

Abstract
Background: Our previous studies demonstrated that a novel quinazoline derivative, DZ-50, inhibited prostate cancer epithelial cell invasion and survival by targeting insulin-like-growth factor binding protein-3 (IGFBP-3) and mediating epithelial-mesenchymal transition (EMT) conversion to mesenchymal-epithelial transition (MET). This study investigated the therapeutic value of DZ-50 agent in in vitro and in vivo models of advanced prostate cancer and the ability of the compound to overcome resistance to antiandrogen (enzalutamide) in prostate tumors. Approach: LNCaP and LNCaP-enzalutamide resistant human prostate cancer (LNCaP-ER) cells, as well as 22Rv1 and enzalutamide resistant, 22Rv1-ER were used as cell models. The effects of DZ-50 and the antiandrogen, enzalutamide (as single agents or in combination) on cell death, EMT-MET interconversion, and expression of IGFBP3 and the androgen receptor (AR), were examined. The TRAMP mouse model of prostate cancer progression was used as a pre-clinical model. Transgenic mice (20-wks of age) were treated with DZ-50 (100 mg/kg for 2 wks, oral gavage daily) and prostate tumors were subjected to immunohistochemical assessment of apoptosis, cell proliferation, markers of EMT and differentiation and IGFBP-3 and AR expression. A tissue microarray (TMA) was analyzed for expression of IGBP-3, the target of DZ-50 and its association with tumor progression and biochemical recurrence. Results: We found that treatment with DZ-50 enhanced the anti-tumor response to the antiandrogen via promoting EMT to MET interconversion, in vitro. This DZ-50-mediated phenotypic reversal to MET leads to prostate tumor re-differentiation in vivo, by targeting nuclear IGFBP-3 expression (without affecting AR). Analysis of human prostate cancer specimens and TCGA patient cohorts revealed that overexpression of IGBP-3 protein correlated with tumor recurrence and poor patient survival. Conclusions: These findings provide significant new insights into (a) the predictive value of IGFBP-3 in prostate cancer progression and (b) the antitumor action of DZ-50, [in combination or sequencing with enzalutamide] as a novel approach for the treatment of therapeutically resistant prostate cancer.
Keywords: Drug-induced phenotypic reversion, therapeutic response, prostate tumors
Introduction
Prostate cancer is the most commonly diagnosed non-skin cancer among American males, and it was estimated that 161,360 new cases will be diagnosed in the United States in 2019 [1]. The widespread screening using serum prostate-specific antigen (PSA) led to the diagnosis of over 95% of new cases, when prostate cancer is localized (within the gland) or regional (beyond the outer layer of the prostate into nearby regions). For localized prostate cancer, primary treatment with surgery or radiation is successful with five-year survival rates over 99% [2,3]. However within ten years of curative-intended treatment, approximately one third of patients experience biochemical recurrence [2]. Among patients who initially present with androgen-sensitive prostate tumors that have already progressed to metastases, the first line therapy is androgen-deprivation therapy (ADT), a mainstay treatment modality pioneered by Charles Huggins more than 70 years ago [4]. ADT dramatically reduces serum testosterone levels, which in turn decreases intratumoral levels of dihydrotestosterone (DHT)-the binding ligand for the androgen receptor (AR) responsible for activation of the androgen signaling in the prostate. Although almost all patients initially respond to ADT, the disease invariably progresses to castration-resistant prostate cancer (CRPC), usually concomitant with metastasis [2,3,5,6]. For hormone-naïve prostate cancer with markers of early metastasis, but lacking symptomatic and radiologic evidence of metastasis, the indicated course of clinical action therapy remains ADT with active surveillance [7,8]. Second line treatments are indicated for mCRPC and include taxane chemotherapy (1st and 2nd line taxane chemotherapy), second-generation antiandrogens, and cellular immunotherapy [5,6,9].
Metastases requires that cancer cells detach from the primary tumor, resist anoikis, invade the basement membrane, and disseminate by blood or lymph to a new location where the cancerous cells embed and proliferate [10]. The cellular plasticity afforded to a fully differentiated epithelium of prostate tumor glands, allows epithelial cells to de-differentiate into mesenchymal cells via epithelial-mesenchymal-transition (EMT) and re-differentiate via reversal to mesenchymal-epithelial transition (MET) within the tumor microenvironment during cancer evolution [10,11]. A characteristic feature of the EMT landscape is the loss of E-cadherin, causing adherens junction breakdown, which circumvents anoikis, promoting metastasis and therapeutic resistance [5,10]. In prostate cancer cells, induction of EMT is regulated by TGF-β and/or androgens, with a threshold AR level determining the phenotypic outcome and invasive properties [12]. A switch from E- to N-cadherin predicts prostate tumor progression, recurrence and mortality [13,14], and therapeutic targeting of N-cadherin in CRPC emerges as an effective strategy blocking metastasis [15].
Despite a growing number of new modalities approved by the US FDA, CRPC remains incurable, mainly due to the tremendous intratumor genomic diversity and cellular heterogeneity that several recent integrative advances were able to define [16-19]. Prostate cancer progression to metastasis and emergence of therapeutic resistance are frequently driven by aberrantly activated kinase signaling pathways regulating EMT that are potentially amenable to pharmacological inhibition [8,10,20]. Ongoing efforts to identify novel targets of therapeutic value towards empowering personalized therapy in patients with CRPC have met with various levels of success. In this study, we examined the effects of the quinazoline-derived agent DZ-50, that was previously developed and characterized in our lab to target anoikis, tight junctions and EMT [21-23]. The impact of DZ-50 in combination with enzalutamide (second generation antiandrogen), on phenotypic alterations within the ecosystem of the prostate tumor microenvironment and expression of insulin-growth factor binding protein-3 (IGFBP-3), was studied in in in vitro and in vivo models of advanced prostate cancer. Our results identified that DZ-50 mediated reversal of EMT to MET resulting in prostate tumor-re-differentiation enhances the response to antiandrogen via targeting IGFBP-3. We also show here that IGFBP-3 has potential predictive value of biochemical recurrence and disease progression in human prostate cancer.
Materials and methods
Cell cultures and drugs
The human prostate cancer cell lines LNCaP and 22Rv1 were obtained from ATCC. The enzalutamide resistant LNCaP-ER and 22Rv1-ER cell lines were generated by Dr. S. Koochekpour as described previously [24]. Briefly, LNCaP and 22Rv1 cells were maintained in RPMI-1640 supplemented with 10% FBS, 1% sodium pyruvate, and 1% antibiotics. For androgen deprivation, normal FBS was replaced with charcoal-stripped FBS in phenol-red free medium [24]. The 22Rv1-ER and LNCaP-ER cell lines were established in androgen-deprived medium with increasing concentrations of enzalutamide (10-40 μM) for a period of 4 months. DZ-50, a first-generation doxazosin quinazoline derivative developed in our laboratory was used as the novel therapeutic agent [21].
