

Abstract
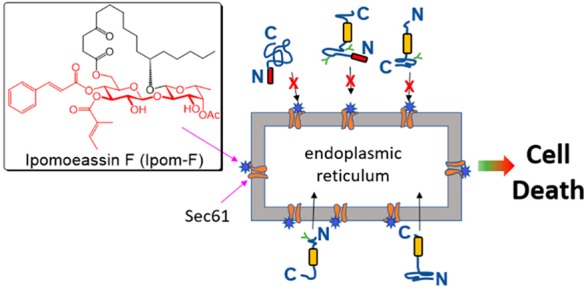
Ipomoeassin F is a potent natural cytotoxin that inhibits growth of many tumor cell lines with single-digit nanomolar potency. However, its biological and pharmacological properties have remained largely unexplored. Building upon our earlier achievements in total synthesis and medicinal chemistry, we used chemical proteomics to identify Sec61α (protein transport protein Sec61 subunit alpha isoform 1), the pore-forming subunit of the Sec61 protein translocon, as a direct binding partner of ipomoeassin F in living cells. The interaction is specific and strong enough to survive lysis conditions, enabling a biotin analogue of ipomoeassin F to pull down Sec61α from live cells, yet it is also reversible, as judged by several experiments including fluorescent streptavidin staining, delayed competition in affinity pulldown, and inhibition of TNF biogenesis after washout. Sec61α forms the central subunit of the ER protein translocation complex, and the binding of ipomoeassin F results in a substantial, yet selective, inhibition of protein translocation in vitro and a broad ranging inhibition of protein secretion in live cells. Lastly, the unique resistance profile demonstrated by specific amino acid single-point mutations in Sec61α provides compelling evidence that Sec61α is the primary molecular target of ipomoeassin F and strongly suggests that the binding of this natural product to Sec61α is distinctive. Therefore, ipomoeassin F represents the first plant-derived, carbohydrate-based member of a novel structural class that offers new opportunities to explore Sec61α function and to further investigate its potential as a therapeutic target for drug discovery.
Introduction
Historically, natural products have significantly contributed to the development of drugs for human disorders,1 most notably as anticancer chemotherapeutics.2 Structurally and functionally unique natural products also provide a spectrum of valuable chemical tools for examining biological systems in translational biomedical research.3,4 To continue our battle against various unsolved problems of human health, including those arising from drug resistance, it is crucial to systematically investigate underexplored areas of existing chemical space such as those offered by bioactive natural products with unique structures and/or mechanisms.5
Resin glycosides, also called glycoresins, are a large collection of amphiphilic glycolipids isolated from the morning glory family of plants,6 and these compounds are considered active ingredients of many morning glory-based traditional medicines that are used worldwide. Resin glycosides consist of a differently acylated oligosaccharide glycosylated with a mono- or dihydroxy C14 or C16 fatty acid, with more than 300 family members discovered to date. Through an ester bond, the fatty acid chain is usually folded back to form a macrolactone ring of various sizes spanning one or more carbohydrate units. Because of their unique macrocyclic architecture with embedded carbohydrates and their broad spectrum of biological activities exhibited in phenotypic screens, resin glycosides have attracted considerable attention from the synthetic chemistry community, but not much beyond.7−9
In 2005, a new family of glycoresins, ipomoeassins A–E, was isolated from the leaves of Ipomoea squamosa found in the Suriname rainforest and shown to inhibit the proliferation of A2780 human ovarian cancer cells.10 Among these compounds, ipomoeassin D (Figure 1) displayed the greatest potency, with an IC50 value of 35 nM. Two years later, a new member of the family, ipomoeassin F (Figure 1), was isolated and showed a cytotoxicity comparable to ipomoeassin D.11 Because of their promising antiproliferative activity and unique molecular skeleton with carbohydrates being part of the macrocycle, these newly discovered natural glycoconjugates quickly inspired synthetic chemists, including ourselves, to tackle their total syntheses.12−16 Subsequently, ipomoeassin F was confirmed to be the most cytotoxic resin glycoside discovered to date with single-digit nanomolar IC50 values against several cancer-derived cell lines.13 Intriguingly, ipomoeassins A and F (see Figure 1) have distinct cytotoxicity profiles, as revealed in the NCI 60-cell-line screen, suggesting that the ipomoeassins may possess an unusual mode of action.16,17 On this basis, ipomoeassin F is a very promising candidate for molecular probe and even chemotherapeutic development; however, the absence of knowledge about the cellular targets of ipomoeassin F has significantly impeded such efforts. To overcome this challenge, after improving our understanding of the structure–activity relationship (SAR) of ipomoeassin F through medicinal chemistry studies,18−20 we employed a chemical proteomics approach to identify its binding partner(s) in human cells. Here we describe the evolution of our chemoproteomic studies that enabled the discovery of Sec61α (protein transport protein Sec61 subunit alpha isoform 1) as a primary molecular target of ipomoeassin F and our subsequent mechanistic investigations that validate this component as a key target mediating cytotoxicity. To this end, we compared the activity of ipomoeassin F to mycolactone, a recently discovered broad-spectrum inhibitor of Sec61-dependent translocation.21,22 This polyketide lactone is the pathogenic exotoxin of Buruli ulcer disease (Mycobacterium ulcerans infection) that has well-established immunosuppressive and cytotoxic activities.23,24 We define the inhibitory effects of ipomoeassin F on Sec61-mediated protein translocation into and across the membrane of the endoplasmic reticulum (ER), corroborating our contemporaneous observations of the potent inhibition of cellular protein secretion by ipomoeassin F.
Figure 1.

Structures of ipomoeassins A, D, and F.
Results and Discussion
Design, Synthesis, and Proteomic Evaluation of Chemical Probes Derived from Ipomoeassin F
Michael acceptor systems are present in many electrophilic natural products, allowing the formation of covalent adducts with bio-macromolecules, which are often responsible for their biological activities.25 Because of significant activity loss (up to 160-fold) after reducing one of the two double bonds in the α,β-unsaturated esters to a single bond,18 we hypothesized that the cinnamate and/or the tiglate (Figure 1) may enable irreversible binding between ipomoeassin F and its biological target(s). Therefore, we initially designed and synthesized a potential activity-based probe,26−283 (Scheme 1), by introducing a small propargyloxy group to the para-position of the benzene ring.
Scheme 1. Synthesis of Ipomoeassin F Probes 3, 5, and 7.

Reagents and conditions: (a) 4-propargyloxycinnamic acid (2), DCC, DMAP, CH2Cl2, 0 °C → RT, overnight, 90%; (b) TBAF, THF, −10 °C, 6 h, 80%; (c) CuSO4, sodium ascorbate, CH2Cl2/t-BuOH, 3:2.
Probe 3 was synthesized in two steps from key intermediate 1 obtained during the total synthesis of ipomoeassin F.15 The alkyne-functionalized cinnamic acid 2 was synthesized as previously described.29N,N′-Dicyclohexylcarbodiimide (DCC)-mediated Steglich esterification was applied to couple 1 with 2 in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP) to give the tert-butylsilyl (TBS)-protected intermediate in 90% yield. Subsequently, exposure of the intermediate to tetra-n-butylammonium fluoride (TBAF) led to the removal of the TBS protecting groups and delivered the final product 3 in good yield. As expected, the small bioorthogonal alkyne tag did not affect cytotoxicity, and 3 was equally potent as compared to the original natural product (Table 1).
Table 1. Cytotoxicity (IC50, nM) of Ipomoeassin F, Cotransin (CT8), and Probes 3, 5, and 7–15a.
| cell line |
||||
|---|---|---|---|---|
| MDA-MB-231b | MCF7 | MCF-10A | HCT116b,c | |
| ipomoeassin F | 6.3 | 36.4 | 5.3 | 8 |
| CT8 | –d | – | – | 243 |
| 3 | 6.3 | 22.2 | 9.6 | – |
| 5 | 29.5 | 187.4 | 21.7 | – |
| 7 | 7.0 | 19.8 | 5.1 | – |
| 8 | >25,000 | >25,000 | – | 2832 |
| 9 | >25,000 | >25,000 | – | – |
| 10 | 8.9 | 172.2 | 11.3 | – |
| 11 | 2.8 | 21.1 | 2.0 | – |
| 12 | 253.4 | 172.2 | 133.8 | – |
| 14 | 98018 | 256018 | – | 1874 |
| 15 | 42918 | 47718 | – | 480 |
The data were obtained from at least two independent experiments, and the standard errors are within 20%.
