

Abstract
Background: The Hedgehog (Hh) pathway is stimulated by inactivating mutations of Patched Homolog 1 (PTCH1) gene. There is accumulating evidence that Hh signaling plays a critical role in the pathogenesis of various haemopoietic malignancies. Particular interest has focused on the role of Hh signaling in chronic myeloid leukemia (CML). The Hh signaling is increased in BCR-ABL+ve progenitor cells and Hh signaling is further up regulated with disease progression. Aim: The aim of this study was to determine the frequency and types of PTCH1 gene mutations in Chronic Myeloid Leukemia (CML) patients and to correlate the effect of these mutations on the prognosis and outcome of CML and for predicting the imatinib response in CML patients. Subjects and methods: The study included fifty newly diagnosed CML patients and ten healthy volunteers (the control group) to verify the presence or absence of PTCH1 gene mutation. The patients were subjected to clinical examination, routine laboratory investigations, bone marrow examination, Cytogenetic evaluations of t(9;22) and molecular study of BCR-ABL fusion gene. All participants in this study were subjected to the assessment for the presence of PTCH1 gene mutation by DNA extraction followed by polymerase chain reaction (PCR) of genomic DNA corresponding to exon 23 of PTCH1 gene, purification of amplified PCR product, followed by sequencing analysis for detection of PTCH1 gene exon 23 mutations and the types of these mutations. Results: Four types of mutations of PTCH1 gene were detected in 24 CML patients (48%), three types of them were missence while the fourth type was frame shift mutation. There was no significant association between PTCH1 gene mutation and percent of BCR-ABL fusion genes at level less than 10% at 3 months of treatment, complete cytogenetic response (CCyR) at one year, disease free survival and overall survival. However there was significant association between PTCH1 gene mutation and imatinib failure (P=0.03). Conclusion: PTCH1 gene mutation should be considered a promising molecular marker for predicting the probability of imatinib response in CML patients. Hedgehog pathway activation in CML patients can raise a possibility that combinations of ABL and Hh inhibitors might offer a new treatment strategy in CML and might help to effectively cure this disease.
Keywords: CML, PTCH1 gene, hedgehog pathways
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder characterized by massive myeloid expansion, accumulation of differe-ntiating granulocytic precursors and terminally differentiated effectors cells leading to the key clinical features at presentation of marked peripheral blood granulocytosis, basophilia, splenomegaly and often thrombocytosis and anemia [1]. In the case of CML, it has been well established that the defining molecular pathogenetic event is a reciprocal chromosomal translocation t(9:22) that results in the production of an oncogenic chimeric fusion protein BCR-ABL [2]. The fusion of BCR with ABL results in constitutive activation of the ABL kinase, which is critical for the transforming properties of BCR-ABL [3]. Hence, the aberrant kinase activity of BCR-ABL promotes the activation of multiple proliferation and anti-apoptotic signaling cascades that contribute to leukemogenesis [4]. Most CML patients are diagnosed with a chronic phase characterized by an uncontrolled proliferation of myeloid elements that retain their ability to differentiate [5,6]. Through the acquisition of additional mutations, the disease progresses to the accelerated and blast crisis phases whereby normal hematopoiesis is severely compromised as the leukemic clone loses its ability to differentiate, eventually leading to the accumulation of immature cells or blasts in the bone marrow and blood [7]. While imatinib treatment has clearly improved the prognosis of CML patients especially in the chronic phase, the occurrence of relapse and resistance have occurred because the BCR-ABL leukemic stem cells can persist despite tyrosine kinas inhibitors (TKI) therapy, and efforts have intensified towards determining the molecular pathways that are critical for the maintenance of such cells [8].
One of the major pathways influencing stem cell self-renewal is the hedgehog (Hh) signaling pathway [9]. The Hh gene was first described by Nusslein-Volhard and Wieschaus (1980) when investigating mutations affecting segment number and polarity in Drosophila melanogaster.
Patched homolog 1 (PTCH1) is a component of the Hedgehog (Hh) signaling pathway, which is a key regulator of cell proliferation, cell surveillance, embryonic development, adult tissue homeostasis, and stem cell quiescence [10,11].
The Cytogenetic Location of PTCH1 is 9q22.32. The PTCH1 gene provides instructions for producing the patched-1 protein, which functions as a receptor. Based on its role in preventing cells from proliferating in an uncontrolled way, PTCH1 is called a tumor suppressor gene [12].