Western blot analysis
Cell lysates from prostate cells were subjected to Western blotting as previously described [23]. Subcellular fractionation was performed using NE-PER nuclear-cytoplasmic fraction kit (Thermo Scientific, Rockford, IL). Protein samples were analyzed by SDS-PAGE and transferred to Hybond-C membranes (Amersham Pharmacia Biotech, Inc; Piscataway, NJ). Membranes were incubated with the respective primary antibody (4°C), and exposed to species-specific peroxidase-labeled secondary antibodies. Signal detection was achieved and visualized using a UVP Imaging System. Protein bands were normalized to GAPDH or H3 expression. Antibodies against specific proteins are summarized on Table S1. The monoclonal antibody against N-cadherin was from Abcam (San Francisco, CA); Antibodies against the IGFBP3 and AR (N-20) protein were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against E-cadherin, GAPDH and H3 were from Cell Signaling Technology (Danvers, MA).
Cell viability assay
Cell viability was evaluated using the Thiazolyl Blue Tetrazolium bromide (MTT, Thermo Fisher Scientific, Waltham, MA) assay. Briefly cells were seeded into 24-well plates and after grown to 60% to 75% confluence, were treated with vehicle or DZ-50 and/or enzalutamide at indicated doses (DMSO, Sigma-Aldrich, St. Louis, MO). Absorbance was measured at 570 nm and 690 nm using μQuant Spectrophotometer (Biotech Instruments Inc., Winooski, VT).
Migration assays
Cells were seeded in 6-well plates and at 65% to 70% density the cell monolayers were wounded. After 72 hrs the number of migrating cells towards center of the wound is counted in three different fields in the absence and presence of DZ-50 and/or enzalutamide.
Tumor microarray construction and immunohistochemistry staining in human prostate specimens
Paraffin-embedded tissue blocks were selectively cored from 165 PCa patients and 34 benign control patients, who underwent radical prostatectomy at the University of Kentucky, to construct a tissue microarray (TMA) through the Markey Biospecimen and Tissue Procurement Shared Resource, University of Kentucky-Lexington (Table 1). Approval for use of human prostate tissue was obtained from the University of Kentucky Institutional Review Board. Tissue cores (2 mm) containing cancerous tissues, adjacent benign epithelial tissues, or human benign prostatic hyperplasia (BPH) were used for the construction of tissue microarrays with duplicate cores from each patient. There were 4 disease groups: Gleason scores ≥ 8, 7, ≤ 6, and benign prostatic hyperplasia (BPH). Prostate cancer TMA slides were deparaffinized in a 60°C oven for 1 h and then rinsed in three changes of xylene. To block endogenous peroxidases, slides were immersed in 0.3% methanol/hydrogen peroxide for 20 min and then rehydrated for 1 min in each of 100%, 95%, 75%, and 50% ethanol and then double-distilled H2O. Heat-induced epitope retrieval was performed using a digital decloaking chamber (BioCare Medical, Concord, CA) (20 mins; 110°C) in DAKO high pH EDTA or low pH citrate antigen retrieval buffer, as indicated. Endogenous peroxidase activity was quenched using reagent from Envision+ Kits (DAKO, Glostrup Municipality, Denmark), followed by incubation with primary antibodies overnight at 4°C (against IGFBP3). Sides were subsequently incubated with Envision+ polymer-bound secondary antibody (DAKO), then visualized with 3,3’-Diaminobenzidine and lightly counterstained with hematoxylin. Slides were subsequently dehydrated and mounted with coverslips; followed by microscopic analysis. The clinicopathological characteristics of prostate-TMA specimens are described on Table 1. Assessment of immunoreactivity and protein expression and localization patterns were quantified by two independent observers as previously described [25]. The threshold intensity for positive areas (weak, medium, and strong positive) ranged from 0-100.
Table 1.
Description of clinical and pathological characteristics of prostate cancer specimens used on TMA
| TMA Description | Clinicopathlogical Parameters | # of Cases |
|---|---|---|
| Specimens | BPH | 34 |
| Prostate cancer | 165 | |
| Tumor grade | Gleason ≤ 6 | 33 |
| Gleason 7 | 100 | |
| Gleason ≥ 8 | 32 | |
| Clinical Stage | Stage I | 3 |
| Stage II | 86 | |
| Stage III | 47 | |
| Stage IV | 18 | |
| Unknown | 11 | |
| Metastatic prostate cancer | Organ Confined | 141 |
| Lymph node/bone Metastasis | 13 | |
| Unknown | 11 | |
| Smoking | Tobacco/cigar/others | 52 |
| Non-smoking | 56 | |
| Unknown | 57 | |
| Recurrence | Local/Distal Recurrence | 31 |
| No Recurrence | 82 | |
| Unknown | 52 | |
| PSA | < 10 ng/ml | 3 |
| 10-20 ng/ml | 4 | |
| > 20 ng/ml | 66 | |
| Age range (years) | BPH | 57-81 |
| Prostate cancer | 41-75 |
Transgenic mouse model of prostate cancer progression (TRAMP)
The transgenic adenocarcinoma of mouse prostate (TRAMP) develops following the expression of SV40 T antigen under the control of the rat probasin promoter [26,27]. Sexual maturity of TRAMP mice contributes to prostate cancer initiation and progression in a pattern resembling the clinical development of androgen independent prostate cancer [26,27]. Mice were maintained under environmentally controlled conditions and subject to a 12-h light/dark cycle with food and water ad libitum. TRAMP Mice (18-20 weeks) were matched with littermates and were treated with either vehicle control or DZ-50 (100 mg/kg for 14 days, oral gavage daily) and harvested on Day 15.
Immuno-histochemical analysis
Tissue specimens transgenic mouse prostate tumors were formalin fixed and paraffin-embedded; serial sections (5 μ), were subjected to immuno-histochemical analysis using antibodies against Ki-67, E-cadherin, N-cadherin, Androgen Receptor (N-20), IGFBP3, cofilin and cytokeratin-18 (as described on Table S1) After blocking nonspecific binding, sections were incubated with primary antibody (overnight, 4°C) and were subsequently exposed to biotinylated goat anti-rabbit IgG (2 hrs, room temperature) and horseradish peroxidase-streptavidin (EMD Millipore, Billerica, MA). Signal/Color detection was achieved with SigmaFast 3, 3’-Diaminobenzidine tablets (Sigma-Aldrich, St. Louis, MO) and counterstained with haematoxylin. The incidence of apoptosis was examined using the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (Chemicon International). Sections were counterstained with methyl green [27]. Images were captured via light microscopy (40× and 100×) using an Olympus BX51 microscope (Olympus America, Center Valley, PA). The intensity and level of immunoreactivity and the number of positive cells were scored by two independent observers as previously described [28].
