

Abstract
Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) is a key mediator of innate immunity. IRAK4 overactivation is linked with several autoimmune diseases. To date, many IRAK4 inhibitors have been developed to block the protein’s kinase activity with the most advanced reaching Phase II clinical trials. Nevertheless, several reports suggest kinase activity is not disease-relevant in certain cell types, so removing scaffolding signaling in addition to IRAK4 kinase activity may offer a better therapeutic outcome. Herein, we describe the design and synthesis of an IRAK4 Proteolysis Targeted Chimera (PROTAC). We show that IRAK4 degradation induced by compound 9 leads to the inhibition of multiple cytokines in PBMCs. However, in IL-1β stimulated human dermal fibroblasts, inhibition of IL-6 and TNF-α release was not observed despite IRAK4 degradation. Nonetheless, the possibility of targeting both IRAK4 kinase and scaffolding function could potentially lead to new therapeutic opportunities to treat autoimmune, inflammatory, and oncological diseases.
Keywords: IRAK4, PROTAC, scaffolding role, degradation
Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) belongs to a family of four kinases (IRAK4, IRAK1, IRAK2, and IRAK-M). IRAK4 is a serine/threonine kinase that is involved in transduction pathways stimulated by the Toll-like receptors (TLRs) and the Interleukin-1 (IL-1) family of receptors.1,2 Recognition of foreign pathogens and inflammatory signals by these receptors promotes IRAK4 binding to the adapter protein myeloid differentiation primary response gene (88) (MyD88) resulting in IRAK4 activation that in turn leads to the production of pro-inflammatory cytokines via the NFκβ pathway.3 IRAK4 deficiency or loss of function has been reported to increase susceptibility to several pathogens, while kinase activation has been linked with various autoimmune diseases such as systemic lupus erythematosus, psoriasis, rheumatoid arthritis, and cancer.4,5 Interestingly, IRAK4 regulation of downstream processes has been mainly associated with its kinase function; however, several reports have indicated a nonkinase function for IRAK4 in several cell types.4,6 Cushing et al. showed that pharmacological inhibition of IRAK4 did not result in IL-6 and TNF-α inhibition despite IRAK4 phosphorylation levels being reduced in IL-1β stimulated human dermal fibroblasts.7 In support of these observations, IRAK4 deficient fibroblasts versus wild-type cells show IRAK4 scaffolding functions were important for IL-1β signaling but that its kinase role was redundant.6 Chiang and co-workers showed that IRAK4 kinase activity was dispensable in human B and T cells, dendritic, and monocytic cells, whereas genetic ablation by siRNA demonstrated a scaffolding role for IRAK4 in these cells.4 In addition, IRAK4 knockout mice show profound impaired cellular responses to IL-1, IL-8, and TLRs ligands.8 Together, these data indicate that the development of strategies to remove IRAK4 protein could offer an attractive therapeutic opportunity for diverse disease indications.
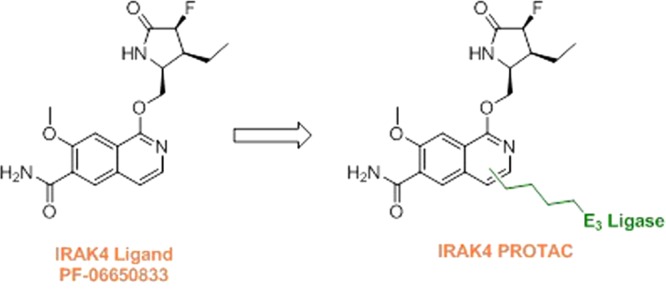
Multiple potent and selective inhibitors of IRAK4 have been reported in the literature targeting autoimmune, inflammatory, and oncological diseases.8,9 To date, three molecules have reached clinical trials with the most advanced being the isoquinoline ligand PF-06650833 (Figure 1).10 However, inhibiting IRAK4 kinase activity alone may not be sufficient to produce a therapeutic effect due to the role of IRAK4 as both a scaffolding protein and an active kinase.
Figure 1.