There are three Hh homologues in humans: Sonic Hedgehog (SHH), Desert Hedgehog (DHH) and Indian Hedgehog (IHH) [13]. The most studied, SHH, plays a pivotal role in a number of essential pleotropic processes, including embryonic development, stem cell maintenance and cellular proliferation [14]. The fundamental components of the Hh pathway are the cell surface Patched-1 (PTCH1) receptor and Smoothened (SMO), a heptahelical transmembrane G-protein coupled-receptor that initiates Hh cytoplasmic signalling. Under resting conditions and the absence of one or more of the three Hh ligands, PTCH1 is localized at the cell surface where it induces catalytic inhibition of SMO. PTCH1 possesses a sterol-sensing domain that appears pivotal for facilitating this tonic SMO inhibition [15,16].
Different alterations in Hh have been described in basocellular carcinoma, medulloblastoma, rhabd-myosarcoma, lung cancer, gastro-intestinal tract cancers, pancreatic cancer, lymphoma, and myeloma [17-24]. There is accumulating evidence that Hh signaling plays a critical role in the pathogenesis and genesis of various haemopoietic malignancies [25]. Particular interest has focused on the role of Hh signaling in CML. Studies have shown that Hh signaling is increased in BCR-ABL+ progenitor cells and Hh signaling is further upregulated with disease progression [26]. The persistence of CD34+38- stem/progen-itor cells that are non-cycling and thus insensitive to BCR-ABL1 inhibitors also plays a significant role. Targeting of the BCR-ABL1+ Leukemic Stem Cell (LSC) compartment in CML remains a rational goal as it is well recognized that these cells are not eliminated by the use of tyrosine kinas inhibitors (TKI), such as imatinib [27]. Therefore, alternative agents are required to eliminate this population, raise the possibility of functional cure and enable TKI discontinuation. Multiple studies have explored the contributory role of deregulated Hh pathway signaling in CML maintenance, progression and response to therapy [28-30].
The aim of this study was to determine the frequency and types of PTCH1 gene mutations in Chronic Myeloid Leukemia (CML) patients and to correlate the effect of these mutations on the prognosis and outcome of CML and for predicting the imatinib response in CML patients.
Subjects and methods
This study included 50 newly diagnosed CML patients ranging in age from 32 to 72 years; 22 were males and 28 were females. They all presented to medical oncology clinics, Zagazig University hospital, during the time period from May 2016 to March 2018.
Inclusion criteria: Newly diagnosed CML patients.
Exclusion criteria: CML patients under treatment and follow up.
Final diagnosis of CML was established according to the 2008 WHO criteria using medical history, physical examination, morphologic examination of peripheral blood and bone marrow smears. The Cytogenetic evaluations of t(9;22) and molecular study of BCR-ABL fusion gene are required for the diagnosis of CML according to the WHO classification. CML patients were divided into three groups: chronic phase (CP) 31 patients, accelerated phase (AP) 11 patients and blast crisis phase (BC) 8 patients. Ten haematologically normal individuals (controls) of matched age and sex were also included in the study to verify the presence or absence of PTCH1 gene mutation. Informed consents were taken from all patients.
Molecular detection of PTCH1 gene mutation
All patients in this study were subjected to the assessment for the presence of PTCH1 gene mutation by DNA extraction followed by polymerase chain reaction (PCR) of genomic DNA corresponding to exon 23 of PTCH1 gene, purification of amplified PCR product, followed by sequencing analysis for detection of PTCH1 gene exon 23 mutations and the types of these mutations.
DNA extraction and PCR
DNA was extracted from peripheral blood or bone marrow samples of CML patients and control group. DNA extraction from the white blood cells was achieved by the use of QIAamp DNA blood mini kit (QIAGEN). To screen the PTCH1 mutations, polymerase chain reaction (PCR) amplification of PTCH1 exon 23 was carried out by using primers PTCH1-F (5’-AACCCAAGGAGGGAAGTGTG-3’) and PTCH1-R (5’-AAGCCGTCACAGTGGTGATG-3’). PCR amplification was performed in a 25 ul reaction mixture, containing 100 ng of genomic DNA, 12.5 pmol of both forward and reverse primers, 10 mM of dNTPs, 0.75 ul of 50 Mm of MgCL2, 0.3 ul of hot start Taq DNA polymerase (5 unit/ul) Qiagen, 2.5 ul of 10 X Taq buffer and purified distilled water. PCR was performed with cycling condition of initial denaturation step at 94°C for 3 min followed by 35 cycles of Denaturation at 94°C for 30 second, Annealing at 58°C for 30 second, Extension at 72°C for 45 second and final extension at 72°C for 5 min. The Taq polymerase efficiently synthesies DNA. Cyclic repetition of the dena-turenting, annealing and extension steps, by changing the temperature of the reaction in an automated thermal cycler, resulted in exponential amplification of the DNA that lied between the two oligopeptides used. PCR products were separated on a 2% agarose gel.