Statistical analysis
For the data from in vitro studies and the in vivo pre-clinical model, Student t-test, one-way, or two-way ANOVA were performed using GraphPad Prism 6 software to determine the statistical significance of difference between means/treatments. All numerical data are presented as mean ± SEM. Statistical significance was set at P < 0.05.
The Spearman’s correlation coefficient was calculated to quantify the correlation between IGFBP3 expression and Gleason score. To assess the association between IGFBP-3 expression and patient’s overall survival/recurrence, samples were classified into high or low IGFBP-3 expression subgroup using the median as the cutoff value. Kaplan-Meier curves and the log rank test were used to compare patient’s survival/disease-free time between high and low IGFBP-3 expression subgroups. The proportional hazards model was used to calculate the hazard ratio (HR) and its associated 95% CI. TCGA analysis: IGFBP-3 expression data and associated clinical information from Nakagawa et al (n=596) (PLoS ONE 2008/05/28), Setlur et al (n=363) (J Natl Cancer Inst 2008/06/04), Taylor et al (n=185) (Cancer Cell 2010/07/13), TCGA PRAD (n=498) (Nature volume 487, pages 330-337 (19 July 2012)) were downloaded from Oncomine (https://www.oncomine.org/) or GDC (https://portal.gdc.cancer.gov/projects/TCGA-PRAD).
Results
The cell viability of the dose response of LNCaP and enzalutamide resistant LNCaP-ER cells to increasing doses of DZ-50 was assessed at 48 hrs. The results shown on Figure 1A, indicate that DZ-50 induced a significant loss of cell viability in a dose-dependent manner in both cell lines regardless of their sensitivity to enzalutamide. There was no difference in cell death induction between the LNCaP and LNCaP-ER cells in response to increasing concentrations of DZ-50 (Figure 1A). To investigate the significance/potential role of the AR variants (AR V7) in the response to DZ-50 we examined the response of 22Rv1 and 22Rv1 enzalutamide resistant, 22Rv1-ER, cell lines. The dose response of cell viability loss by DZ-50 (1-10 µM), was similar in both the 22Rv1 parental and 22Rv1-ER enzalutamide-resistant cells, with a significant loss detected at a concentration of DZ-50 as low as 2 µM, compared to untreated control cells (Figure 1B). To determine the antitumor effect by the combination of DZ-50 with the antiandrogen, we subsequently analyzed the response of LNCaP and LNCaP-ER cells to enzalutamide alone and in combination with DZ-50 (Figure 1C and 1D). The results shown on Figure 1 revealed that DZ-50 increased the sensitivity of LNCaP-ER cells to the antiandrogen. In response to increasing doses of enzalutamide (3-10 µM) in combination with DZ-50 (5 µM), there was a significant increase in cell death in both the LNCaP and LNCaP-ER cells, compared to untreated controls or DZ-50-treated only (Figure 1D). Furthermore, loss of cell viability in LNCaP parental cells in response to enzalutamide (100 µM) and DZ-50 combination was significantly higher than that observed for the LNCaP-ER cells.
Figure 1.
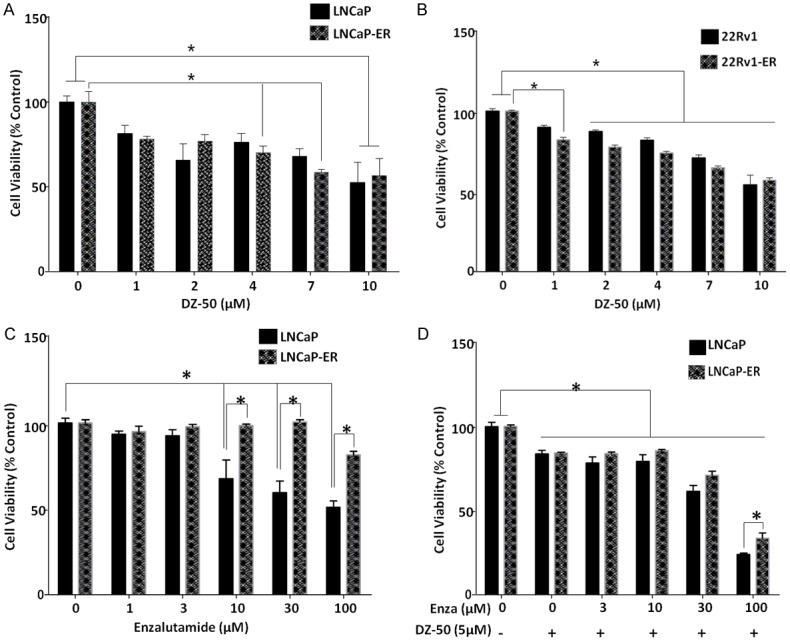
Response of Antiandrogen-Resistant Prostate Cancer Cell to Novel Agent DZ-50. A and B. Reveal the dose-response effect of DZ-50 on LNCaP/LNCaP-ER and 22Rv/22Rv1-ER prostate cancer cell viability, respectively. Cells were treated for 48 hrs with increasing drug concentrations as indicated and cell viability was assessed. DZ-50 treatment resulted in a significant loss of cell viability in the enzalutamide resistant prostate cancer cells (both LNCaP-ER and 22Rv1-ER) at 4 µM. At higher concentrations there was a further induction of cell death that was comparable to the parental LNCaP and 22RV1 cell lines. Numerical values are the average of the percentage of cell viability relative to untreated controls from three independent experiments in duplicate +/- SEM (standard error of the mean). Statistical significance is at *P < 0.05. C and D. Indicate the response of LNCaP and LNCaP-ER prostate cancer cells to enzalutamide alone or in the presence of DZ-50 (5 µM) for 72 hrs. Cell viability was determined and values represent the average from three independent experiments performed in duplicate +/- SEM. *P < 0.05.
Assessment of the effect of the novel quinazoline agent DZ-50, on prostate cancer cell migration, revealed that the LNCaP-ER cells exhibited a significantly increased migration potential compared to LNCaP cells, indicating the aggressive behavior of the enzalutamide resistant cells (P < 0.05) (Figure 2A and 2B). Treatment with DZ-50 and the antiandrogen (enzalutamide), either as single agents or in combination, led to a significant reduction in migratory capacity of both LNCaP and LNCaP-ER cells, compared to untreated controls (P < 0.05). There was no significant difference in the effect among the three treatments (Figure 2B).
Figure 2.
