribute to tumorigenesis. Indeed, mutations in PTPN3, including the L232R substitution in ICC, frequently occur. Thus, an interesting question is whether/how the L232R substitution in PTPN3 loses its tumor suppressor activity in cholangiocarcinoma. PTPN3 (L232R) retains its phosphatase activity (Fig 2B), but loses ability to augment TGF‐β tumor‐suppressive responses (Figs 6 and 7). Mechanistically, PTPN3 (L232R) loses the ability to antagonize Smurf2 in targeting TβRI for degradation (Fig 5). Therefore, mutations in the PTPN3 gene may disable TGF‐β signaling to favor tumorigenesis. Indeed, analysis of gene expression data in the TCGA database reveals that PTPN3 was significantly repressed in liver cancers samples (Fig 7H) and that low expression of PTPN3 predicted poorer survival of liver cancers patients (Fig EV5G). These confirm the tumor‐suppressive role of PTPN3 in liver cancer.
TGF‐β signaling plays dual roles in the regulation of tumor progression (Massague, 2008). TGF‐β induces growth inhibitory and apoptosis to suppress tumorigenesis and also stimulates cell migration and EMT as a tumor promoter (Akhurst & Derynck, 2001; Massague, 2008; Drabsch & ten Dijke, 2012). Since PTPN3 acts at the TGF‐β receptor level, it should enhance TGF‐β‐induced growth inhibition as well as TGF‐β tumor‐promoting properties. Indeed, PTPN3 could promote TGF‐β‐induced cell invasiveness and EMT, indicative of tumor promotion. It awaits further investigation on whether PTPN3 promotes tumor metastasis.
In summary, our study adds PTPN3 as a novel regulator of TGF‐β signaling and elucidates an essential function of PTPN3 in regulating tumor progression. Understanding how cancer‐derived mutants of PTPN3 function in tumorigenesis has important clinical implications in cancers and other diseases.
Materials and Methods
Plasmids
Expression plasmids for CAGA‐luc, Renilla‐luc, HA‐TβRI, Myc‐TβRI, Myc‐ubiquitin, FLAG‐Smurf2, FLAG‐Smurf2 (C716A), and pSRG‐Smurf2 were described previously (Wrighton et al, 2008). Human PTPN3 ORF cDNA was obtained by PCR. C‐terminally FLAG‐tagged and HA‐tagged PTPN3 were constructed by inserting the ORF into CMV‐driven expression vector pRK5F and pRK3HA, respectively, which are derivatives of pRK5 (Genentech). PTPN3 mutants, i.e., L232R, D811A, and C842S, were made by PCR‐based mutagenesis and confirmed by sequencing. Truncations PTPN3‐FERM, PTPN3‐PDZ, PTPN3‐PTP, and PTPN3‐FERM‐PDZ were generated by PCR and constructed into pRK3HA. Full‐length or mutated PTPN3 cDNA was also sub‐cloned into pWPI‐puro vector. To construct shRNA plasmids, we used pLKO.1‐puro vector. We made the following shRNA constructs against PTPN3: shRNA PTPN3‐1 (target sequence GCATTCCTAAGCTGAACGAAG) and shRNA PTPN3‐2 (target sequence GCTAACCTTGTGAACAAGTAC).
Antibodies and reagents
Antibodies were obtained as follows: anti‐p15 (C0287) antibody from Assay Biotech; anti‐PTPN3 (sc‐515181), anti‐GAPDH (FL‐335), anti‐Myc (sc‐40), Ub (sc‐8017), and anti‐Smad4 (sc‐7966) antibodies from Santa Cruz; anti‐beta‐actin (A5441), anti‐TβRI (ab‐31013) from Abcam; anti‐FLAG (F3165) and mouse IgG (I5381) from Sigma‐Aldrich; anti‐HA (3724), anti‐p21 (2947), anti‐PAI‐1 (11907), anti‐phospho‐Smad2 (3108), anti‐phospho‐Smad3 (9520), anti‐Smad2/3 (8685), anti‐vimentin (5741), and anti‐E‐cadherin (3195s) antibodies from Cell Signaling Technology; N‐cadherin (1610920) from BD Biosciences; horseradish peroxidase‐conjugated goat anti‐rabbit (111‐035‐045) and rabbit anti‐mouse (315‐035‐048) from Jackson ImmunoResearch.