DNA sequencing
Purification of amplified PCR product was achieved by the use of QIA quick PCR Purification Kit (QIAGEN). The concentration of purified PCR product was measured by nano drop spectrophotemeter to determine the concentration and purity of amplified DNA, then cycle sequencing was performed to purified PCR product according to the manufacturer’s protocol and directly sequenced using the same forward primer as for the original PCR amplification. Sequencing analysis was performed on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, USA). The sequencing results were examined at National Center for Biotechnology Information (NCBI), nucleotide BLAST (Basic Local Alignment Search Tool) web site for sequencing analysis services and for detection of PTCH1 gene mutations.
Statistical analysis
Data were analyzed using Statistical Package for the Social Sciences (SPSS version 20.0) software. According to the type of data qualitative represent as number and percentage, quantitative continues group represent by mean ± SD, the following tests were used to test differences for significance Difference and association of qualitative variable by Chi square test (X2). Differences between quantitative independent groups by t test. Kaplan Meier for survival curve was used.
P value was set at <0.05 for significant results.
Results
The present study included two groups: control group (10 normal individuals) and patients group (50 newly diagnosed CML patients). Patients were diagnosed at clinical pathology and medical oncology departments, faculty of medicine, Zagazig university during the time period from May 2016 to March 2018 with the following characteristics (Table 1).
Table 1.
The patients characteristics
| Clinicl charachteristic | Total No=50 | PTCH1 mutation No (%) | PTCHI normal No (%) |
|---|---|---|---|
| CML chronic phase | 31 (62) | 12 (38.7) | 19 (61.3) |
| CML accelerated phase | 11 (22) | 7 (63.6) | 4 (36.4) |
| CML blastic phase | 8 (16) | 5 (62.5) | 3 (37.5) |
| Response at 3 months No (%) | |||
| <10% BCR-ABL | 21 (44.7) | 10 (47.6) | 11 (52.4) |
| >10% BCR-ABL | 26 (55.3) | 13 (50) | 13 (50) |
| Response at 1 year | |||
| CCyR | 22 (51.2) | 11 (50) | 11 (50) |
| No CCyR | 21 (48.8) | 8 (38.1) | 13 (61.9) |
| Patients on Imatinip at last follow up | 39 | 20 (51.3) | 19 (48.7) |
| Imatinip failure | 6 | 5 (83.3) | 1 (16.7) |
| No imatinip failure | 37 | 14 (37.8) | 23 (62.2) |
| Imatinip discontinuation due to side effect | 4 | 3 (75) | 1 (25) |
| Evolution of chronic phase to AP/BC | 11 | 6 (54.5) | 5 (45.5) |
| Evolution of accelerated phase to Blastic crises | 2 | 1 (50) | 1 (50) |
| Deaths related to CML | 4 | 3 (75) | 1 (25) |
| Allo bone marrow transplantation | 0 | 0 | 0 |
| Patients lost during follow up | 3 | 2 (66.7) | 1 (33.3) |
CCyR: Complete cytogenetic response. AP: Accelerated phase. BC: Blastic crisis.
The ages of studied CML patients ranging from 32 to 72 years with a median of 50 years. Gender distribution in this study group was 22 males and 28 females with a female predominance. CML patients were divided into three groups: chronic phase, accelerated phase (AP) and blast crisis phase (BC). All patients were treated with imatinib.
Sex patients discontinued imatinib due to lack of efficacy. Four patients presented with primary resistance (i.e., failed to achieve CCyR with imatinib), and two patients with secondary resistance (i.e., positive response followed by a loss of response). Four patients discontinued imatinib due to side effects. CML-related death occurred in four patients, three of them (75%) had Ptch1 mutation.