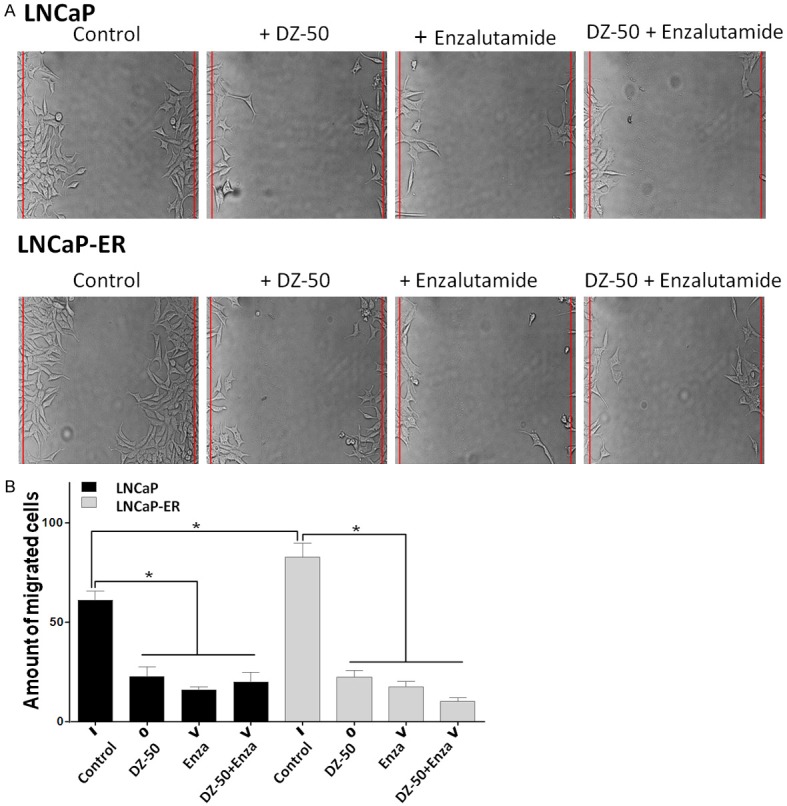
Effect of DZ-50 and Enzalutamide Combination on Prostate Cancer Cell Migration. A. Representative images of cell migration; cell monolayers were subjected to wounding, cultures were treated with either DZ-50 (5 µM) or enzalutamide (30 µM) for 72 hrs. B. Indicates the data from the quantitative analysis of cell migration, values represent the average from three independent experiments in duplicate +/- SEM. *P < 0.05.
Epithelial protein markers lost are adherens and tight junction proteins (E-cadherin, ZO-1), while there is gain of mesenchymal markers including vimentin and N-cadherin [14]. We next profiled the EMT protein markers by Western blot analysis. As shown on Figure 3, DZ--50 treatment for 72 hrs led to a marked increase in the epithelial marker (E-cadherin), as well as the mesenchymal marker (N-cadherin) expression in both LNCaP and LNCaP-ER cells (Figure 3A). Treatment of LNCaP cells with enzalutamide (alone) led to a decrease in N-cadherin levels, with no major effect on E-cadherin (E/N=1.2), reflecting an epithelial phenotype, while the combination treatment of DZ-50 and enzalutamide reversed the phenotypic ratio (E/N=1.0; Figure 2A). The LNCaP-ER cells exhibited an increase in N-cadherin concurrent with a modest increase of E-cadherin levels after treatment with either drug alone, pointing to an epithelial characteristics; significantly enough, the combination of DZ-50 and enzalutamide resulted in a dramatic overexpression of N-cadherin, reflecting reversal to the original mesenchymal phenotype (Figure 3A).
Figure 3.
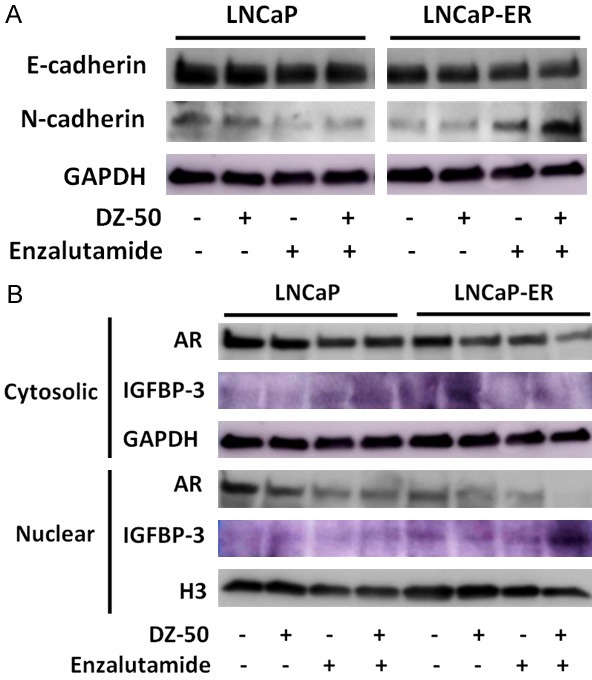
DZ-50 Treatment Mediates Phenotypic EMT-MET Interconversion and IGFBP3 Expression in Vitro. A. Reveals the E-cadherin and N-cadherin expression profile by Western blot analysis in prostate cancer LNCaP and LNCaP-ER cells in response to DZ-50 (5 µM), or enzalutamide (30 µM) as single agents or in combination (72 hrs). No marked changes in E-cadherin are detected, while there is a significant reduction in N-cadherin in response to the combination treatment. B. Reveals the expression profile of AR and IGBP-3 in the cytosolic and nuclear fractions of prostate cancer cells after the various treatments. Subcellular fractionation was performed as in “Materials and Methods”. Western blots are each representative of three independent experiments. Protein expression was assessed by densitometric analysis of band intensity (numerical values shown at the bottom of blot).
Representative analysis of subcellular fractions of the LNCaP and LNCaP-ER cell lines before and after treatment is indicated on Figure 3B. As shown DZ-50 (alone) results in marked reduction of nuclear IGFBP-3 in LNCaP cells. Enzalutamide treatment exerted a similar effect by decreasing nuclear IGFBP-3, while increasing cytosolic IGFBP-3 levels; the combination of DZ-50 and enzalutamide led to depletion of nuclear IGFBP-3 and paralleled by increased cytoplasmic accumulation in LNCaP cells (Figure 3B). In LNCaP-ER cells, DZ-50 decreases nuclear IGFBP-3 with a simultaneous increase of cytosolic IGFBP-3. In the LNCaP-ER cells, enzalutamide decreased nuclear AR with no apparent effect on cytosolic AR. Interestingly, the combination treatment led to a further decrease of both cytosolic and nuclear AR levels compared to DZ-50, or enzalutamide alone. Treatment with enzalutamide (as single agent) decreased nuclear AR levels in both the LNCaP and LNCaP-ER cells (Figure 3B).