The following chemical compounds were commercially obtained: SB431542 (S4317) from Sigma‐Aldrich; TGF‐β (TGFB1‐100) from StemRD. If not specified, the following concentrations of the reagents or chemicals were generally used in cell culture: TGF‐β at a final concentration of 2 ng/ml, SB431542 at 5 μM, and MG132 (Calbiochem) at 20 μM.
Cell culture and transfection
HaCaT and HepG2 cells were cultured in EMEM (Lonza) supplemented with 10% fetal bovine serum (FBS) (Invitrogen). A549 cells were maintained in RPMI 1640 (Corning) with 10% FBS. HEK293T and Huh7 cells were maintained with DMEM supplemented with 10% FBS. HaCaT and A549 cells were transfected with X‐tremeGENE (Roche Applied Science), while HEK293T cells with PEI (Polyscience).
RNA interference
For transient knockdown of PTPN3, siRNAs targeting human PTPN3 were transfected into cells for 48 h using Lipofectamine® RNAiMAX Reagent (Invitrogen). siRNAs were made by RiboBio Co: siPTPN3 (Human), #1 target sequence: CCTTATCAGTGGAGCACTT (Gao et al, 2014), #2 target sequence: CGACTTCTATGGAGTAGAA; siPTPN3 (Mouse), target sequence: GTGTGAAGCGATCCTTCGA. For stable knockdown of PTPN3, an shRNA against PTPN3 was expressed via lentiviral vector pLKO.1‐shRNA‐puro. Lentiviral production was made according to standard methods as described below.
Lentivirus production and stable cell line generation
Lentiviral vector pWPI‐puro carrying PTPN3 cDNA or pLKO.1‐shRNA‐puro harboring shPTPN3 was transfected into HEK293T cells together with packaging plasmid psPAX2 and envelope plasmid pMD2. After 48 h, supernatants of lentiviruses were collected and used to infect host cells. Stable cells were selected in the presence of puromycin (Sigma).
Transcription reporter assay
Sixteen hours after transfection, cells were treated with TGF‐β (2 ng/ml, 8 h) as described (Dai et al, 2009). Cells were then harvested and analyzed with the Dual Luciferase Reporter Assay system (Promega) and Luc‐PairTM Duo‐Luciferase HS Assay Kit (# LF006). All assays were done in triplicates, and all values were normalized for transfection efficiency against Renilla luciferase activities.
Quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
Total RNAs were obtained by TRIzol method (Invitrogen). RNAs were reverse‐transcribed to cDNA using the PrimeScript® RT reagent kit (TaKaRa). qRT–PCR was performed on an ABI PRISM 7500 Sequence Detector System (Applied Biosystems) using gene‐specific primers and SYBR Green Master Mix (Invitrogen). For the amplification, gene‐specific primers were used as follows (5′–>3′):
Human p21 ACCATGTGGACCTGTCACTGT (Forward),
TTAGGGCTTCCTCTTGGAGAA (Reverse);
Human PAI‐1 CAAGAGTGATGGCAATGTGAC (Forward),
TTTGCAGGATGGAACTACGG (Reverse);
Human GAPDH CGACCACTTTGTCAAGCTCA (Forward),
TTACTCCTTGGAGGCCATGT (Reverse);
Human PTPN3 TGTAAACTATGTGAGGTCTCTG (Forward),
AGCTCCACCTAGAAGCACAGAA (Reverse);
Human TβRI ACGGCGTTACAGTGTTTCTG (Forward),
GCACATACAAACGGCCTATCTC (Reverse).