Evolution of CML patients in chronic phase to accelerated and blastic phases were higher in patients with mutated PTCH1 (54.5%) than patients with normal PTCH1 (45.5%) (P=0.13).
PTCH1 exon 23 mutation was studied in all CML patients and control groups. It was detected by DNA sequencing analysis of purified PCR product of PTCH1 exon 23 DNA. There was no detectable mutation in PTCH1 gene in control group. PTCH1 gene mutations were identified in 24 CML patients (48%) Figure 1. PTCH1 mutilation was detected in 12 patients of CML in chronic phase (38.7%), in 7 patients of CML in accelerated phase (63.6%) and in 5 patients of CML in 5 (62.5%) patients of blastic crisis with no significant difference of PTCH1 mutation in different CML phases, as the frequency of PTCH1 mutation is higher in accelerated and blastic phases than chronic phase (P=0.24), Figure 2; Table 2.
Figure 1.
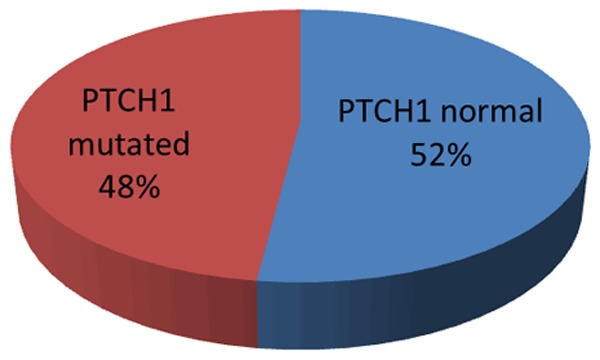
Frequency of PTCH1 gene mutation among 50 CML patients.
Figure 2.
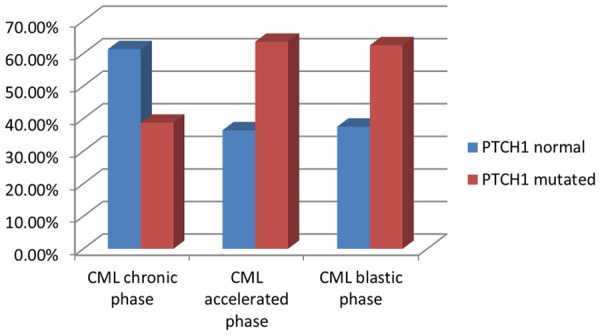
PTCH1 gene mutation in CML phases.
Table 2.
PTCH1 mutation in different CML phases
| PTCH1 gene | X2 | P | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mutated | Normal | |||||
| CML phases | Accelerated | N | 7 | 4 | 2.82 | 0.24 |
| % | 63.6% | 36.4% | ||||
| Blasic crises | N | 5 | 3 | |||
| % | 62.5% | 37.5% | ||||
| Chronic | N | 12 | 19 | |||
| % | 38.7% | 61.3% | ||||
Table 3 and Figures 3, 4, 5, 6 and 7 show description of identified PTCH1 mutation in the CML patients. Four types of PTCH1 mutations were detected in CML patients, the most evident one was substitution of nucleotide (c. 3944 T>C) which was detected in nine patients (39.2%), (c. 3936 G>A) was detected in seven patients (30.4%), (c. 3915 C>T) was detected in three patients (13%), the previous three types of mutation were missence mutations while the fourth type was heterozygous deletion of nucleotide C (c. C 3921 del) which cause frame shift mutation and was detected in four patients (17.4%).
Table 3.
Description of identified PTCH1 mutation in the CML patients
| Nucleotide change | Type | Identified cases | |
|---|---|---|---|
|
| |||
| No | % | ||
| c. 3944 T>C. | Missence | 9 | 39.2 |
| c. 3936 G>A. | Missence | 7 | 30.4 |
| c. C 3921 del | Frame shift | 4 | 17.4 |
| c. 3915 C>T. | Missence | 3 | 13 |
Figure 3.
PTCH1 wild type.
Figure 4.
Substitution of nucleotide. C. 3944 [T>C].
Figure 5.
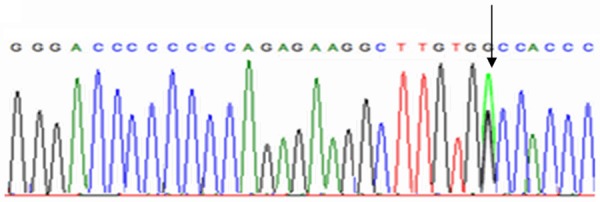
Substitution of nucleotide. C. 3936 [G>A].