See Figure S1 for IC50 curves.
Assayed separately.
“–” indicates “not determined”.
With probe 3 in hand, we conducted activity-based protein profiling experiments for target identification both in the soluble fraction of cell lysates made from MDA-MB-231 breast cancer cells and in live MDA-MB-231 cells. Unfortunately, after in situ click chemistry with a fluorescent rhodamine azide, S27(30) (see Supporting Information), we detected no differences in labeled proteins between negative control (lane 1), probe 3 (lane 2), and probe 3 plus ipomoeassin F (competition; lane 3) (Figure S2A), while an unrelated probe used as a positive control for pulldown gave clear signals (Figure S2A, lanes 4 and 5). We considered two possible explanations for this negative result: first, ipomoeassin F is not a covalent protein modifier; second, target proteins of ipomoeassin F are in low abundance. Due to the dramatic loss in activity with alkene-reduced compounds, we favored the second possibility.
In order to enrich for relatively low abundance cellular proteins, we next synthesized the biotin-labeled analogue 5 of ipomoeassin F by reacting the alkyne probe 3 with biotin azide 4(31) under chemoselective copper-catalyzed alkyne–azide coupling (CuAAC) conditions (Scheme 1).32 The synthesis was successful in producing biotin probe 5, which remained cytotoxic (Table 1), showing only a ∼5-fold decrease in potency. After incubating 5 with either detergent-solubilized cell lysate or intact cells, we used streptavidin beads to recover the biotin probe 5-bound proteins followed by electrophoresis and silver staining of the captured material. However, no proteins were selectively enriched by probe 5 (Figures S3A and C, lanes 1 to 3), nor was any enrichment apparent when these samples were probed using a fluorophore-labeled streptavidin (Figure S3B, lanes 1 to 3).
We hypothesized that if the target protein(s) of ipomoeassin F is of relatively low abundance, then endogenously biotinylated proteins could outcompete, and thereby mask, any positive signal. To minimize interference from endogenously biotinylated proteins, we synthesized a new biotin probe, 7 (Scheme 1), containing a photocleavable nitrobenzene moiety,33 by coupling the alkyne analogue 3 with the biotin azide fragment 6, containing an o-nitrobenzyl photolabile linker, the latter synthesized by modifying previously reported procedures31−33 (see Supporting Information). Notably, the capacity of 7 to inhibit cancer cell proliferation was close to that of parental ipomoeassin F (Table 1), despite the large size of the appendage at the para-position of the cinnamate benzene ring. We then used probe 7 to label target proteins in live MDA-MB-231 cells, performed a streptavidin pulldown on the resulting cell lysate, and selectively released bound proteins by UV photocleavage. When the resulting products were analyzed by SDS-PAGE, probe 7 strongly enriched a ∼40 kDa protein and, to a lesser extent, a second protein of ∼100 kDa (Figure 2A, lane 2, red arrows), both of which were absent from the negative control (Figure 2A, lane 1). In support of their bona fide interaction with 7, these two proteins were lost when a 100-fold excess of unmodified ipomoeassin F was included during the incubation (Figure 2A, lane 3). Because no proteins were selectively recovered with probe 7 when it was incubated with detergent-solubilized (1% NP40 and 0.25% sodium deoxycholate) extracts of MDA-MB-231 cells (Figure S3C, lanes 1, 4, and 5), we believe that the interaction between ipomoeassin F and its target protein(s) may require a particular subcellular environment that is detergent sensitive.
Figure 2.

SDS-PAGE images (silver staining) for affinity pulldown using probe 7 with (A) or without (B) photocleavage. Red arrows indicate specifically pulled down protein species. (C) Target validation by Western blot with a Sec61α antibody. Ipom-F, ipomoeassin F; AP, affinity pulldown.
We next investigated whether photocleavage is essential for the selective recovery of the two putative target protein bands using probe 7. Strikingly, the ∼40 kDa protein was also recovered without photocleavage. It was substantially enriched by 7 when total streptavidin-bound products were analyzed and was lost in the presence of excess ipomoeassin F (Figure 2B, lanes 1–3). In contrast, an enrichment of the ∼100 kDa component under these conditions was not apparent (Figure 2A and B). Since photocleavage was not essential for probe 7 to selectively recover the ∼40 kDa protein, it is very likely that the increased linker length between the biotin moiety and the ipomoeassin F region, but not its capacity for selective cleavage, significantly improved the performance of probe 7 over probe 5. Our data also suggest that, once formed, the complex between probe 7 and the ∼40 kDa protein is tight and can survive detergent-containing cell lysis. This is further supported by pulldown of the ∼40 kDa target protein with low concentrations of probe 7 (10 nM) that was outcompeted by a 5-fold excess of ipomoeassin F (50 nM) (Figure S4A, lanes 1–3). Fluorescent streptavidin staining suggested that a stable covalent adduct between the ∼40 kDa protein and probe 7 was not formed under our experimental conditions (Figure S3B, lanes 4 and 5). We next submitted both the ∼40 kDa protein(s) (Figure 2B, lane 2) and the corresponding regions of the gel for the negative control (Figure 2B, lane 1) and the ipomoeassin F competition sample (Figure 2B, lane 3) for mass spectrometry analysis. When spectral counts were compared, Sec61α (protein transport protein Sec61 subunit alpha isoform 1) stood out as the only protein that was both substantially enriched in the pulldown with probe 7 and showed a corresponding reduction when excess ipomoeassin F was present (Table S1). The selective recovery of Sec61α by probe 7 was further confirmed by Western blotting (Figure 2C, cf. signals between input and AP).
To further support Sec61α as a target protein responsible for the biological activity of ipomoeassin F, we synthesized two inactive reference compounds, 8 and 9 (Figure 3 and Table 1), by removing both double bonds in cinnamate and tiglate (see Supporting Information). As predicted, pulldown experiments with inactive biotin probe 9 revealed a loss of the ∼40 kDa protein, even with 500 nM of 9 (Figure S4B, lanes 1 and 4). Additionally, inactive analogue 8 was ineffective at competing with probe 7 binding, even when 8 was present in 50-fold excess (Figure S4B, cf. lanes 1 to 3). Taken together, these findings further support the specificity of Sec61α as a primary cellular target of ipomoeassin F.
Figure 3.

Structures of two inactive derivatives of ipomoeassin F.
Cell Imaging Studies
The Sec61 protein translocon plays an essential role in translocating newly synthesized membrane and secretory polypeptides into and across the membrane of the ER.34,35 Sec61α forms the membrane conduit that nascent secretory polypeptides pass through. To examine the subcellular localization of ipomoeassin F, we performed live cell imaging studies using three fluorescent analogues, 10–12 (Figure 4), prepared from alkyne probe 3 using CuAAC (see Supporting Information). Both coumarin-coupled probe 10 and NBD-coupled probe 11 retained substantial cytotoxicity, with 11 being even more potent than ipomoeassin F (see Table 1). Unfortunately, 11 underwent photobleaching very easily and could not be used for imaging studies. In contrast, 10 gave a rather weak fluorescent signal, presumably due to an internal photoinduced electron transfer effect combined with its shorter excitation maximum (365 nm). To overcome these technical limitations, rhodamine analogue 12 was synthesized from the rhodamine azide precursor S27. Although 12 is between 5- and 40-fold less potent than ipomoeassin F depending on the cell type analyzed, it retains substantial cytotoxicity when compared to inactive analogues 8 and 9 (see Table 1), and we therefore used it for our imaging studies.
Figure 4.