We subsequently investigated the in vivo antitumor effect of DZ-50 in the pre-clinical model of androgen-sensitive advanced prostate tumor model. Prostate tumors were removed from intact TRAMP mice after 2 wks of treatment with DZ-50 (100 mg/kg/day) (Figure 4). Panel A reveals representative images of prostate of 6-pair of littermates and the weight of the respective prostate glands/tumors. Panel B reveals characteristic images of Ki67 immunoreactivity (cell proliferation) and TUNEL (apoptosis) positive cells, in serial sections of prostate tumors derived from 20 wk-old male TRAMP transgenic mice treated with DZ-50. The results on Figure 4 indicate that treatment DZ-50 decreases the overall prostate tumor weight in these transgenic mice (Figure 4A). Treatment with DZ-50, led to a significant reduction in cell proliferation and a parallel significant increase in tumor cell apoptosis (Figure 4B). Quantitative analysis of the Ki-67 immunoreactivity among the prostate tumor epithelial cells indicated a significantly lower proliferative index in DZ-50 treated tumors compared to controls (*P < 0.05). There was a significant reduction in number of apoptotic cells among the prostate tumor cell populations after treatment with DZ-50 (100 mg/kg), indicating a marked induction of apoptosis, compared to vehicle controls (Figure 4B; *P < 0.05).
Figure 4.
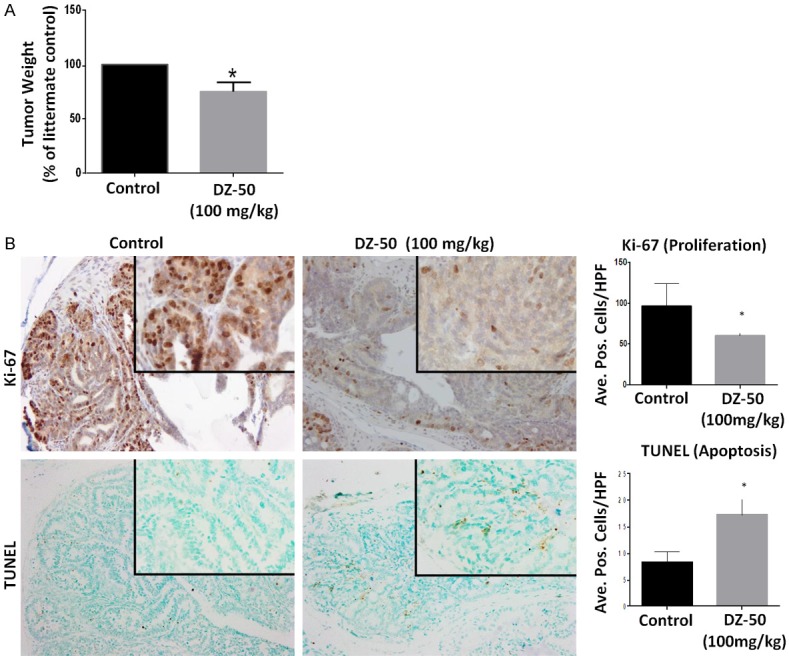
Effect of DZ-50 on Prostate Tumor Growth Kinetics in TRAMP Model. Prostate tumors were surgically removed from TRAMP mice after 2 wks of treatment with DZ-50 (100 mg/kg/day). A. The average values of prostate weight from 6-pairs of littermates (+/- SEM). B. Shows representative images of Ki-67 immunoreactivity (cell proliferation) and TUNEL (apoptosis) positive cells, in serial sections of prostate tumors derived from (20 wk-old) TRAMP mice in response to DZ-50 treatment. Magnification ×100, insert ×400. The numerical data from the quantitative analysis are shown on the left respectively. Scoring was performed as described in “Materials and Methods” in littermates (n=6). Quantitative analysis of Ki-67 immunoreactivity among the prostate tumor epithelial cells indicated a significantly lower proliferative index in prostate tumors from DZ-50 treated compared to controls (*P < 0.05). There was a significant increase in the number of apoptotic (TUNEL-positive) cells among the prostate tumor cell populations after DZ-50 treatment (100 mg/kg), compared to controls (untreated mice). Numerical data represent the average scoring of three fields per individual section, (assessed by two independent observers). Values represent the average +/- SEM (standard error of the mean). Statistically significant difference between groups is set at *P < 0.05.
To determine the impact of treatment with DZ-50 on the phenotypic landscape of the tumors in vivo, we next examined the EMT features of the TRAMP prostate tumors, before and after treatment. Figure 5A, reveals characteristic immunostaining images from profiling the EMT landscape in serial sections of the tumors, as well the expression of AR and IGFBP3 in prostate tumor specimens from TRAMP mice. Our findings show that in vivo exposure to DZ-50 leads to a considerable reduction in IGFBP-3 and N-cadherin, while it increases E-cadherin, indicating EMT conversion to MET. Interestingly enough, DZ-50 exerted an effect on neither the immunoreactivity nor the localization of AR (Figure 5A). Profiling of cytokeratin-18, indicated a markedly increased expression after DZ-50 treatment, suggesting that the DZ-50 mediated reversal to MET among prostate tumor epithelial cells (TRAMP model) results in prostate tumor- re-differentiation (Figure 5B). Previous work from our group identifying cofilin, the primary actin cytoskeleton regulator as a potential marker of radioresistance in human prostate cancer [25], prompted us to investigate the impact of DZ-50 on cofilin expression in serial sections of prostate tumors. As shown on Figure 5B, DZ-50 treatment of TRAMP mice decreases cofilin immunoreactivity compared to prostate tumors from control mice.
Figure 5.
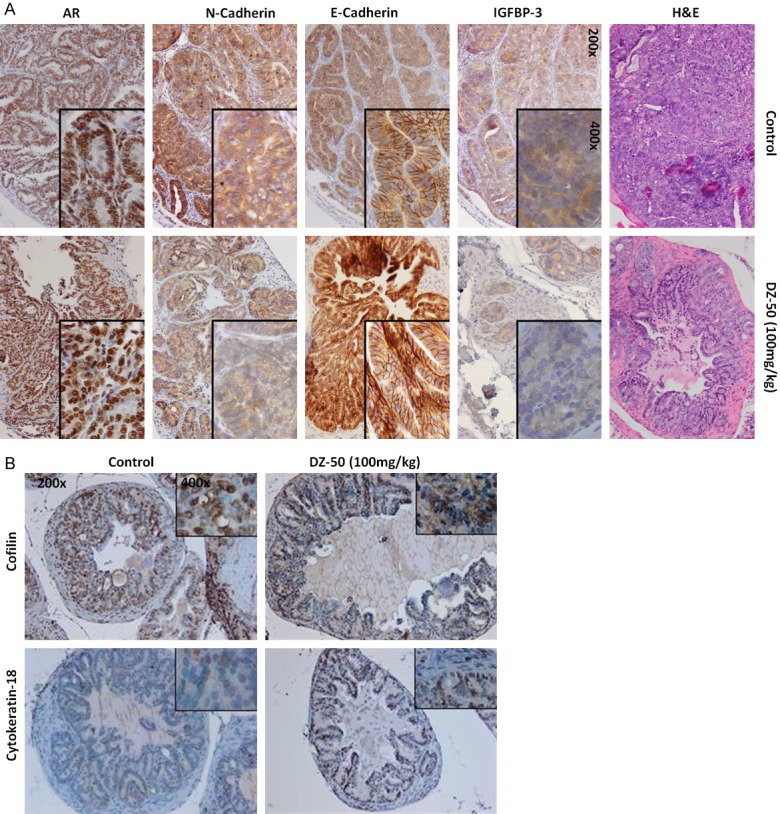
DZ-50 Promotes EMT-MET and Re-differentiation in Prostate Tumors in Vivo. A. Reveals representative images of prostate tumor serial sections from untreated control and DZ-50 treated prostate tumors stained with H&E, and assessment of the expression of IGFBP3, E-cadherin, N-cadherin and the AR. B. Reveals characteristic patterns of expression of cofilin (the actin cytoskeleton organization effector) and cytokeratin-18 differentiation marker among prostate tumor cells consequential to treatment with DZ-50. (Magnification ×200, insert ×1000).