Figure 6.
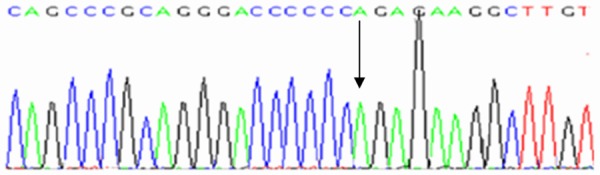
Deletion of nucleotide C [Del 3921 C].
Figure 7.
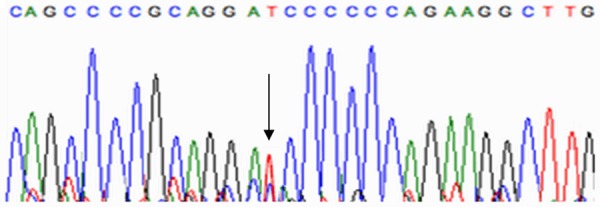
Substitution of nucleotide. C. 3915 [C>T].
Comparative analysis of percent of BCR-ABL fusion genes at level less than 10% at 3 months of treatment between CML patients with normal and mutated PTCH1 showed no statistically significant difference between two groups (P=0.8), (Table 4). complete cytogenetic response (CCyR) at one year revealed that there was no statistically significant difference between CML patients with normal and mutated PTCH1 (P=0.43), (Table 5).
Table 4.
Comparative analysis of percent of BCR-ABL fusion genes at level less than 10% at 3 months of treatment between CML patients with normal and mutated PTCH1
| PTCH1 gene | X2 | P | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mutated 23 | Normal 24 | |||||
| BCR_ABL3_months | <10% | N | 10 | 11 | 0.96 | 0.8 |
| % | 47.6% | 52.4% | ||||
| >10% | N | 13 | 13 | |||
| % | 50.0% | 50.0% | ||||
Table 5.
Comparative analysis of complete cytogenetic response (CCyR) at one year of treatment between CML patients with normal and mutated PTCH1
| PTCH1 gene | X2 | P | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Mutated 19 | Normal 24 | |||||
| CCyR at 1 year | NO | N | 11 | 11 | 0.61 | 0.43 |
| % | 50.0% | 50.0% | ||||
| YES | N | 8 | 13 | |||
| % | 38.1% | 61.9% | ||||
The response of treatment by imatinib revealed that there was statistically significant difference between two groups of CML patients as the imatinip failure was higher in CML patients with mutated PTCH1 (P=0.03) (Table 6). Calculated risk estimation revealed that the mutated group conferred two fold increased risk of no response to imatinib than the wild group (OR=2.2, 95% CI=1.2-3.8) (Figure 8).
Table 6.
Comparative analysis of the response of treatment by imatinib between CML patients with normal and mutated PTCH1
| PTCH1 gene | X2 | P | RR (CI 95%) | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Mutated 19 | Normal 24 | ||||||
| Imatinip failure | NO | N | 14 | 23 | 4.33 | 0.03* | 2.20 (1.2-3.8)* |
| % | 37.8% | 62.2% | |||||
| YES | N | 5 | 1 | ||||
| % | 83.3% | 16.7% | |||||
*RR (Relative risk), *CI (confidence interval).
Figure 8.
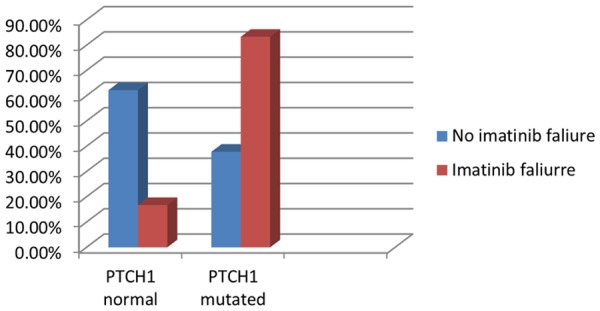
Comparison between wild and mutated groups as regards imatinib failure.