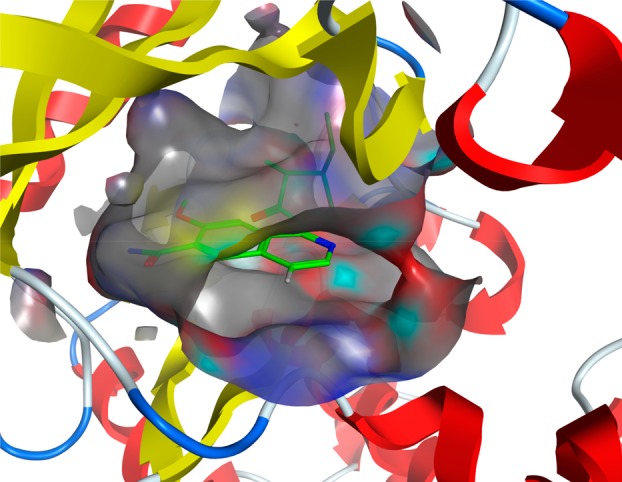
Cocrystal structure of PF-06650833 with IRAK4 kinase domain showing solvent exposed vector at 4-position of isoquinoline core (PDB 5UIU).
The heterobifunctional molecules referred to as proteolysis targeting chimeras (PROTACs) were identified as promoters of cellular protein degradation in 2001.11,12 PROTACs contain one moiety that binds an E3 ligase and another that binds a desired cellular target protein of interest. PROTAC-induced proximity results in ubiquitination of the target followed by its degradation by the proteasome. One of the key differences of a PROTAC over an inhibitor is that by removing all protein function, there is the potential to achieve more profound pharmacology than by inhibiting a functional binding site.13 Herein, we describe the design and development of PROTACs that induce the degradation of IRAK4.
Results and Discussion
We evolved a synthetic strategy for PROTAC design that facilitated variations in both E3 ligase ligands and linker properties and allowed rapid assessment of the degradation profiles of PROTAC analogues. To prepare IRAK4 degrader compounds, we chose to incorporate a chemical template related to PF-06650833 that was known to potently bind the IRAK4 kinase domain10 and to attach this to either a Von Hippel Lindau (VHL) E3 ligase ligand, a Cereblon (CRBN) E3 ligase ligand, or a ligand to the Inhibitor of Apoptosis (IAP) E3 ligases. Three separate E3 ligases were employed, as engagement of a given ligase does not always translate into functional degradation.14 Using published crystal structures of IRAK4 in complex with kinase inhibitors, we were able to perform small molecule docking studies to predict the best position to attach our linkers such that critical binding interactions with the target were not disrupted. Based on our modeling, we initially evaluated two flexible 12 atom linkers as this was predicted to be sufficient length to exit the IRAK4 binding site, and in an initial survey of the potential physicochemical space, we chose the hydrophilic polyethylene glycol (PEG) as well as the hydrophobic all-carbon chain (Figure 2a).15,16
Figure 2.

Approach to IRAK4 PROTACs. (a) Linking VHL, CRBN, and IAP E3 ligase ligands to simplified IRAK4 ligand through either a carbon or PEG chain to generate IRAK4 PROTACs. (b) Table comprising PROTAC binding affinity and lipophilicity as determined by Chrom LogD. nt = not tested, D max = maximum degradation observed.
The six heterobifunctional compounds were then assayed to confirm retention of IRAK4 kinase domain binding, as well as selectivity versus the closely related IRAK1 protein (Figure 2b).17 Gratifyingly, all compounds tested maintained affinity and selectivity for IRAK4, with the assay variability confounded by poor physicochemical properties.
The ability of these PROTACs to reduce IRAK4 protein levels was then tested in peripheral blood mononuclear cells (PBMCs). The carbon linked VHL PROTAC (compound 3) induced 50% degradation at 3 μM (Figure 3a). Importantly, the E3 ligase dependence upon degradation was confirmed using a VHL-nonbinding enantiomeric control, which after inverting the three stereocenters is unable to recruit the VHL ligase18 and, as expected, afforded no degradation of IRAK4 (Figure 3b). The other five PROTACs tested (compounds 2, 4–7) had no effect on cellular IRAK4 levels (see Figure S1). The PEG-linked VHL PROTAC (compound 2) is less lipophilic than the carbon analogue (compound 3), which may suggest that poor permeability was the reason for the lack of degradation observed with this molecule. For the CRBN and IAP PROTACs, there are many potential reasons why these were not found to degrade IRAK4. For example, the linker may be the wrong length to facilitate efficient ternary complex formation. The orientation of the protein–CRBN/IAP E3 ligase ternary complex may also not be able to promote efficient ubiquitin transfer onto an IRAK4 surface lysine residue. Finally, even though these compounds may bind to the protein, this does not always translate into degradation, as has been observed previously in promiscuous kinase PROTAC experiments with both VHL and CRBN E3 ligase binders.14
Figure 3.