Western blotting and IP analysis
Cells were incubated with TGF‐β (2 ng/ml) or chemical inhibitors for the indicated time periods, washed twice with ice‐cold PBS, and suspended in ice‐cold lysis buffer (150 mM NaCl, 20 mM Tris–HCl [pH 7.4], 5 mM EDTA, and 1% Triton X‐100) containing a protease inhibitor mixture (Sigma‐Aldrich). Proteins were solubilized in a sample buffer at 95°C for 5 min and resolved by 10% SDS–PAGE. For Western blotting, proteins were electro‐transferred onto a PVDF membrane and blocked with 5% Blotting‐Grade Blocker (#170‐6404, Bio‐Rad) in TBS‐0.10% Tween‐20 (TBST) at room temperature for 1 h. Then, the membrane was incubated with primary antibody at 4°C overnight. After washing in TBST and subsequent blocking, the blot was incubated with HRP‐conjugated secondary antibody (1:10,000, Jackson ImmunoResearch) for 1 h at room temperature. After washing, antibody binding was detected with the ECL detection reagent (Thermo Fisher).
For IP, 24 h after transfection, cell lysates were harvested by NET lysis buffer (150 mM NaCl, 20 mM Tris–HCl [pH7.4], 5 mM EDTA, and 0.5% NP‐40) and incubated with protein A Sepharose CL‐4B (GE Healthcare) and appropriate antibodies for 4 h. After extensive washes, immunoprecipitated proteins were eluted in SDS sample loading buffer, separated by SDS–PAGE, transferred onto PVDF membranes (Millipore), and detected by Western blotting analysis.
RNA‐Seq and data analysis
Cells were harvested for total RNA extraction with TRIzol (Invitrogen). Then, the RNA samples were subjected to transcriptome sequencing (RNA‐Seq) with the VAHTS™ mRNA‐seq V2 Library Prep Kit for Illumina (Vazyme). Sequencing reads were trimmed to 50 bp and mapped to the human genome (hg19) using Tophat v2.1.1. Only uniquely mapped reads (~90% of total reads) were subsequently assembled into transcripts guided by the reference annotation (UCSC gene models) with Cufflinks v2.2.1. Expression level of each gene was quantified with normalized FPKM (fragments per kilobase of exon per million mapped fragments). Genes with FPKM < 1 in all samples were excluded in subsequent analyses. For the remaining genes, all FPKM values that are less than 1 were set to 1. Venn diagrams and heat maps were generated using “VennDiagram” and “gplots” packages in R, respectively.
Immunofluorescence
Cells were cultured on glass coverslips for 24 h. After washing twice with PBS, cells were incubated with 4% PFA for 20 min and then incubated with 0.5% Triton X‐100 for 15 min at room temperature. The cells were then blocked in 5% BSA and incubated with primary antibody at 4°C overnight, washed three times with PBS, and incubated with Alexa Fluor 488 or Alexa Fluor 546 goat anti‐rabbit antibody (1:1,000, Invitrogen) or donkey anti‐mouse antibody (1:1,000, Invitrogen) for 1 h at room temperature. After three washing steps, all the slides and coverslips were mounted with ProLong Gold antifade with DAPI reagent (Invitrogen). Fluorescence images were captured by Zeiss Axiovert 200M microscope (Carl Zeiss).
MTS assay
MTS assay (Promega, Madison, WI, USA) was used to determine relative cell growth. Cells were seeded into 96‐well plates at a density of 2,000 cells in a final volume of 100 μl/well, incubated without serum at 37°C for 24 h, and then treated with TGF‐β (1 ng/ml). Upon indicated days of treatment, 20 μl of MTS was added into each well and cells were then incubated for 4 h at 37°C. Finally, the absorbance was measured at 490 nm using a microplate reader (Bio‐Rad iMark, Hercules, CA, USA). All experiments were performed in triplicate.
Clonogenic assay
Equal numbers of cells were trypsinized, resuspended, and seeded at a density of 2,000 cells/ml into 35‐mm culture plates and incubated at 37°C for the days as indicated. At the end of the experiments, the cells were stained with 0.1% crystal violet for 15 min. The positive colonies with > 50 cells were counted under a microscope.