Structures of fluorescent derivatives of ipomoeassin F.
Subsequently, we confirmed that 12 could penetrate the cell membrane in a concentration- and time-dependent manner and produced a strong intracellular fluorescent signal. Hence, 12 stained cells within 30 min when present at 2 μM, but took >3 h to give clear images when used at 20 nM. When cells labeled with 12 were costained with markers for either the ER or the nucleus, it was apparent that 12 strongly labeled the ER (Figure 5), but not the nucleus (Figure S5A). To confirm the specificity of ER labeling by 12, competition experiments were performed using ipomoeassin F or inactive compound 8. The fluorescence signal of 12 was almost completely abolished when the cells were preincubated for 30 min with a 100-fold excess of free ipomoeassin F (Figure S5B), yet preincubation with compound 8 had little effect (Figure S5C). The apparent ER localization of the cellular interacting partner(s) of ipomoeassin F strongly supports the notion that its target is Sec61α.
Figure 5.
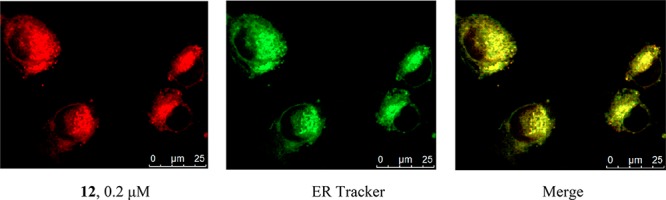
Cell imaging studies with fluorescent probe 12 in MDA-MB-231 cells. Rhodamine-conjugated ipomoeassin F analogue 12 (0.2 μM) was added to cells, and after 1 h, cells were imaged to analyze localization of 12 relative to ER staining.
Pulldown from ER Microsomes
As evidenced by the negative results from our pulldown experiments using cell lysates prepared with detergent, we propose that the membrane environment of the ER may be required to maintain Sec61α in a conformation that can bind to ipomoeassin F effectively. In this regard, it would be unlikely that purified Sec61α alone could recapitulate a direct interaction with the natural product. However, since the biological function of Sec61α is maintained in isolated ER microsomes22 and once prebound to probe 7 in intact cells the resulting complex appears stable to detergent treatment (Figures 2A and 2B), we attempted to pull down Sec61α from purified ER-derived microsomes. After incubation with probe 7 and detergent solubilization (1% n-dodecyl-β-d-maltoside) to release biotin-bound proteins from the phospholipid bilayer, only a single protein was visible after pulldown (Figure S6A). The identity of this product was confirmed as Sec61α by Western blotting (Figure S6B). This finding strongly supports our previous conclusion made with live cells, namely, that Sec61α directly interacts with ipomoeassin F.
Competition of Ipomoeassin F with Cotransin for Binding to Sec61α
To test whether ipomoeassin F competes for binding to Sec61α with known Sec61 ligands, such as cotransin and mycolactone,36−38 we exploited CT7,39 a cotransin photoaffinity probe that can covalently label Sec61α. As reported earlier,36 mycolactone efficiently competes with CT7 and prevents its photo-cross-linking to Sec61α (Figure 6). A similarly potent competition was observed with ipomoeassin F, suggesting it binds directly to Sec61α, potentially at the same or a partially overlapping interaction site as that used by cotransin. Ipomoeassin F competes with CT7 photo-cross-linking with similar potency to mycolactone, consistent with its ability to inhibit ER translocation with high potency.
Figure 6.

Sheep rough microsomes were preincubated with ipomoeassin F (Ipom-F) or mycolactone (Myco) at 10 μM. Subsequently 100 nM photocotransin CT7 was added and samples were photolyzed. The covalent CT7/Sec61α adduct was detected using click chemistry to install a rhodamine-azide reporter and in-gel fluorescent scanning.
In Vitro Inhibition of Protein Translocation by Ipomoeassin F
In order to understand the consequences of ipomoeassin F binding for Sec61α function, we used a well-established in vitro system to study the Sec61-dependent translocation of radiolabeled precursor proteins into ER microsomes derived from canine pancreas.22,23 In the presence of 1 μM ipomoeassin F, the translocation of two model secretory proteins, bovine preprolactin and yeast prepro-alpha-factor, was almost completely prevented. The blockade of protein translocation by ipomoeassin F was comparable to that achieved using 1 μM mycolactone and resulted in a loss of signal sequence cleavage and protein N-glycosylation (see Figure 7A and B; cf. lanes 1, 3, and 4), both events specific to the ER lumen. The unusual ability of tail-anchored membrane proteins to insert into the ER membrane independently of the Sec61 complex provides a useful control for validating the specificity of ER translocation inhibitors.40 The in vitro integration of two model tail-anchored proteins bearing C-terminal N-glycosylation reporters was unaffected by ipomoeassin F treatment (Figure 7C and D, cf. lanes 1 and 3), mirroring the specificity of the Sec61-selective inhibitor mycolactone21,22 (see also Figure 7C and D, lane 4). Furthermore, despite the presence of carbohydrate moieties in ipomoeassin F (see Figure 1), it has no effect on the N-glycosylation of our model tail-anchored proteins, and we conclude that protein N-glycosylation of model substrates provides a robust reporter for the ipomoeassin F-mediated inhibition of Sec61-dependent protein translocation.
Figure 7.

Ipomoeassin F inhibits Sec61-mediated protein translocation into and across the ER in a substrate-selective manner. Phosphorimages of membrane-associated in vitro products resolved by SDS-PAGE together with outline structures of the protein substrates generated in the presence or absence of ipomoeassin F (Ipom-F) or mycolactone (Myco): secretory proteins (A) bovine preprolactin, PPL, and (B) yeast prepro-alpha-factor, ppαF; tail-anchored proteins (C) C-terminally tagged Sec61β subunit, Sec61βOPG2, and (D) C-terminally tagged cytochrome b5, Cytb5OPG2; type I single pass transmembrane protein (TMP) (E) vascular cell adhesion molecule 1, VCAM1; type II single pass TMP (F) isoform 2 (residues 17 to 232) of the short form of HLA class II histocompatibility antigen gamma chain, Ii; type III single pass TMP (G) glycophorin C, GypC; short secretory protein (H) C-terminally tagged Hyalophora cecropia preprocecropin A, ppcecAOPG2. Samples were treated with endoglycosidase H (Endo H) where indicated to distinguish N-glycosylated (XGly) from nonglycosylated (0Gly) products. nc, signal sequence not cleaved; sc, signal sequence cleaved; TM1, transmembrane domain 1. Proteins are of human origin unless otherwise stated.
Known small-molecule inhibitors of Sec61-mediated protein translocation show varying degrees of substrate selectivity,23,41 and we therefore studied the effects of ipomoeassin F on the Sec61-mediated translocation of four other classes of precursor proteins (see Figure S7) into and across the ER membrane using the in vitro system outlined above.22,42 We find that ipomoeassin F potently inhibits the membrane insertion of representative type I and type II integral membrane proteins that have a single transmembrane spanning domain (Figures 7E and F). In contrast, the compound has no effect on the membrane insertion of the single-spanning type III integral membrane protein glycophorin C and only a marginal impact on the translocation of the short secretory protein prepro-cecropin A (Figure 7G and H). Interestingly, it has recently been shown that type III membrane proteins have the unusual ability to exploit an alternative, Sec61-independent, mechanism for ER insertion, which relies on the ER membrane protein complex acting as an integrase.43 On this basis, we conclude that the integration of glycophorin C is completely refractive to ipomoeassin F (this study) and mycolactone (see ref (42)) because it can insert into the ER via a Sec61-independent mechanism. Based on the model membrane and secretory proteins that we have studied here, the substrate selectivity for the ipomoeassin F-mediated inhibition of ER protein translocation appears remarkably similar to that of mycolactone.42
To further investigate ipomoeassin F specificity, we used the ER insertion of the type II membrane protein Ii (HLA class II histocompatibility antigen gamma chain; cf. Figure 7F) to compare the effectiveness of a subset of structural variants. The differences we observe in this in vitro assay (see Figure S8) are strikingly similar to the variations in cytotoxicity that the same compounds display in cell-based assays (Table 1). Hence, ipomoeassin F and the open-chain analogue 13(18) (Figure 8) strongly inhibit membrane insertion, while 14(18) (Figure 8) is a far less effective inhibitor, and analogue 8 (Figure 3) has no measurable inhibitory activity in vitro (see Figure S8A and B). Ii membrane insertion was also used to estimate IC50 values for ipomoeassin F (∼50 nM) and 13 (∼120 nM) (see Figure S8C and D). The fact that we observe IC50 values in the nM range using in vitro translocation inhibition and cell-based cytotoxicity assays, combined with the similarity in the structure–activity relationship observed in both systems, leads us to conclude that the inhibitory effect of ipomoeassin F on Sec61-mediated protein translocation is most likely the principal molecular basis for its cytotoxic activity. Furthermore, since the ER acts as the entry point into the eukaryotic secretory pathway, our in vitro findings predict that if cells are exposed to ipomoeassin F, their capacity to produce and deploy membrane and/or secretory proteins is likely compromised.