Profiling the expression of IGBP3 in human prostate TMA tissue specimens consisting of normal prostate, benign prostate (BPH) and prostate cancer of increasing Gleason grade (Table 1), revealed a significant increase in IGFBP-3 levels in prostate cancer compared to normal or benign prostate (P < 0.01). Furthermore a strong association between high IGBP-3 expression with clinical tumor aggressiveness as well as prostate cancer recurrent after ADT (Figure 6A-C). We also found a significant correlation between elevated IGFBP-3 levels with tumor recurrence and poor survival/lethal disease (Figure 6D and 6E, respectively).
Figure 6.

Clinical Value of IGFBP3 as a Biomarker in Human Prostate Cancer. A. The human prostate cancer specimens on the TMA were subjected to immunostaining profiling the IGFBP3 expression. Progression through through stage and grade, there was a significant increase in IGFBP3 levels; there was markedly higher expression detected in prostate tumor specimens from a patient who had received treatment with neoadjuvant ADT. (Magnification ×200, insert ×1000). B and C. Indicate the numerical analysis of IGFBP3 scoring in the human TMA. A significant increase in IGFBP3 levels was detected in prostate tumors with increasing Gleason grade compared to normal and BPH prostate (P < 0.001). Elevated IGFBP3 levels were observed in prostate cancer specimens from recurrent disease after ADT. D. Reveals there was a significant increase in IGFBP3 expression among tumors from patients who exhibited both local and distant recurrence. E. Indicates that a significant increase in IGFBP3 levels is associated with poor survival and death of patients with prostate cancer (P < 0.001).
TCGA analysis was performed to assess the association between IGFBP-3 expression and overall survival/recurrence of prostate cancer patients. As shown on Figure 7A, IGFBP-3 expression data and associated clinical information from Nakagawa et al (n=596) (PLoS ONE, 2008), Setlur et al (n=363) (JNCI 2008), Taylor et al (n=185) (Cancer Cell 2010), TCGA PRAD (n=498) (Nature, 487: 330-337, 2012) were downloaded from Oncomine (https://www.oncomine.org/) or GDC (https://portal.gdc.cancer.gov/projects/TCGA-PRAD). The Spearman’s correlation coefficient was calculated, to quantify the correlation between IGFBP-3 expression and Gleason score. Kaplan-Meier curves and the log rank test were used to compare patient’s survival/disease-free time between high and low IGFBP-3 expression subgroups. The hazard ratio (HR) and its associated 95% CI was calculated using the proportional hazards model and identified a significant correlation between IGFBP3 high expression levels and disease-free survival (Figure 7B and 7C). Genomic analysis of IGFBP isoforms (including IGFBP3) using several cohorts of prostate cancer patients (MSKCC, TCGA, FH, Michigan, SU2C), showed no significant difference in IGFBP3 mRNA expression levels between primary and metastatic prostate tumors or therapeutic response to ADT (Figure 7D).
Figure 7.
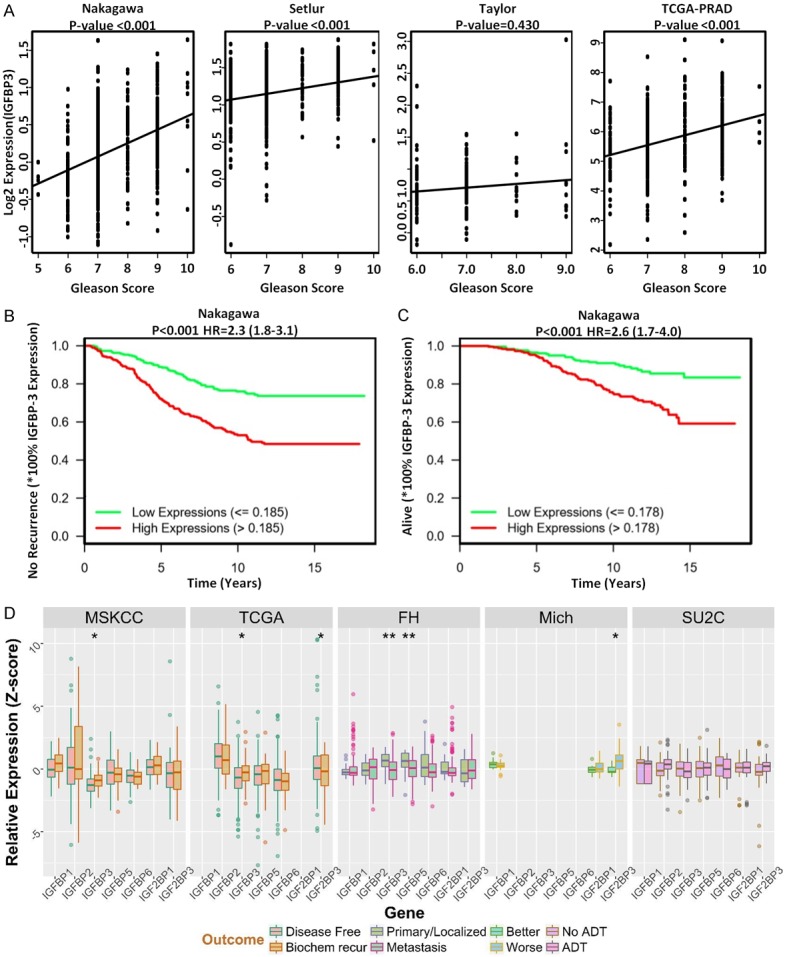
TCGA Analysis of IGBP3 Expression in Human Prostate Cancer Patient Cohorts. (A) Reveals IGFBP3 expression data and associated clinical information from Nakagawa et al (n=596), Setlur et al (n=363), Taylor et al (n=185), TCGA PRAD (n=498) were downloaded from Oncomine (https://www.oncomine.org/) or GDC (https://portal.gdc.cancer.gov/projects/TCGA-PRAD). The Spearman’s correlation coefficient was calculated, to quantify the correlation between IGFBP3 expression and Gleason score. (B and C) Indicate Kaplan-Meier curves comparing patients’ survival/disease-free time between high and low IGFBP3 expression subgroups. The proportional hazards model was used to calculate the hazard ratio (HR) and its associated 95% CI. (D) Reveals the genomic analysis of IGFBP proteins (IGFBP1, IGFBP3, IGFBP4, IGFBP5, IGFBP6), using several cohorts of prostate cancer patients (MSKCC, TCGA, FH, Michigan, SU2C); there was no significant difference in IGFBP3 mRNA expression levels between primary and metastatic prostate tumors or therapeutic response to ADT (D).