IRAK4 degradation induced by IRAK4-VHL PROTAC. Representative image of Western blotting analysis of PBMC cells treated with increasing amounts of compound 3 (a) and the VHL nonbinding enantiomeric control, compound 3-ve (b) over 24 h treatment.
Subsequently, shorter four and six carbon atom linkers to VHL were evaluated (see Figure S2). However, these compounds did not afford IRAK4 degradation, presumably as a result of being too short to form a stable ternary complex between IRAK4 and VHL proteins.
We hypothesized that a more potent IRAK4 binder could be used to achieve greater levels of degradation for the next iteration of PROTAC design.15 Through a previously reported synthesis,19 we attached the fully functionalized fluoro/ethyl lactam present in PF-06650833 onto the IRAK4 PROTAC warhead to prepare compound 8 (Figure 4a). This compound afforded more potent IRAK4 degradation compared to 3 as observed by Western blot, with a DC50 in PBMC cells of 259 nM (Figure 4b and Figure S3). Following a short optimization effort focusing on polarity and flexibility, we modified the 12-atom carbon linker to the VHL E3 ligase binder to a more rigid, polar spirocyclic pyrimidine. Pleasingly, compound 9 demonstrated increased potency with a DC50 of 151 nM in PBMC cells in addition to lower in vitro clearance in human and rat liver microsomes (Figure 4a,b). As previously shown (Figure 3), no IRAK4 degradation was observed when cells were treated with a VHL-nonbinding enantiomeric control (9-ve)18 (Figure S4b). In addition, based on the kinome selectivity profile of the IRAK4 ligand,10 the extent of degradation of IRAK1 and LRRK2 was measured with compound 9. Our data shows that similar effects were observed with both the ligand and compound 9, supporting literature evidence that ligand binding does not necessarily translate into degradation14 (Figure S4c,d).
Figure 4.
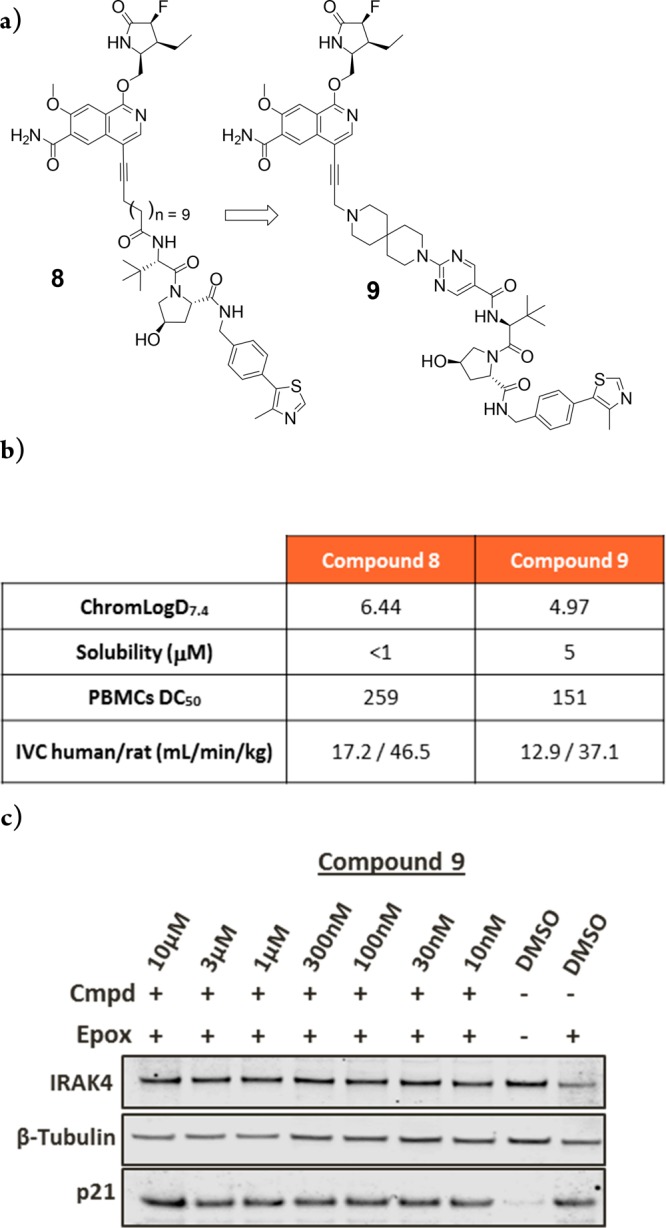
IRAK4 PROTAC-mediated degradation requires ternary complex formation and proteasome activity. (a) PROTAC optimization of IRAK4 warhead and linker to identify compound 9. (b) Comparison of compound properties between compound 8 and 9. (c) PBMCs treated with 10 μM epoxomicin for 2 h prior to the addition of compound 9 for a further 22 h.