Figure 8.

Structures of ipomoeassin F analogues 13–15.
Inhibition of Protein Secretion by Ipomoeassin F in Live Cells
Through a partnership with the National Center for Advancing Translational Sciences (NCATS), ipomoeassin F was included within a collection of natural products for screening against a panel of assays to interrogate a variety of biological pathways and assess cell-type-dependent toxicity.44 Ipomoeassin F was identified as an inhibitor of protein secretion in two distinct quantitative high-throughput screening (qHTS) assays using different cell lines and secretory reporters.45 In U2-OS human osteosarcoma cells, a protein reporter consisting of secreted NanoLuc (secNLuc) fused to the Z mutant of alpha-1 antitrypsin (secNLuc-ATZ) was designed to monitor the extracellular accumulation of this protein.46 Ipomoeassin F demonstrated acute, highly potent secretion inhibition (EC50 = ∼5 nM) in U2-OS cells prior to the onset of cytotoxicity, as assessed by decreasing cellular ATP after 24 h (Figure 9A). A second reporter of protein secretion was also examined, using SH-SY5Y human neuroblastoma cells expressing a secreted Gaussia luciferase (GLuc). Ipomoeassin F inhibited secretion of the reporter protein with a lower potency (EC50 = ∼120 nM) than in U2-OS cells (Figure 9B), reflecting a potential cell-type-dependent functional effect. The secretion phenotype in cells treated with ipomoeassin F is strongly consistent with Sec61α as its inhibitory protein target, since most secretory proteins must pass through the Sec61 translocon into the ER prior to traversing the secretory pathway for extracellular secretion. The difference in potency between different cell lines is, again, in line with previous observations with mycolactone.47,48
Figure 9.

Differential effect of acute ipomoeassin F treatment on protein secretion and cell viability. Bioluminescence outputs from secretion sensor proteins (●) and cellular viability based on total well ATP content (○). (A) secNLuc-ATZ secretion and cell viability measured in U2-OS cells after 24 h. (B) GLuc secretion and cell viability measured in SH-SY5Y cells after 48 h. Error bars represent the SEM for n = 2 or 3 replicates.
Selective Prevention of Biogenesis of Secreted Proteins by Ipomoeassin F
To assess ipomoeassin F effects on cellular protein production, we employed a metabolic labeling strategy. Cells were pretreated with ipomoeassin F or its analogue 8 (Figure 3), 14, or 15 (Figure 8), for 30 min before addition of 35S-labeled methionine/cysteine for a further 90 min. The lack of inhibition of nascent protein biogenesis observed in total cellular lysates and cytosolic fractions (Figure 10A and B) demonstrates that ipomoeassin F does not acutely influence global cellular processes such as transcription or translation and is in line with previous observations using mycolactone.21,49 In contrast, strong inhibition was observed for the production of secreted proteins for cells treated with 100 nM ipomoeassin F (Figure 10C). Importantly, ipomoeassin F inhibits the production of most newly synthesized secretory proteins, suggesting its mechanism of action leads to an inhibition of biogenesis of a broad range of Sec61 substrate proteins that is comparable to known Sec61 inhibitors such as mycolactone21,36 and apratoxin A.38,50 Ipomoeassin F analogues 8, 14, and 15 all inhibit the production of secreted proteins less potently when tested at 1 μM concentration. Furthermore, the rank order of potencies with which ipomoeassin F and its analogues inhibit protein secretion (Ipom-F > 15 > 14 > 8) correlates with their observed cytotoxicity against HCT-116 cells (cf. Table 1 and Figure S1C), suggesting a link between cytotoxicity and inhibition of cellular protein secretion, as previously suggested for other Sec61 inhibitors.36,38,51,52
Figure 10.

HCT-116 cells were washed twice with PBS and incubated in the presence of indicated concentrations of compounds under media lacking methionine and cysteine for 30 min. 35S-labeled methionine and cysteine were then introduced to the media, and cells incubated for a further 90 min. Cells were harvested by scraping into ice cold PBS and analyzed by SDS-PAGE and autoradiography. (A) The collected cells were subsequently homogenized and analyzed by SDS-PAGE and autoradiography. (B) As for (A) but the samples are the cytosolic contents of harvested cells following partial permeabilization with 0.15% digitonin. (C) As for (A) but the samples are from TCA-precipitated culture medium from the same experiment. Ipom-F denotes ipomoeassin F. AprA denotes control samples treated with apratoxin A to block protein translocation into the ER. CHX denotes samples treated with cycloheximide and chloramphenicol to inhibit total cellular protein synthesis. Cotransin CT8 is a substrate-selective inhibitor of protein translocation into the ER.
HepG2 cells secrete a range of endogenous proteins, but these products are almost completely absent following treatment with the ER translocation inhibitor eeyarestatin I or the vesicular transport inhibitor brefeldin A.40 To extend our studies with HCT116 cells (Figure 10), we therefore treated HepG2 cells with ipomoeassin F or mycolactone overnight and then followed the fate of newly synthesized proteins by pulse chase analysis. When the cell media was analyzed for secretory proteins, a substantial reduction was observed for cells treated with ipomoeassin F or mycolactone when compared to control cells treated with DMSO or compound 8 (see Figure S9, media, lanes 1 to 4). While the effect of these Sec61 inhibitors was akin to that of brefeldin A, treatment with the proteasome inhibitor bortezomib had no effect (see Figure S9, media, lanes 3 to 6). When the levels of total radiolabeled proteins recovered from the same cells were analyzed, they were directly comparable across all treatments, confirming that ipomoeassin F did not inhibit global protein synthesis during the treatment used (Figure S9, whole cell extract, cf. lanes 1 to 4). In fact, overnight exposure to ipomoeassin F increases at least one prominent cellular component (Figure S9, whole cell extract, lane 3, ∼80 kDa), perhaps reflecting the early stages of a cellular stress response.51,53
Immunomodulatory Effects of Ipomoeassin F
While ultimately cytotoxic in most cell types, mycolactone has immediate immunosuppressive effects.23 Recent studies have indicated that these effects are a direct consequence of mycolactone’s inhibitory activity on Sec61,21,36,53,54 and we find the effects of ipomoeassin F and mycolactone are equivalent in cell-free assays of Sec61-dependent protein translocation (Figure 7). By blocking Sec61, mycolactone prevents the production of both secreted proteins and membrane receptors that are key to the generation of immune responses,21 as is clearly illustrated by the biogenesis of CD62L, a cell surface receptor mediating naive lymphocyte homing to secondary lymphoid organs, which is highly susceptible to mycolactone.36 In the human Jurkat T cell line, we find that ipomoeassin F and mycolactone downregulated CD62L expression with comparable potency (Figure 11A). Likewise, they were also equivalent in capacity to prevent interleukin (IL)-2 production by activated T cells (Figure 11B). Together, these data suggest that, like mycolactone,55 ipomoeassin F may display systemic immunosuppressive effects in vivo.