Discussion
The cellular composition of the prostate tumor microenvironment is critical for progression to metastatic disease and patients’ prognosis and response to treatment [11,29]. Emerging single cell tools can profile the individual cell types comprising the tumor ecosystem to reconstruct mechanisms of cell-cell interactions that can potentially be targeted to overcome therapeutic resistance [30]. There is rapidly growing interest in the phenotypic manipulation of the EMT dynamic process towards overcoming therapeutic resistance in advanced metastatic cancer. Evidence by our team and other investigators supported the therapeutic targeting value of androgen and/or TGF-β regulated EMT-MET interconversion in pre-clinical models of prostate cancer progression, with potential applications in clinical advanced disease [15,28,31].
This study demonstrates the ability of a novel-quanizoline analogue, DZ-50, to overcome resistance to enzalutamide in human prostate cancer cells in vitro and in the TRAMP transgenic mouse preclinical model in vivo. The ability of DZ-50 to inhibit prostate cancer cell migration, shown here in the enzalutamide-resistant cells, is consistent with our previous characterization of the action of the drug in blocking prostate tumor migration and invasion via anoikis induction [21,22]. The DZ-50 induced phenotypic reversal of EMT to MET, results in prostate tumor re-differentiation; importantly there was no apparent impact on AR expression or cellular localization by DZ-50. Profiling the expression of EMT markers revealed a marked reduction in N-cadherin driving an epithelial phenotype after treatment with DZ-50 in in vitro and in vivo prostate tumor models. These results support that phenotypic reversion of prostate tumor EMT to MET, consequential to DZ-50 treatment, results in the acquisition of the well-differentiated phenotype, ultimately overcoming resistance to the antiandrogen. Indirect support for our findings stems from recent evidence suggesting different mechanisms and responses utilized by prostate cancer cells in the acquisition of enzalutamide resistance. Beyond the canonical AR-mediated mechanisms of resistance, a broader approach across several cell lines implicates a significant contribution from non-AR mediated mechanisms [24,32,33]. The present findings indicate DZ-50 alone had no impact on cytosolic AR, but it decreased nuclear AR expression levels in the enzalutamide resistant prostate cancer cells (LNCaPER) to the same degree as in the enzalutamide sensitive cells LNCaP. In the LNCaP-ER cells, enzalutamide decreased nuclear AR with no apparent effect on cytosolic AR levels. Interestingly, the combination treatment led to a further decrease of both cytosolic and nuclear AR levels compared to DZ-50, or enzalutamide alone. Treatment with enzalutamide (as single agent) decreased nuclear AR levels in both the LNCaP and LNCaP-ER cells (Figure 3B). This downregulation of AR-protein could be due to protein degradation as recently shown in response to other drugs [24], without involving changes in mRNA expression levels.
Our previous studies showed that TGF-β resistant LNCaP cells exhibit higher sensitivity to DZ-50 compared to TGF-β responsive LNCaPTβRII cells, implicating EMT in this temporal resistance to DZ-50 [23]. The present results provide proof-of principle that DZ-50 mediates (via TGF-β), conversion of prostate cancer cell EMT to MET by targeting IGFBP-3 in vitro and in vivo. In accord with recent studies supporting that TGFβ signaling engages the actin cytoskeleton via cofilin as the main driver to dictate EMT and invasive properties in prostate cancer [34,35], our original findings link targeting cofilin to acquisition of prostate tumor re-differentiation by DZ-50. This is the first time that changes in the phenotypic landscape and the actin cytoskeleton induced by the novel agent DZ-50, were found to lead to tumor re-differentiation and sensitization of advanced prostate tumors to the antiandrogen therapy. Identification of the EMT effector IGFBP-3 as the primary target of DZ-50 (independent of AR) is of major translational significance, as overexpression of this protein correlates with poor clinical outcomes and disease recurrence in CRPC patients (Figure 7). Taken together with recent evidence suggesting that IGFBP-3 promotes TGFβ-mediated EMT and cell motility in other human cancer cells [36], our study supports a potential therapeutic value of IGFBP-3 as a target of DZ-50 towards the development of optimized strategies in combination with enzalutamide for the treatment of therapeutically resistant prostate cancer, as well as a predictive value of IGFBP-3 in progression to recurrent disease.
In conclusion, the present study demonstrates that DZ-50 treatment-induced dramatic phenotypic alterations, dictate the interconversion of mesenchymal cells into epithelial cells entering re-differentiation. These treatment-induced phenotypic changes sensitize prostate tumor cell populations, by compromising their survival, to overcome therapeutic resistance to antiandrogens. Further exploitation of high-throughput datasets interrogating multiple cellular functions in human prostate cancer specimens will enable identification not only of druggable signaling events, but also of predictive biomarkers, like IGFBP-3 for tailoring therapy optimization towards personalized approaches for the treatment of patients with advanced prostate cancer.
Acknowledgements
These studies were supported by the James F. Hardymon Endowment to the Urology Department at the University of Kentucky (PJH, HP, NK); Urology Care Foundation Scholarship (ZC); the Appalachian Cancer Training for Undergraduate Students Program, NCI R25CA221765 (HD) and the Biostatistics and Bioinformatics Core of Markey Cancer Center, NCI P30CA177558 (DH, CW).
Disclosure of conflict of interest
None.