In vivo tumor formation assay
A total of 2 × 106 cells were suspended in 100 μl cell culture media with Matrigel and then injected subcutaneously into 5‐week‐old nude mice. Twenty days later, tumorigenesis was determined by bioluminescent imaging on a Xenogen IVIS‐200 (Caliper Life Sciences, Hopkinton, MA), or after 50 days post‐injection, mice were sacrificed and tumors were excised and measured.
Statistical analysis
Kaplan–Meier method was used to calculate survival curves and used a log‐rank test to check whether gene levels were significantly associated with overall patient survival. Patient samples were grouped into high and low expression groups based on medium expression of PTPN3. t‐test was used to compare PTPN3 mRNA levels between normal and tumor samples from The Cancer Genome Atlas (TCGA) liver cancer.
Author contributions
X‐HF conceived and coordinated the study. X‐HF, XL, LS, JJ, YL, JX, and XC supervised experimental design. BY designed and performed most experiments. JL helped with some biochemical/cellular experiments and participated in manuscript revision. JC performed in vivo tumor formation assay. HZ, FW, YZ, and LS conducted RNA‐Seq analysis. YQY performed TCGA database analysis in Figs 7H and EV5G. LM did tumor collection and analysis. DX and NX conducted initial screen. BZ and PX provided essential reagents. BY, JL, YY, XL, and X‐HF analyzed data; BY, XL, and X‐HF wrote the manuscript. MX, YY, SL, BZ, and JX participated in manuscript writing.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 4
Acknowledgements
We thank Xianghuo He for HCC‐9810, Huh28, and RBE cells, Junfang Ji for database analysis and Huh7‐luc cells, Kaiyi Li for HepG2, Huh7, SNU423, and SNU449 cells, and Peter ten Dijke for CAGA‐luc reporter plasmid. We thank the laboratory members for helpful discussion and technical assistance. This research was partly supported by grants from National Natural Science Foundation of China (NSFC) (31730057, 91540205, 31571447), MOST 973 Program (2015CB553803), and the Fundamental Research Funds for the Central Universities.
The EMBO Journal (2019) 38: e99945
Data availability
RNA‐seq data have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE127903 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127903).
References
- Afrakhte M, Moren A, Jossan S, Itoh S, Sampath K, Westermark B, Heldin CH, Heldin NE, ten Dijke P (1998) Induction of inhibitory Smad6 and Smad7 mRNA by TGF‐beta family members. Biochem Biophys Res Commun 249: 505–511 [DOI] [PubMed] [Google Scholar]
- Akhurst RJ, Derynck R (2001) TGF‐beta signaling in cancer–a double‐edged sword. Trends Cell Biol 11: S44–S51 [DOI] [PubMed] [Google Scholar]
- Arpin M, Algrain M, Louvard D (1994) Membrane‐actin microfilament connections: An increasing diversity of players related to band 4.1. Curr Opin Cell Biol 6: 136–141 [DOI] [PubMed] [Google Scholar]
- Bauler TJ, Hendriks WJ, King PD (2008) The FERM and PDZ domain‐containing protein tyrosine phosphatases, PTPN4 and PTPN3, are both dispensable for T cell receptor signal transduction. PLoS ONE 3: e4014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell DM, Roulot D, George J (2001) Transforming growth factor beta and the liver. Hepatology 34: 859–867 [DOI] [PubMed] [Google Scholar]
- Chen KE, Li MY, Chou CC, Ho MR, Chen GC, Meng TC, Wang AH (2015) Substrate specificity and plasticity of FERM‐containing protein tyrosine phosphatases. Structure 23: 653–664 [DOI] [PubMed] [Google Scholar]
- Dai F, Lin X, Chang C, Feng XH (2009) Nuclear export of Smad2 and Smad3 by RanBP3 facilitates termination of TGF‐beta signaling. Dev Cell 16: 345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta PK, Blake MC, Moses HL (2000) Regulation of plasminogen activator inhibitor‐1 expression by transforming growth factor‐beta ‐induced physical and functional interactions between smads and Sp1. J Biol Chem 275: 40014–40019 [DOI] [PubMed] [Google Scholar]
- Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF (1995) Transforming growth factor beta induces the cyclin‐dependent kinase inhibitor p21 through a p53‐independent mechanism. Proc Natl Acad Sci USA 92: 5545–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM (1998) Direct binding of Smad3 and Smad4 to critical TGF beta‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J 17: 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ (2007) Differentiation plasticity regulated by TGF‐beta family proteins in development and disease. Nat Cell Biol 9: 1000–1004 [DOI] [PubMed] [Google Scholar]
- Dooley S, ten Dijke P (2012) TGF‐beta in progression of liver disease. Cell Tissue Res 347: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabsch Y, ten Dijke P (2012) TGF‐beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 31: 553–568 [DOI] [PubMed] [Google Scholar]
- Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K (2001) Smurf1 interacts with transforming growth factor‐beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem 276: 12477–12480 [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R (2005) Specificity and versatility in tgf‐beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693 [DOI] [PubMed] [Google Scholar]
- Gao Q, Zhao YJ, Wang XY, Guo WJ, Gao S, Wei L, Shi JY, Shi GM, Wang ZC, Zhang YN et al (2014) Activating mutations in PTPN3 promote cholangiocarcinoma cell proliferation and migration and are associated with tumor recurrence in patients. Gastroenterology 146: 1397–1407 [DOI] [PubMed] [Google Scholar]
- Gordon KJ, Blobe GC (2008) Role of transforming growth factor‐beta superfamily signaling pathways in human disease. Biochim Biophys Acta 1782: 197–228 [DOI] [PubMed] [Google Scholar]
- Hsu EC, Lin YC, Hung CS, Huang CJ, Lee MY, Yang SC, Ting LP (2007) Suppression of hepatitis B viral gene expression by protein‐tyrosine phosphatase PTPN3. J Biomed Sci 14: 731–744 [DOI] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P et al (2013) Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 45: 1470–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi‐Googheri M, Daneshvar H, Nosratabadi R, Zare‐Bidaki M, Hassanshahi G, Ebrahim M, Arababadi MK, Kennedy D (2014) Important roles played by TGF‐beta in hepatitis B infection. J Med Virol 86: 102–108 [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6: 1365–1375 [DOI] [PubMed] [Google Scholar]
- Lawler S, Feng XH, Chen RH, Maruoka EM, Turck CW, Griswold‐Prenner I, Derynck R (1997) The type II transforming growth factor‐beta receptor autophosphorylates not only on serine and threonine but also on tyrosine residues. J Biol Chem 272: 14850–14859 [DOI] [PubMed] [Google Scholar]
- Li MY, Lai PL, Chou YT, Chi AP, Mi YZ, Khoo KH, Chang GD, Wu CW, Meng TC, Chen GC (2015) Protein tyrosine phosphatase PTPN3 inhibits lung cancer cell proliferation and migration by promoting EGFR endocytic degradation. Oncogene 34: 3791–3803 [DOI] [PubMed] [Google Scholar]
- Lin X, Liang M, Feng XH (2000) Smurf2 is a ubiquitin E3 ligase mediating proteasome‐dependent degradation of Smad2 in transforming growth factor‐beta signaling. J Biol Chem 275: 36818–36822 [DOI] [PubMed] [Google Scholar]
- Llovet JM, Zucman‐Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G (2016) Hepatocellular carcinoma. Nat Rev Dis Primers 2: 16018 [DOI] [PubMed] [Google Scholar]
- Ma S, Yin N, Qi X, Pfister SL, Zhang MJ, Ma R, Chen G (2015) Tyrosine dephosphorylation enhances the therapeutic target activity of epidermal growth factor receptor (EGFR) by disrupting its interaction with estrogen receptor (ER). Oncotarget 6: 13320–13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Curley SA, Wu X, Brown P, Hwang JP, Shetty K, Yao ZX, He AR, Li S, Katz L et al (2012) Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 9: 530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2008) TGFbeta in cancer. Cell 134: 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13: 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Nikolic‐Paterson DJ, Lan HY (2016) TGF‐beta: the master regulator of fibrosis. Nat Rev Nephrol 12: 325–338 [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Miyazono K (2017) Regulation of TGF‐beta family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol 9: a022095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Pradere JP, Affo S, Dapito DH, Friedman R, Lefkovitch JH, Schwabe RF (2016) Epithelial transforming growth factor‐beta signaling does not contribute to liver fibrosis but protects mice from cholangiocarcinoma. Gastroenterology 150: 720–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH et al (2012) Exome sequencing of liver fluke‐associated cholangiocarcinoma. Nat Genet 44: 690–693 [DOI] [PubMed] [Google Scholar]