Figure 11.

Compared effects of ipomoeassin F and synthetic mycolactone (A/B form) in assays of CD62L expression (A) and activation-induced production of IL-2 (B) by Jurkat T cells. Data are means ± SEM of biological duplicates and are representative of two independent experiments.
Resistance of Sec61α Mutants to Ipomoeassin F Cytotoxicity
Having established that binding of ipomoeassin F to Sec61α potently impairs protein translocation at the ER both in vitro and in vivo, we sought further evidence that this interaction also underlies the cytotoxic effects of ipomoeassin F (cf. Table 1).
Chemogenetic screening has identified mutations in the SEC61A1 gene that confer resistance to known inhibitors of Sec61α-dependent protein translocation56 and established that these mutations can confer resistance when engineered into a naïve cell background.37,38 We therefore compared the effects of ipomoeassin F and mycolactone on the growth of HEK293 and HCT116 cells expressing either wild-type Sec61α or a previously characterized point mutant of Sec61α that is resistant to mycolactone.36,41 As previously observed using mycolactone in HEK293 cells engineered to express mutant alleles of Sec61α,36,41 point mutations R66I and S82P confer significant and dominant desensitization to ipomoeassin F in a viability assay of HEK293 cells, yet T86M confers resistance to mycolactone but not to ipomoeassin F (see Figure 12A and B). Furthermore, while isolated drug-resistant HCT116 cells carrying mutations to Sec61α of R66K and D60G were strongly resistant to mycolactone (Figure 12D) as previously reported,51 any decrease in sensitivity to ipomoeassin F was comparatively modest (Figure 12C). Taken together, these results strongly suggest that ipomoeassin F and mycolactone bind to a region similar to other known inhibitors of Sec61, but indicate that the interaction of ipomoeassin F may be partially dependent upon the basic side chain of residue R66 but does not require conservation of D60 or T86.
Figure 12.
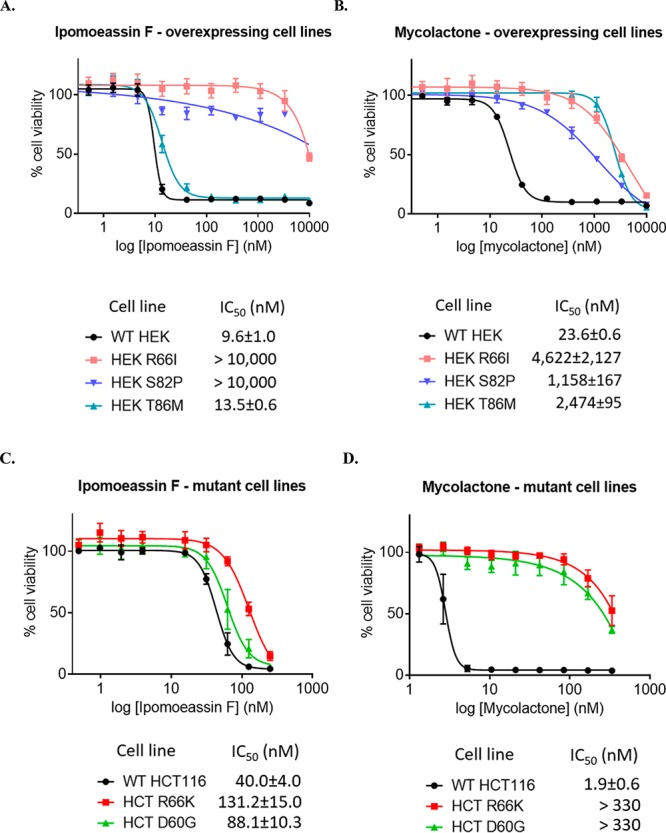
Human HEK293 cells or cells engineered to express Sec61α alleles bearing indicated point mutations were treated with increasing concentrations of ipomoeassin F (A) or mycolactone (B), and cell viability was assayed at 72 h with the Alamar Blue assay (mean ± SD; n = 4 technical quadruplicates). Parental or HCT116 cells containing mutations in Sec61A1 were seeded at 3 × 104/well in 96-well plates in triplicate. Ipomoeassin F (C) or mycolactone (D) were added at various concentrations with the carrier DMSO used as a control. Cell viability was measured at 96 h with the Alamar Blue assay (mean ± SEM of n = 3 independent experiments performed in triplicate).
Strong Yet Reversible Binding of Ipomoeassin F to Sec61α
Although our SAR studies suggested that a covalent adduct may be formed between ipomoeassin F and Sec61α, no such adduct could be detected by fluorescent streptavidin staining of a Western blot. In order to further address the nature of the interaction between ipomoeassin F and Sec61α, we designed a delayed competition experiment by adding a 100-fold excess of ipomoeassin F only after a preincubation with biotin probe 7 (lane 4 in Figure 13A and B). Our cytotoxicity data show that probe 7 closely resembles ipomoeassin F, and we reasoned that if a stable covalent complex was formed between 7 and Sec61α, the later addition of ipomoeassin F would not significantly decrease the signal of Sec61α in an affinity pulldown. When this experiment was carried out, we observed a time-dependent decrease in the probe 7-dependent recovery of Sec61α in the presence of excess ipomoeassin F, such that after 60 min, the signal was substantially reduced (Figure 13A and B, cf. lanes 2 and 4; see also Supporting Information, Figure S10, for complete gel images).
Figure 13.
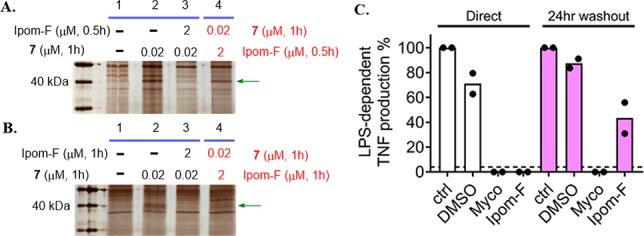
SDS-PAGE images (silver staining) for affinity pulldown using probe 7 with 0.5 h (A) or 1 h (B) competition in both regular (lane 3) and reverse (lane 4) order. Green arrows indicate the protein band for Sec61α. (C) TNF abundance in RAW264.7 cell supernatants 4 h after stimulation with 100 ng/mL LPS in the presence of medium alone (ctrl), DMSO, 125 ng/mL mycolactone (Myco), or 250 nM ipomoeassin F (Ipom-F). Cells were either preincubated for 1 h prior to addition of LPS (“direct”) or incubated for 1 h, washed, and allowed to recover for 24 h in complete medium before addition of LPS (“24 h washout”). TNF was quantified by ELISA in triplicate, and the dashed line shows the detection limit of the assay. Controls for each condition without LPS stimulation did not reach this detectability threshold. For each replicate, data were normalized to control levels and the mean of n = 2 independent cellular assays is presented.
Independently, the reversibility of ipomoeassin F activity was also investigated using a cellular assay. This measured Sec61-dependent production of tumor necrosis factor (TNF) by lipopolysaccharide (LPS)-stimulated macrophage-like RAW264.7 cells. Mycolactone’s ability to completely block TNF production directly after a 1 h preincubation (Figure 13C, white bars) was previously shown not to be restored following a 24 h washout,21 and this was confirmed here (Figure 13C, purple bars). In contrast, while ipomoeassin F similarly blocked TNF production in the direct assay, after extensive washing and 24 h recovery, LPS-dependent TNF production was restored, albeit at a reduced level. Therefore, in contrast to mycolactone, the effects of ipomoeassin F on cells are, at least partially, reversible under these conditions.
On the basis of these results, we conclude that the binding of ipomoeassin F to Sec61α is strong yet apparently reversible, suggesting that it does not involve a stable covalent interaction.