Kaplan-Meier analysis of our data after a follow up period of 20 months revealed that the overall survival (OS) was (96.2%) in CML patients with normal PTCH1 which is better than CML patients with mutated PTCH1 (87.5%). In addition, the mean survival time with 95% confidence interval was 11.69 ± 0.7 months in CML patients with mutated PTCH1 versus 14.46 ± 0.52 months in CML patients with normal PTCH1. The difference was statistically insignificant (P=0.26) (Figure 9).
Figure 9.
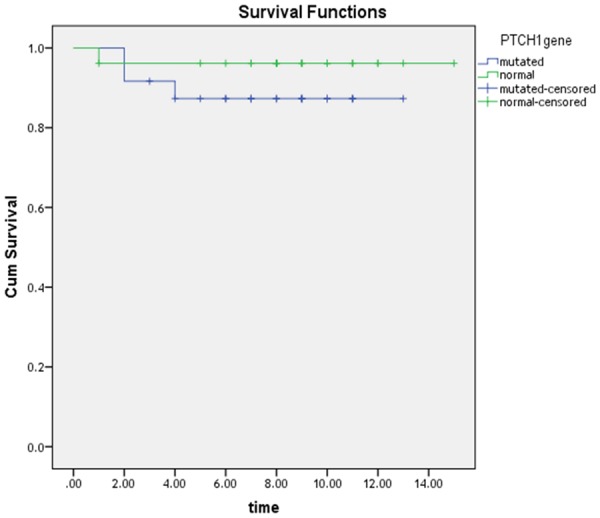
Kaplan Meier curve for OS for wild and mutated groups.
Disease free survival (DFS) was estimated after 15 months. The mean DFS with 95% confidence interval was 9.92 ± 0.83 months in CML patients with mutated PTCH1 versus 12.19 ± 0.85 months in CML patients with normal PTCH1. The difference was not statistically significant (P=0.8). The DFS was (69.9%) in CML patients with normal PTCH1 and it was (58.2%) in CML patients with mutated PTCH1 (Figure 10).
Figure 10.
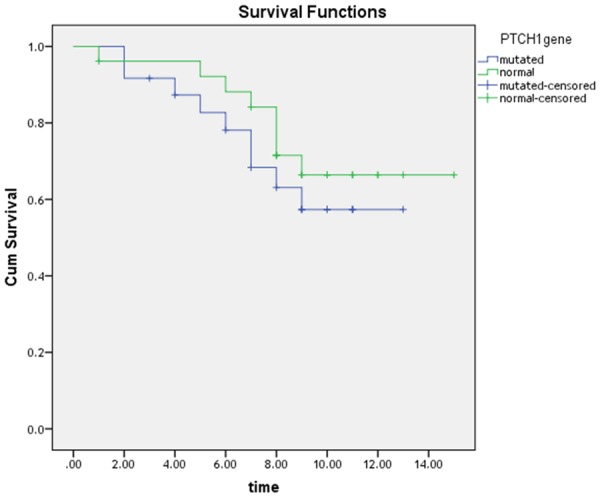
Kaplan Meier curve for DFS for wild and mutated groups.
Discussion
Hedgehog (Hh) signaling pathway is involved in regulation of many tissues development and oncogenesis. Hh signaling has been identified as a required functional pathway for leukemia stem cells (LSCs). Several mechanisms have been described that lead to the activation of the Hh signaling pathway in tumor cells, such as activating point mutations of SMO or inactivating point mutations in PTCH1 [31-35]. Mutation of PTCH1 leads to remove of its inhibitory effect of SMO and signaling transduction is activated which is promoting cellular proliferation and survival [36]. SO deregulation of the Hh pathway may also contribute to CML disease progression and imatinib failure. Patched homolog 1 (PTCH1) had a significant impact on predicting the response to imatinib [37].
In the present study, we evaluated the frequency and prognostic impact of (PTCH1) mutations among a group of Egyptian CML patients. This is the first time to study mutation of PTCH1 in CML patients, in the previous studies the expression only of the PTCH1 gene was studied.
In our study the median age of the patients was 50 years and the range was 32 to 72 years and the male to female ratio was 1:1.3. These findings are comparable to the study of Safaa and Nashwa, 2018 [38] including 180 CML patients at Assiut University Hospital-Egypt with a median age of 42 years and male to female ratio of 1:1.7.