Abbreviations
- AR
androgen receptor
- CRPC
castration-resistant prostate cancer
- PSA
prostate specific antigen
- LNCaP-ER
LNCAP enzalutamide resistant
- 22Rv1-ER
22Rv1 enzalutamide resistant
- EMT
epithelial-mesenchymal-transition
- MET
mesenchymal epithelial-transition
- TGF-β
transforming growth factor-β
- DHT
dihydrotestosterone
- ADT
androgen deprivation therapy
- CDK
cyclin dependent kinases
- ECM
extracellular matrix
- CAFs
Cancer-associated fibroblasts
- IGF
insulin growth factor
- IGFBP-3
Insulin growth factor binding protein-3
- TRAMP
transgenic adenocarcinoma of the mouse prostate
- MTT
Mitochondrial Thiazolyl Tetrazolium
- TMA
tissue microarray
- TUNEL
deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Fakhrejahani F, Madan RA, Dahut WL. Management options for biochemically recurrent prostate cancer. Curr Treat Options Oncol. 2017;18:26–31. doi: 10.1007/s11864-017-0462-4. [DOI] [PubMed] [Google Scholar]
- 3.Crawford ED, Petrylak D, Sartor O. Navigating the evolving therapeutic landscape in advanced prostate cancer. Urol Oncol. 2017;35S:S1–S13. doi: 10.1016/j.urolonc.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 4.Huggins C, Stevens RE Jr, Hodges CV. Studies on prostatic cancer: II. the effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209–223. [Google Scholar]
- 5.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO TROPIC Investigators. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 6.Nakazawa M, Paller C, Kyprianou N. Mechanisms of therapeutic resistance in prostate cancer. Curr Oncol Rep. 2017;19:13–20. doi: 10.1007/s11912-017-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaiswing L, Weiss HL, Jayswal RD, Clair DKS, Kyprianou N. Profiles in prostate cancer radioresistance mechanisms in radiation and cancer. Crit Rev Oncog. 2018;23:39–67. doi: 10.1615/CritRevOncog.2018025946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Z, Livas T, Kyprianou N. Anoikis and EMT: lethal “Liaisons” during cancer progression. Crit Rev Oncog. 2016;21:155–168. doi: 10.1615/CritRevOncog.2016016955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor hetergeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Zhu M, Kyprianou N. Role of androgens and the androgen receptor in epithelial- mesenhymal transition and invasion of prostate cancer cells. FASEB J. 2010;24:769–77. doi: 10.1096/fj.09-136994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolijn K, Verhoef EI, van Leenders GJ. Morphological and immunohistochemical identification of epithelial-mesenchymal transition in clinical prostate cancer. Oncotargert. 2015;6:24488–24497. doi: 10.18632/oncotarget.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progression of prostate cancer. Clin Cancer Res. 2007;13:7003–7011. doi: 10.1158/1078-0432.CCR-07-1263. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, An J, Horvath S, Gleave M, Rettig MB, Wainberg ZA, Reiter RE. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16:1414–1420. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, Lucas JM, Brown LG, Dumpit RF, DeSarkar N, Higano C, Yu EY, Coleman R, Schultz N, Fang M, Lange PH, Shendure J, Vessella RL, Nelson PS. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;4:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peacock JW, Takeuchi A, Hayashi N, Liu L, Tam KJ, Al Nakouzi N, Khazamipour N, Tombe T, Dejima T, Lee KC, Shiota M, Thaper D, Lee WC, Hui DH, Kuruma H, Ivanova L, Yenki P, Jiao IZ, Khosravi S, Mui AL, Fazli L, Zoubeidi A, Daugaard M, Gleave ME, Ong CJ. SEMA3C drives cancer growth by transactivating multiple receptor tyrosine kinases via plexin B1. EMBO Mol Med. 2018;10:219–238. doi: 10.15252/emmm.201707689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrison JB, Shaw YJ, Chen CS, Kyprianou N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007;67:11344–11352. doi: 10.1158/0008-5472.CAN-07-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hensley PJ, Desiniotis A, Wang C, Stromberg A, Chen CS, Kyprianou N. Novel pharmacologic targeting of tight junctions in prostate cancer cells. PLoS One. 2014;9:e86238. doi: 10.1371/journal.pone.0086238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao Z, Koochekpoor S, Strup ES, Kyprianou N. Reversion of epithelial-mesenchymal-transition (EMT) by DZ-50 via targeting IGBP3 leads to anoikis of prostate cancer cells. Oncotarget. 2017;8:78507–78519. doi: 10.18632/oncotarget.19659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wadosky KM, Shourideh M, Goodrich DW, Koochekpour S. Riluzole induces AR degradation via endoplasmic reticulum stress pathway in androgen-dependent and castration-resistant prostate cancer cells. Prostate. 2019;79:140–150. doi: 10.1002/pros.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stark TW, Hensley PJ, Spear A, Pu H, Strup SS, Kyprianou N. Predictive value of EMT signature and cofilin expression in prostate cancer response to radiotherapy. Prostate. 2017;77:1583–1591. doi: 10.1002/pros.23435. [DOI] [PubMed] [Google Scholar]
- 26.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of utochthonous prostate cancer in the TRAMP model. Prostate Cancer Prostatic Dis. 1999;2:70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 27.Pu H, Collazo J, Jones E, Gayheart D, Sakamoto S, Vogt A, Mitchell B, Kyprianou N. Dysfunctional TGF-β receptor II accelerates prostate tumorigenesis in the TRAMP mouse model. Cancer Res. 2009;69:7366–7374. doi: 10.1158/0008-5472.CAN-09-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paller C, Pu H, Begemann DE, Wade CA, Hensley PJ, Kyprianou N. TGF-β signaling blockade enhances therapeutic response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate. 2019;79:31–43. doi: 10.1002/pros.23708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wadosky KM, Koochekpour S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget. 2016;7:64447–64470. doi: 10.18632/oncotarget.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson AD, Kessenbrock K, Davis RT, Pervolaraks N, Webb Z. Tumor hetergeneity and metastasis at single-cell resolution. Nature Cell Biol. 2018;20:1349–1360. doi: 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin SK, Pu H, Penticuff J, Cao Z, Horbinski C, Kyprianou N. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res. 2016;76:912–926. doi: 10.1158/0008-5472.CAN-15-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, Fessenden TB, Isikbay M, Paner GP, Szmulewitz RZ, Vander Griend DJ. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016;7:26259–26265. doi: 10.18632/oncotarget.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai YC, Chen WY, Abou-Kheir W, Zeng T, Yin JJ, Bahmad H, Lee YC, Liu YN. Androgen deprivation therapy-induced epithelial-mesenchymal transition of prostate cancer through downregulating SPDEF and activating CCL2. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1717–1727. doi: 10.1016/j.bbadis.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Collazo J, Zhu B, Larkin S, Pu H, Koochekpour S, Martin SK, Horbinski C, Kyprianou N. Cofilin regulates cellular invasion responses to TGF-β towards prostate cancer metastasis. Cancer Res. 2014;74:2362–2373. doi: 10.1158/0008-5472.CAN-13-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin SK, Kamelgarn M, Kyprianou N. The cytoskeleton targeting value in prostate cancer treatment. Am J Clin Exper Urol. 2014;2:15–26. [PMC free article] [PubMed] [Google Scholar]
- 36.Natsuizaka N, Obashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, Klein-Szanto AJ, Herlyn M, Diehl JA, Nakagawa H. Insulin-like growth factor binding protein-3 promotes transforming growth factor-beta1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31:1344–1353. doi: 10.1093/carcin/bgq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.