Conclusions
Here, we report comprehensive chemical biology studies of ipomoeassin F to understand the molecular basis for its potent cytotoxicity. By designing and evolving our chemical probes on an empirical basis, Sec61α was ultimately identified and validated as a direct and physiologically relevant target of ipomoeassin F. The results from fluorescent streptavidin staining, delayed competition in affinity pulldown, and TNF-production washout experiments indicate that ipomoeassin F is unlikely to form a stable covalent complex with Sec61α, despite the presence of two Michael acceptor systems in its structure (cinnamate and tiglate, Figure 1). We therefore conclude that the pulldown of Sec61α with probe 7 represents a successful biotin affinity enrichment of a reversible ligand–membrane protein complex formed in live cells, most likely reflecting a slow disassociation rate of ipomoeassin F from Sec61α. The binding of ipomoeassin F to cellular Sec61α is supported by the ER staining seen with a fluorescent version of the compound, the highly selective pulldown of Sec61α from ER-derived microsomes, and the ability of ipomoeassin F to compete with cotransin for Sec61α binding.
The impact of ipomoeassin F on Sec61 function was first investigated in vitro, revealing a strong inhibition of protein translocation into the ER that affected secretory, type I and type II membrane proteins. The in vitro effect of ipomoeassin F translated into potent secretion inhibition in two cellular models utilizing independent luciferase secretory reporters. Likewise, ipomoeassin F treatment downregulated the release of a broad range of endogenous secretory proteins from cultured cells, acting at a stage after their translation. At this point in our studies, it was striking that, although structurally quite distinct, the molecular mechanisms of ipomoeassin F and the functional consequences of its application closely resemble those seen with the immunosuppressive macrolide mycolactone that is released by the human pathogen Mycobacterium ulcerans.(23) We therefore studied the effects of ipomoeassin F on immune cells and found that its inhibition of membrane receptor expression and cytokine production is comparable to that achieved with mycolactone. Previous studies have illustrated the translational potential of mycolactone against inflammatory disorders,55 but further exploitation of mycolactone-inspired molecules has been limited by their intrinsic structural complexity that required extensive synthetic efforts.57−62 Given that ipomoeassin variants are easier to generate by synthetic chemistry and equally inhibitory in cellular assays of inflammation, future studies of their pharmacokinetics and therapeutic efficacy against inflammatory diseases and inflammatory pain are an obvious area to pursue in the future.
Lastly, we could establish an excellent correlation between the functional Sec61 inhibition and the cytotoxic potency of ipomoeassin F using cell lines expressing inhibitor-resistant mutants of Sec61α. The loss of cytotoxicity we observed in HEK293 cells expressing R66I and S82P mutants is consistent with the notion that ipomoeassin F binds to a location that overlaps with the interaction site for other small-molecule inhibitors of Sec61α, most likely at or near its so-called lateral gate.41 However, differences between ipomoeassin F and mycolactone in response to other mutant cell lines strongly suggest that the mode of ipomoeassin F binding to Sec61α is distinctive from other known Sec61α inhibitors.41 The sustained sensitivity of the mycolactone-resistant T86M (in HEK293) and D60G (in HCT116) mutant cell lines to ipomoeassin F that we report is particularly intriguing, highlighting that ER protein translocation can be complex63 and our current understanding is far from complete.43
In short, we provide compelling evidence that the core α subunit of the Sec61 translocation complex is the major target for ipomoeassin F in a cellular context, thereby opening the way for the exploitation of ipomoeassin F for target-based drug discovery. More broadly, given its comparatively advantageous synthesis, we anticipate that ipomoeassin F and its analogues/probes will provide accessible new molecular tools that will help our efforts to fully define the molecular basis for Sec61-mediated protein translocation at the ER membrane.
Acknowledgments
We thank the Arkansas Nano & Bio Materials Characterization Facility at the Institute for Nano Sciences & Engineering for our imaging studies, and Prof Yoshito Kishi (Harvard University) for the kind gift of synthetic mycolactone A/B used by S.H. and R.S. W.S. is supported by Grant No. R15GM116032 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and startup funds from the University of Arkansas. This work was also supported in part by Grant No. P30 GM103450 from the National Institute of General Medical Sciences of the NIH and by seed money from the Arkansas Biosciences Institute (ABI). S.O’K. is the recipient of a Biotechnology and Biological Sciences Research Council (BBSRC) Doctoral Training Programme Award (BB/J014478/1), and S.H. holds a Welcome Trust Investigator Award in Science (204957/Z/16/Z). The alpha-1 antitrypsin work was supported by the Alpha-1 Foundation (J.I. and M.J.I.). J.I. and M.J.H. were supported by the intramural program of NCATS, National Institutes of Health, projects 1ZIATR000048-03 (J.I.) and ZIATR000063-04 (M.J.H.). R.S. holds a Welcome Trust Investigator Award in Science (202843/Z/16/Z). C.D. received funding from the Institut Pasteur, the Institut National de la Santé et de la Recherche Médicale, and the Fondation Raoul Follereau. N.B.’s synthesis and chemical biology studies of mycolactone were supported by CNRS, Université de Strasbourg, Fondations Potier et Follereau, and the Investissement d’Avenir (Idex Unistra). V.O.P. is supported by the Academy of Finland (Grants 289737 and 314672) and the Sigrid Juselius Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b13506.
Author Contributions
▽ G. Zong and Z. Hu contributed equally as first authors.
Author Contributions
▼ S. O’Keefe, D. Tranter, M. J. Iannotti, L. Baron, B. Hall, K. Corfield , A. O. Paatero, M. J. Henderson, and P. Roboti contributed equally as second authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Cragg G. M.; Grothaus P. G.; Newman D. J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- Carlson E. E. Natural Products as Chemical Probes. ACS Chem. Biol. 2010, 5, 639–653. 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo S.; Waldmann H. Development of a Natural-Product-Derived Chemical Toolbox for Modulation of Protein Function. Chem. Rev. 2014, 114, 4621–4639. 10.1021/cr400442v. [DOI] [PubMed] [Google Scholar]
- Pye C. R.; Bertin M. J.; Lokey R. S.; Gerwick W. H.; Linington R. G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 5601–5606. 10.1073/pnas.1614680114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda-Miranda R.; Rosas-Ramirez D.; Castaneda-Gomez J. Resin glycosides from the morning glory family. Prog. Chem. Org. Nat. Prod. 2010, 92, 77–153. 10.1007/978-3-211-99661-4_2. [DOI] [PubMed] [Google Scholar]
- Furukawa J.-i.; Sakairi N. Synthetic Studies on Resin Glycosides. Trends Glycosci. Glycotechnol. 2001, 13, 1–10. 10.4052/tigg.13.1. [DOI] [Google Scholar]
- Fürstner A. Total Syntheses and Biological Assessment of Macrocyclic Glycolipids. Eur. J. Org. Chem. 2004, 2004, 943–958. 10.1002/ejoc.200300728. [DOI] [Google Scholar]
- Jarikote D. V.; Murphy P. V. Metathesis and Macrocycles with Embedded Carbohydrates. Eur. J. Org. Chem. 2010, 26, 4959–4970. 10.1002/ejoc.201000491. [DOI] [Google Scholar]
- Cao S.; Guza R. C.; Wisse J. H.; Miller J. S.; Evans R.; Kingston D. G. I. Ipomoeassins A–E, Cytotoxic Macrocyclic Glycoresins from the Leaves of Ipomoea squamosa from the Suriname Rainforest1. J. Nat. Prod. 2005, 68, 487–492. 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]
- Cao S.; Norris A.; Wisse J. H.; Miller J. S.; Evans R.; Kingston D. G. I. Ipomoeassin F, a new cytotoxic macrocyclic glycoresin from the leaves of Ipomoea squamosa from the Suriname rainforest. Nat. Prod. Res. 2007, 21, 872–876. 10.1080/14786410600929576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürstner A.; Nagano T. Total Syntheses of Ipomoeassin B and E. J. Am. Chem. Soc. 2007, 129, 1906–1907. 10.1021/ja068901g. [DOI] [PubMed] [Google Scholar]
- Nagano T.; Pospíšil J.; Chollet G.; Schulthoff S.; Hickmann V.; Moulin E.; Herrmann J.; Müller R.; Fürstner A. Total Synthesis and Biological Evaluation of the Cytotoxic Resin Glycosides Ipomoeassin A–F and Analogues. Chem. - Eur. J. 2009, 15, 9697–9706. 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]
- Postema M. H. D.; TenDyke K.; Cutter J.; Kuznetsov G.; Xu Q. Total Synthesis of Ipomoeassin F. Org. Lett. 2009, 11, 1417–1420. 10.1021/ol900086b. [DOI] [PubMed] [Google Scholar]
- Zong G.; Barber E.; Aljewari H.; Zhou J.; Hu Z.; Du Y.; Shi W. Q. Total Synthesis and Biological Evaluation of Ipomoeassin F and Its Unnatural 11R-Epimer. J. Org. Chem. 2015, 80, 9279–9291. 10.1021/acs.joc.5b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong G.; Whisenhunt L.; Hu Z.; Shi W. Q. Synergistic Contribution of Tiglate and Cinnamate to Cytotoxicity of Ipomoeassin F. J. Org. Chem. 2017, 82, 4977–4985. 10.1021/acs.joc.7b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston D. G. I.; Cao S. U.S. Patent US 7,560,536 B2, 2009.