In our study sequencing analysis of the PTCH1 exon 23 was done for all CML patients in different phases and for control group. It revealed that 24 out of 50 CML patients (48%) harbored PTCH1 gene mutations and there was no detectable mutation in PTCH1 gene in control group. Four types of PTCH1 mutations were detected in CML patients, three types of mutation were missence mutations while the fourth type was frame shift mutation, in the previous studies the expression only of the PTCH1 gene in CML was studied not mutations [39,40].
Xie et al., 1998 [33] reported that mutation of PTCH1 is Loss-of-funzction mutations, also called inactivating mutations, result in the gene product having less or no function (being partially or wholly inactivated) and leads to decrease the expression of the gene.
In the current study Comparative analysis of percent of BCR-ABL fusion genes at level less than 10% at 3 months and complete cytogenetic response (CCyR) at one year revealed that there was no statistically significant difference between CML patients with normal and mutated PTCH1. In the study carried out by Juan et al., 2017 [39] who studied expression of PTCH1 in 101 CML patients in chronic phase found that there was no significant differences were found when CCyR probabilities were compared between the high and low PTCH1 expression groups but they found that a greater proportion of patients in the low PTCH1 expression group presented a non-optimal molecular response of percent of BCR-ABL fusion genes at level less than 10% at 3 months.
In our study, CML patients with normal PTCH1 had statistically significant better response to imatinib than CML patients with mutated PTCH1 (P=0.03).
In the study carried out by Juan et al., 2017 [39] reported that the CML patients with high PTCH1 expression had significant better response to imatinib (P=0.011) and Hasford J et al., 2011 [41] reported that the PTCH1 expression had the ability to predict imatinib failure and had a sensitivity, specificity of 86%, 56% respectively, On the other hand Bing et al., 2011 [40] reported that there were no statistically significant differences of response to imatinib between CML patients with high and low PTCH1 expression.
As regard to overall and disease free survival, no significant statistically differences was detected between CML patients with normal and mutated PTCH1, but overall survival was better (96.2%) in CML patients with normal PTCH1 than CML patients with mutated PTCH1 (87.5%). The disease free survival was (69.9%) in CML patients with normal PTCH1 and it was (58.2%) in CML patients with mutated PTCH1.
In conclusion, we found that CML patients with normal PTCH1 had statistically significant better response to imatinib than CML patients with mutated PTCH1. So PTCH1 gene mutation should be considered a promising molecular marker for predicting the probability of imatinib response in CML patients. Hedgehog pathway activation in CML patients can raise a possibility that combinat-ions of ABL and Hh inhibitors might offer a new treatment strategy in CML and might help to effectively cure this disease. Further studies on large number of CML patients are needed to validate this assumption.
Disclosure of conflict of interest
None.
References
- 1.Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385:1447–1459. doi: 10.1016/S0140-6736(13)62120-0. [DOI] [PubMed] [Google Scholar]
- 2.Ravi B, Melissa H, Ning N. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenic remission following imatinib mesylate treatment. Blood. 2003;101:4701–4707. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- 3.Graham SM, Jorgensen HG, Allan E. Primitive, quiescent, philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 4.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 5.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–17. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 6.Faderl S, Talpaz M, Estrov Z. Chronic myelogenous leukemia: biology and therapy. Ann Intern Med. 1999;131:207–219. doi: 10.7326/0003-4819-131-3-199908030-00008. [DOI] [PubMed] [Google Scholar]
- 7.Sattler M, Griffin JD. Mechanisms of transformation by the BCR/ABL oncogene. Int J Hematol. 2001;73:278–291. doi: 10.1007/BF02981952. [DOI] [PubMed] [Google Scholar]
- 8.Kumari A, Brendel C, Hochhaus A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119:530–539. doi: 10.1182/blood-2010-08-303495. [DOI] [PubMed] [Google Scholar]
- 9.Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem cell fate. Blood. 2008;111:492–503. doi: 10.1182/blood-2007-07-075168. [DOI] [PubMed] [Google Scholar]
- 10.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 11.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–12. doi: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindström E, Shimokawa T, Toftgård R. PTCH mutations: distribution and analyses. Hum Mutat. 2006;3:215–219. doi: 10.1002/humu.20296. [DOI] [PubMed] [Google Scholar]