To confirm that IRAK4 degradation was proteasome dependent, PBMCs were pretreated with epoxomicin before compound addition. Figure 4c shows that IRAK4 levels are unchanged vs DMSO-treated cells, suggesting that IRAK4 PROTAC-mediated degradation is occurring in a proteasome-dependent manner.13 Inhibition of the proteasome by epoxomicin is confirmed by the accumulation of p21 protein levels.
Next, we sought to investigate the IRAK4 potential kinase independent role in TLR mediated signaling. For this purpose, the IRAK4 ligand (PF-06650833), the tool IRAK4 PROTAC (compound 9), and the inactive control (compound 9-ve) were monitored for cytokine inhibition upon TLR7/8 stimulation in PBMCs. All compounds were capable of completely blocking IL-6 secretion (Figure 5a) as well as a wider panel of cytokines (Figure 5b). PF-06650833 exhibited the highest potency (Figure 5a,b), consistent with its higher binding potency, indicating that there was no additional potency benefit achieved through IRAK4 PROTAC-mediated degradation over kinase inhibition.
Figure 5.

IRAK4 phenotypic signaling. (a,b) PBMCs were pretreated for 18 h with PF-06650833, compound 9, or compound 9-ve followed by 8 h stimulation with R848 (2.5 μg/mL), and cytokine levels were determined using MSD technology. (c) Western blotting of dermal fibroblast treated with compound 9 for 24 h. (d) Secreted IL-6 levels from dermal fibroblast following 18 h pretreatment with 300 nM of each compound followed by 8 h stimulation with IL-1β (10 ng/mL). n = 4 ± SE.
Finally, we sought to investigate whether our PROTAC could replicate the reported findings that IRAK4 nonkinase functions were critical for cytokine secretion in human dermal fibroblasts.7 First, we determined that compound 9 was able to induce a reduction of IRAK4 protein levels with a DC50 = 36 nM in this cell type (Figure 5c). Compound 9, compound 9-ve, and PF-06650833 were then added to dermal fibroblasts followed by IL-1β stimulation. However, we did not observe any inhibition of IL-6 secretion upon PROTAC treatment (Figure 5d). This result contrasts with previous reports that showed that IL-6 levels were completely abrogated in IRAK4 deficient dermal fibroblasts.7 A plausible explanation could be related to the origin of the fibroblast cells tested, as different donors may respond differently to similar stimulation. Additionally, since IRAK4-null fibroblasts never expressed IRAK4, we could hypothesize that signaling patterns such as MyD88 and IRAK1 or its downstream pathways such as NF-κβ and MAPK are not responsive to IL-1R signaling in these cells, whereas temporary removal of IRAK4 protein by a PROTAC is not sufficient to attenuate this pathway. There is also the possibility that the remaining portion of IRAK4 in the cell that is not degraded by a PROTAC is sufficient to drive IL-1R responses.
In summary, we report a small molecule PROTAC approach achieving potent intracellular degradation of the kinase IRAK4. The PROTAC-induced IRAK4 degradation is dependent on binding to VHL and is reversed upon blocking proteasome activity. However, compound 9 was not found to possess a differentiated pharmacological profile over PF-06650833 in a phenotypic assay measuring a variety of inflammatory cytokines (Figure 5b). In addition, the IRAK4 knockout phenotype reported with dermal fibroblasts could not be reproduced with compound 9 (Figure 5d). Therefore, more work is required to understand the biology of this target. It is clear, however, that development of new modalities such as PROTACs to target IRAK4 may not only support the understanding of IRAK4 biology but could also lead to the development of new therapeutic agents to treat inflammatory and oncological disease.
Acknowledgments
We would like to thank Afjal Miah and Ian Smith for helpful chemistry discussions, Jane Denyer and Zuni Bassi for biology support, and Sebastien Campos and Andrew Benowitz for reviewing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00219.
General experimental information; full synthetic procedures and characterization data for all new compounds; NMR and LC–MS data. Experimental procedures for cell culture, PBMC isolation from blood, degradation assay, Western blotting and cytokine analysis (PDF)
Author Contributions
The manuscript was written by N.A.A., J.N., and G.A.M. All authors have given approval to the final version of the manuscript.