- Zong G.; Aljewari H.; Hu Z.; Shi W. Q. Revealing the Pharmacophore of Ipomoeassin F through Molecular Editing. Org. Lett. 2016, 18, 1674–1677. 10.1021/acs.orglett.6b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong G.; Hirsch M.; Mondrik C.; Hu Z.; Shi W. Q. Design, synthesis and biological evaluation of fucose-truncated monosaccharide analogues of ipomoeassin F. Bioorg. Med. Chem. Lett. 2017, 27, 2752–2756. 10.1016/j.bmcl.2017.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong G.; Sun X.; Bhakta R.; Whisenhunt L.; Hu Z.; Wang F.; Shi W. Q. New insights into structure–activity relationship of ipomoeassin F from its bioisosteric 5-oxa/aza analogues. Eur. J. Med. Chem. 2018, 144, 751–757. 10.1016/j.ejmech.2017.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. S.; Hill K.; McKenna M.; Ogbechi J.; High S.; Willis A. E.; Simmonds R. E. The Pathogenic Mechanism of the Mycobacterium ulcerans Virulence Factor, Mycolactone, Depends on Blockade of Protein Translocation into the ER. PLoS Pathog. 2014, 10, e1004061 10.1371/journal.ppat.1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M.; Simmonds R. E.; High S. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J. Cell Sci. 2016, 129, 1404–1415. 10.1242/jcs.182352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demangel C.; High S. Sec61 blockade by mycolactone: A central mechanism in Buruli ulcer disease. Biol. Cell 2018, 110, 237–248. 10.1111/boc.201800030. [DOI] [PubMed] [Google Scholar]
- Chany A.-C.; Tresse C.; Casarotto V.; Blanchard N. History, biology and chemistry of Mycobacterium ulcerans infections (Buruli ulcer disease). Nat. Prod. Rep. 2013, 30, 1527–1567. 10.1039/c3np70068b. [DOI] [PubMed] [Google Scholar]
- Gersch M.; Kreuzer J.; Sieber S. A. Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012, 29, 659–682. 10.1039/c2np20012k. [DOI] [PubMed] [Google Scholar]
- Cravatt B. F.; Wright A. T.; Kozarich J. W. Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- Li X.; Hu Y. Identifying the Cellular Targets of Bioactive Small Molecules with Activity-Based Probes. Curr. Med. Chem. 2010, 17, 3030–3044. 10.2174/092986710791959747. [DOI] [PubMed] [Google Scholar]
- Heal W. P.; Dang T. H. T.; Tate E. W. Activity-based probes: discovering new biology and new drug targets. Chem. Soc. Rev. 2011, 40, 246–257. 10.1039/C0CS00004C. [DOI] [PubMed] [Google Scholar]
- Park K.; Lee S. Additive-Free Decarboxylative Coupling of Cinnamic Acid Derivatives in Water: Synthesis of Allyl Amines. Org. Lett. 2015, 17, 1300–1303. 10.1021/acs.orglett.5b00303. [DOI] [PubMed] [Google Scholar]
- Kii I.; Shiraishi A.; Hiramatsu T.; Matsushita T.; Uekusa H.; Yoshida S.; Yamamoto M.; Kudo A.; Hagiwara M.; Hosoya T. Strain-promoted double-click reaction for chemical modification of azido-biomolecules. Org. Biomol. Chem. 2010, 8, 4051–4055. 10.1039/c0ob00003e. [DOI] [PubMed] [Google Scholar]
- Tantama M.; Lin W.-C.; Licht S. An Activity-Based Protein Profiling Probe for the Nicotinic Acetylcholine Receptor. J. Am. Chem. Soc. 2008, 130, 15766–15767. 10.1021/ja805868x. [DOI] [PubMed] [Google Scholar]
- Haldon E.; Nicasio M. C.; Perez P. J. Copper-catalysed azide-alkyne cycloadditions (CuAAC): an update. Org. Biomol. Chem. 2015, 13, 9528–9550. 10.1039/C5OB01457C. [DOI] [PubMed] [Google Scholar]
- Holmes C. P. Model Studies for New o-Nitrobenzyl Photolabile Linkers: Substituent Effects on the Rates of Photochemical Cleavage. J. Org. Chem. 1997, 62, 2370–2380. 10.1021/jo961602x. [DOI] [PubMed] [Google Scholar]
- Linxweiler M.; Schick B.; Zimmermann R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target Ther. 2017, 2, 17002. 10.1038/sigtrans.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield J. J.; High S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 1999, 112, 1477–1486. [DOI] [PubMed] [Google Scholar]
- Baron L.; Paatero A. O.; Morel J.-D.; Impens F.; Guenin-Macé L.; Saint-Auret S.; Blanchard N.; Dillmann R.; Niang F.; Pellegrini S.; Taunton J.; Paavilainen V. O.; Demangel C. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 2016, 213, 2885–2896. 10.1084/jem.20160662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon A. L.; Paavilainen V. O.; Sharma A.; Hegde R. S.; Taunton J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife 2014, 3, e01483 10.7554/eLife.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paatero A. O.; Kellosalo J.; Dunyak B. M.; Almaliti J.; Gestwicki J. E.; Gerwick W. H.; Taunton J.; Paavilainen V. O. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem. Biol. 2016, 23, 561–566. 10.1016/j.chembiol.2016.04.008. [DOI] [PubMed] [Google Scholar]
- MacKinnon A. L.; Garrison J. L.; Hegde R. S.; Taunton J. Photo-Leucine Incorporation Reveals the Target of a Cyclodepsipeptide Inhibitor of Cotranslational Translocation. J. Am. Chem. Soc. 2007, 129, 14560–14561. 10.1021/ja076250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B. C. S.; McKibbin C.; Callan A. C.; Roboti P.; Piacenti M.; Rabu C.; Wilson C. M.; Whitehead R.; Flitsch S. L.; Pool M. R.; High S.; Swanton E. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 2009, 122, 4393–4400. 10.1242/jcs.054494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Puyenbroeck V.; Vermeire K. Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cell. Mol. Life Sci. 2018, 75, 1541–1558. 10.1007/s00018-017-2743-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M.; Simmonds R. E.; High S. Mycolactone reveals substrate-driven complexity of Sec61-dependent transmembrane protein biogenesis. J. Cell Sci. 2017, 130, 1307–1320. 10.1242/jcs.198655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood P. J.; Juszkiewicz S.; Guna A.; Shao S.; Hegde R. S. EMC Is Required to Initiate Accurate Membrane Protein Topogenesis. Cell 2018, 175, 1507. 10.1016/j.cell.