- 13.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 14.Hooper JE, Scott MP. Communicating with hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–17. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 15.Johnson RL, Scott MP. New players and puzzles in the hedgehog signaling pathway. Curr Opin Genet Dev. 1998;8:450–6. doi: 10.1016/s0959-437x(98)80117-2. [DOI] [PubMed] [Google Scholar]
- 16.Murone M, Rosenthal A, de Sauvage FJ. Hedgehog signal transduction from flies to vertebrates. Exp Cell Res. 1999;253:25–33. doi: 10.1006/excr.1999.4676. [DOI] [PubMed] [Google Scholar]
- 17.Taipale J, Beachy PA. The hedgehog and wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 18.Bermudez O, Hennen E, Koch I. Gli1 mediates lung cancer cell proliferation and sonic hedgehog dependent mesenchymal cell activation. PLoS One. 2013;8:e63226. doi: 10.1371/journal.pone.0063226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn H, Wojnowski L, Zimmer AM. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of gorlin syndrome. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- 20.Goodrich LV, Milenković L, Higgins KM. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 21.Berman DM, Karhadkar SS, Maitra A. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–849. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 22.Park KS, Martelotto LG, Peifer M. A crucial requirement for hedgehog signaling in small cell lung cancer. Nat Med. 2011;17:1504–1508. doi: 10.1038/nm.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thayer SP, di Magliano MP, Heiser PW. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–656. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–951. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- 25.Queiroz KC, Ruela-de-Sousa RR, Fuhler GM. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. 2010;29:6314–6322. doi: 10.1038/onc.2010.375. [DOI] [PubMed] [Google Scholar]
- 26.Zhao C, Chen A, Jamieson C. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458:776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graham S, Jorgensen H, Allan E. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 28.Jagani Z, Dorsch M, Warmuth M. Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle. 2010;9:3449–3456. doi: 10.4161/cc.9.17.12945. [DOI] [PubMed] [Google Scholar]
- 29.Long J, Li B, Rodriguez-Blanco J. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened to abrogate the growth of hedgehog driven cancers. J Biol Chem. 2014;289:35494–502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su W, Meng F, Huang L. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through b-catenin signaling. Exp Hematol. 2012;40:418–27. doi: 10.1016/j.exphem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Johnson RL, Rothman AL, Xie J. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 32.Hahn H, Wicking C, Zaphiropoulous PG. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 33.Xie JW, Murone M, Luoh SM. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 34.Xie JW, Johnson RL, Zhang XL. Mutations of the patched gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997;57:2369–2372. [PubMed] [Google Scholar]
- 35.Karhadkar SS, Hallahan AR, Pritchard JI. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 36.Bermudez O, Hennen E, Koch I, Lindner M, Eickelberg O. Gli1 mediates lung cancer cell proliferation and sonic hedgehog dependent mesenchymal cell activation. PLoS One. 2013;8:e63226. doi: 10.1371/journal.pone.0063226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alonso-Dominguez JM, Grinfeld J, Alikian M. PTCH1 expression at diagnosis predicts imatinib failure in chronic myeloid leukaemia patients in chronic phase. Am J Hematol. 2015;90:20–26. doi: 10.1002/ajh.23857. [DOI] [PubMed] [Google Scholar]
- 38.Safaa AA, Nashwa MA. Demographic, clinical, and hematologic characteristics of patients with chronic myeloid leukemia in Upper Egypt: association with treatment responses. Egyptian Journal of Haematology. 2015;40:195–200. [Google Scholar]
- 39.Alonso-Dominguez JM, Casado LF, Anguita E, Gomez-Casares MT, Buño I, Ferrer-Marín F, Arenas A, Del Orbe R, Ayala R, Llamas P, Salgado RN, Osorio S, Sanchez-Godoy P, Burgaleta C, Mahíllo-Fernández I, Garcia-Gutierrez V, Steegmann JL, Martinez-Lopez J. PTCH1 is a reliable marker for predicting imatinib response in chronic myeloid leukemia patients in chronic phase. PLoS One. 2017;12:e0181366. doi: 10.1371/journal.pone.0181366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long B, Zhu HL, Zhu CX. Activation of the hedgehog pathway in chronic myelogeneous leukemia patients. J Exp Clin Cancer Res. 2011;30:8. doi: 10.1186/1756-9966-30-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasford J, Baccarani M, Hoffmann V, Guilhot J, Saussele S, Rosti G, Guilhot F, Porkka K, Ossenkoppele G, Lindoerfer D, Simonsson B, Pfirrmann M, Hehlmann R. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118:686–692. doi: 10.1182/blood-2010-12-319038. [DOI] [PubMed] [Google Scholar]