- Parker EJ (2015) The molecular basis for the substrate specificity of protein tyrosine phosphatase PTPN3. Structure 23: 608–609 [DOI] [PubMed] [Google Scholar]
- Ponting CP, Phillips C, Davies KE, Blake DJ (1997) PDZ domains: targeting signalling molecules to sub‐membranous sites. BioEssays 19: 469–479 [DOI] [PubMed] [Google Scholar]
- Reynisdottir I, Polyak K, Iavarone A, Massague J (1995) Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF‐beta. Genes Dev 9: 1831–1845 [DOI] [PubMed] [Google Scholar]
- Shi W, Sun C, He B, Xiong W, Shi X, Yao D, Cao X (2004) GADD34‐PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol 164: 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia D, Villanueva A, Friedman SL, Llovet JM (2017) Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152: 745–761 [DOI] [PubMed] [Google Scholar]
- Sozio MS, Mathis MA, Young JA, Walchli S, Pitcher LA, Wrage PC, Bartok B, Campbell A, Watts JD, Aebersold R et al (2004) PTPH1 is a predominant protein‐tyrosine phosphatase capable of interacting with and dephosphorylating the T cell receptor zeta subunit. J Biol Chem 279: 7760–7769 [DOI] [PubMed] [Google Scholar]
- Tonks NK (2013) Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J 280: 346–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108 [DOI] [PubMed] [Google Scholar]
- Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS et al (2004) Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304: 1164–1166 [DOI] [PubMed] [Google Scholar]
- Warner BJ, Blain SW, Seoane J, Massague J (1999) Myc downregulation by transforming growth factor beta required for activation of the p15(Ink4b) G(1) arrest pathway. Mol Cell Biol 19: 5913–5922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton KH, Lin X, Feng XH (2008) Critical regulation of TGFbeta signaling by Hsp90. Proc Natl Acad Sci USA 105: 9244–9249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MY, Hill CS (2009) Tgf‐beta superfamily signaling in embryonic development and homeostasis. Dev Cell 16: 329–343 [DOI] [PubMed] [Google Scholar]
- Xu P, Liu J, Derynck R (2012) Post‐translational regulation of TGF‐beta receptor and Smad signaling. FEBS Lett 586: 1871–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Lin X, Feng XH (2016) Posttranslational regulation of Smads. Cold Spring Harb Perspect Biol 8: a022087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Liu Z, Chen Y (2009) Regulation of TGF‐beta signaling by Smad7. Acta Biochim Biophys Sin 41: 263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Tonks NK (1991) Isolation of a cDNA clone encoding a human protein‐tyrosine phosphatase with homology to the cytoskeletal‐associated proteins band 4.1, ezrin, and talin. Proc Natl Acad Sci USA 88: 5949–5953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Liu J, Kobayashi R, Tonks NK (1999) Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1‐related protein‐tyrosine phosphatase PTPH1. J Biol Chem 274: 17806–17812 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Xiao M, Gu S, Xu Y, Liu T, Li H, Yu Y, Qin L, Zhu Y, Chen F et al (2019) ALK phosphorylates SMAD4 on tyrosine to disable TGF‐beta tumour suppressor functions. Nat Cell Biol 21: 179–189 [DOI] [PubMed] [Google Scholar]
- Zhi HY, Hou SW, Li RS, Basir Z, Xiang Q, Szabo A, Chen G (2011) PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene 30: 1706–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 4
Data Availability Statement
RNA‐seq data have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE127903 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE127903).