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney S. E.; Zahoranszky-Kohalmi G.; Brimacombe K. R.; Henderson M. J.; Lynch C.; Zhao T.; Wan K. K.; Itkin Z.; Dillon C.; Shen M.; Cheff D. M.; Lee T. D.; Bougie D.; Cheng K.; Coussens N. P.; Dorjsuren D.; Eastman R. T.; Huang R.; Iannotti M. J.; Karavadhi S.; Klumpp-Thomas C.; Roth J. S.; Sakamuru S.; Sun W.; Titus S. A.; Yasgar A.; Zhang Y. Q.; Zhao J.; Andrade R. B.; Brown M. K.; Burns N. Z.; Cha J. K.; Mevers E. E.; Clardy J.; Clement J. A.; Crooks P. A.; Cuny G. D.; Ganor J.; Moreno J.; Morrill L. A.; Picazo E.; Susick R. B.; Garg N. K.; Goess B. C.; Grossman R. B.; Hughes C. C.; Johnston J. N.; Joullie M. M.; Kinghorn A. D.; Kingston D. G. I.; Krische M. J.; Kwon O.; Maimone T. J.; Majumdar S.; Maloney K. N.; Mohamed E.; Murphy B. T.; Nagorny P.; Olson D. E.; Overman L. E.; Brown L. E.; Snyder J. K.; Porco J. A. Jr.; Rivas F.; Ross S. A.; Sarpong R.; Sharma I.; Shaw J. T.; Xu Z.; Shen B.; Shi W.; Stephenson C. R. J.; Verano A. L.; Tan D. S.; Tang Y.; Taylor R. E.; Thomson R. J.; Vosburg D. A.; Wu J.; Wuest W. M.; Zakarian A.; Zhang Y.; Ren T.; Zuo Z.; Inglese J.; Michael S.; Simeonov A.; Zheng W.; Shinn P.; Jadhav A.; Boxer M. B.; Hall M. D.; Xia M.; Guha R.; Rohde J. M. Canvass: A Crowd-Sourced, Natural-Product Screening Library for Exploring Biological Space. ACS Cent. Sci. 2018, 4, 1727–1741. 10.1021/acscentsci.8b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J.; Auld D. S.; Jadhav A.; Johnson R. L.; Simeonov A.; Yasgar A.; Zheng W.; Austin C. P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11473–11478. 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannotti M. J.; MacArthur R.; Jones R.; Tao D.; Singec I.; Michael S.; Inglese J. Detecting Secretory Proteins by Acoustic Droplet Ejection in Multiplexed High-Throughput Applications. ACS Chem. Biol. 2019, 14, 497–505. 10.1021/acschembio.9b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbechi J.; Ruf M.-T.; Hall B. S.; Bodman-Smith K.; Vogel M.; Wu H.-L.; Stainer A.; Esmon C. T.; Ahnström J.; Pluschke G.; Simmonds R. E. Mycolactone-Dependent Depletion of Endothelial Cell Thrombomodulin Is Strongly Associated with Fibrin Deposition in Buruli Ulcer Lesions. PLoS Pathog. 2015, 11, e1005011 10.1371/journal.ppat.1005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfo F. S.; Phillips R.; Wansbrough-Jones M.; Simmonds R. E. Recent advances: role of mycolactone in the pathogenesis and monitoring of Mycobacterium ulcerans infection/Buruli ulcer disease. Cell. Microbiol. 2016, 18, 17–29. 10.1111/cmi.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds R. E.; Lali F. V.; Smallie T.; Small P. L. C.; Foxwell B. M. Mycolactone Inhibits Monocyte Cytokine Production by a Posttranscriptional Mechanism. J. Immunol. 2009, 182, 2194–2202. 10.4049/jimmunol.0802294. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Law B. K.; Luesch H. Apratoxin A Reversibly Inhibits the Secretory Pathway by Preventing Cotranslational Translocation. Mol. Pharmacol. 2009, 76, 91–104. 10.1124/mol.109.056085. [DOI] [PubMed] [Google Scholar]
- Ogbechi J.; Hall B. S.; Sbarrato T.; Taunton J.; Willis A. E.; Wek R. C.; Simmonds R. E. Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis. 2018, 9, 397. 10.1038/s41419-018-0427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison J. L.; Kunkel E. J.; Hegde R. S.; Taunton J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 2005, 436, 285. 10.1038/nature03821. [DOI] [PubMed] [Google Scholar]
- Morel J.-D.; Paatero A. O.; Wei J.; Yewdell J. W.; Guenin-Macé L.; Van Haver D.; Impens F.; Pietrosemoli N.; Paavilainen V. O.; Demangel C. Proteomics Reveals Scope of Mycolactone-mediated Sec61 Blockade and Distinctive Stress Signature. Mol. Cell. Proteomics 2018, 17, 1750–1765. 10.1074/mcp.RA118.000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzke J. E.; Kozik P.; Morel J.-D.; Impens F.; Pietrosemoli N.; Cresswell P.; Amigorena S.; Demangel C. Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E5910–E5919. 10.1073/pnas.1705242114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenin-Macé L.; Baron L.; Chany A.-C.; Tresse C.; Saint-Auret S.; Jönsson F.; Le Chevalier F.; Bruhns P.; Bismuth G.; Hidalgo-Lucas S.; Bisson J.-F.; Blanchard N.; Demangel C. Shaping mycolactone for therapeutic use against inflammatory disorders. Sci. Transl. Med. 2015, 7, 289ra285–289ra285. 10.1126/scitranslmed.aab0458. [DOI] [PubMed] [Google Scholar]
- Wacker S. A.; Houghtaling B. R.; Elemento O.; Kapoor T. M. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol. 2012, 8, 235. 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chany A.-C.; Casarotto V.; Schmitt M.; Tarnus C.; Guenin-Macé L.; Demangel C.; Mirguet O.; Eustache J.; Blanchard N. A Diverted Total Synthesis of Mycolactone Analogues: An Insight into Buruli Ulcer Toxins. Chem. - Eur. J. 2011, 17, 14413–14419. 10.1002/chem.201102542. [DOI] [PubMed] [Google Scholar]
- Chany A.-C.; Veyron-Churlet R.; Tresse C.; Mayau V.; Casarotto V.; Le Chevalier F.; Guenin-Macé L.; Demangel C.; Blanchard N. Synthetic Variants of Mycolactone Bind and Activate Wiskott–Aldrich Syndrome Proteins. J. Med. Chem. 2014, 57, 7382–7395. 10.1021/jm5008819. [DOI] [PubMed] [Google Scholar]
- Saint-Auret S.; Abdelkafi H.; Le Nouen D.; Bisseret P.; Blanchard N. Synthetic strategies towards mycolactone A/B, an exotoxin secreted by Mycobacterium ulcerans. Org. Chem. Front. 2017, 4, 2380–2386. 10.1039/C7QO00608J. [DOI] [Google Scholar]
- Saint-Auret S.; Abdelkafi H.; Le Nouen D.; Guenin-Macé L.; Demangel C.; Bisseret P.; Blanchard N. Modular total syntheses of mycolactone A/B and its [2H]-isotopologue. Org. Biomol. Chem. 2017, 15, 7518–7522. 10.1039/C7OB01943B. [DOI] [PubMed] [Google Scholar]
- Gersbach P.; Jantsch A.; Feyen F.; Scherr N.; Dangy J.-P.; Pluschke G.; Altmann K.-H. A Ring-Closing Metathesis (RCM)-Based Approach to Mycolactones A/B. Chem. - Eur. J. 2011, 17, 13017–13031. 10.1002/chem.201101799. [DOI] [PubMed] [Google Scholar]
- Scherr N.; Gersbach P.; Dangy J.-P.; Bomio C.; Li J.; Altmann K.-H.; Pluschke G. Structure-Activity Relationship Studies on the Macrolide Exotoxin Mycolactone of Mycobacterium ulcerans. PLoS Neglected Trop. Dis. 2013, 7, e2143 10.1371/journal.pntd.0002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraneni P. K.; Conti B.; Matsumura Y.; Yang Z.; Johnson A. E.; Skach W. R. Stepwise Insertion and Inversion of a Type II Signal Anchor Sequence in the Ribosome-Sec61 Translocon Complex. Cell 2011, 146, 134–147. 10.1016/j.cell.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.