All animal studies were ethically reviewed and carried out in accordance with Animals (Scientific Procedures) Act 1986 and the GSK Policy on the Care, Welfare and Treatment of Animals.
The authors declare the following competing financial interest(s): The authors are employees and shareholders of GlaxoSmithKline.
Supplementary Material
References
- Akira S.; Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4 (7), 499–511. 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Brzezinska A. A.; Johnson J. L.; Munafo D. B.; Ellis B. A.; Catz S. D. Signalling mechanisms for Toll-like receptor-activated neutrophil exocytosis: key roles for interleukin-1-receptor-associated kinase-4 and phosphatidylinositol 3-kinase but not Toll/IL-1 receptor (TIR) domain-containing adaptor inducing IFN-beta (TRIF). Immunology 2009, 127 (3), 386–97. 10.1111/j.1365-2567.2008.02980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill L. A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–8. 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Chiang E. Y.; Yu X.; Grogan J. L. Immune complex-mediated cell activation from systemic lupus erythematosus and rheumatoid arthritis patients elaborate different requirements for IRAK1/4 kinase activity across human cell types. J. Immunol. 2011, 186 (2), 1279–88. 10.4049/jimmunol.1002821. [DOI] [PubMed] [Google Scholar]
- Ngo V. N.; Young R. M.; Schmitz R.; Jhavar S.; Xiao W.; Lim K. H.; Kohlhammer H.; Xu W.; Yang Y.; Zhao H.; Shaffer A. L.; Romesser P.; Wright G.; Powell J.; Rosenwald A.; Muller-Hermelink H. K.; Ott G.; Gascoyne R. D.; Connors J. M.; Rimsza L. M.; Campo E.; Jaffe E. S.; Delabie J.; Smeland E. B.; Fisher R. I.; Braziel R. M.; Tubbs R. R.; Cook J. R.; Weisenburger D. D.; Chan W. C.; Staudt L. M. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470 (7332), 115–9. 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Jiang Z.; Qian Y.; Casanova J. L.; Li X. IRAK4 kinase activity is redundant for interleukin-1 (IL-1) receptor-associated kinase phosphorylation and IL-1 responsiveness. J. Biol. Chem. 2004, 279 (25), 26748–53. 10.1074/jbc.M400785200. [DOI] [PubMed] [Google Scholar]
- Cushing L.; Stochaj W.; Siegel M.; Czerwinski R.; Dower K.; Wright Q.; Hirschfield M.; Casanova J. L.; Picard C.; Puel A.; Lin L. L.; Rao V. R. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J. Biol. Chem. 2014, 289 (15), 10865–75. 10.1074/jbc.M113.544809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Wesche H.; Stevens T.; Walker N.; Yeh W. C. IRAK-4 inhibitors for inflammation. Curr. Top. Med. Chem. 2009, 9 (8), 724–37. 10.2174/156802609789044407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary D.; Robinson S.; Romero D. L. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J. Med. Chem. 2015, 58 (1), 96–110. 10.1021/jm5016044. [DOI] [PubMed] [Google Scholar]
- Lee K. L.; Ambler C. M.; Anderson D. R.; Boscoe B. P.; Bree A. G.; Brodfuehrer J. I.; Chang J. S.; Choi C.; Chung S.; Curran K. J.; Day J. E.; Dehnhardt C. M.; Dower K.; Drozda S. E.; Frisbie R. K.; Gavrin L. K.; Goldberg J. A.; Han S.; Hegen M.; Hepworth D.; Hope H. R.; Kamtekar S.; Kilty I. C.; Lee A.; Lin L. L.; Lovering F. E.; Lowe M. D.; Mathias J. P.; Morgan H. M.; Murphy E. A.; Papaioannou N.; Patny A.; Pierce B. S.; Rao V. R.; Saiah E.; Samardjiev I. J.; Samas B. M.; Shen M. W. H.; Shin J. H.; Soutter H. H.; Strohbach J. W.; Symanowicz P. T.; Thomason J. R.; Trzupek J. D.; Vargas R.; Vincent F.; Yan J.; Zapf C. W.; Wright S. W. Discovery of Clinical Candidate 1-{[(2S,3S,4S)-3-Ethyl-4-fluoro-5-oxopyrrolidin-2-yl]methoxy}-7-methoxyisoquinoli ne-6-carboxamide (PF-06650833), a Potent, Selective Inhibitor of Interleukin-1 Receptor Associated Kinase 4 (IRAK4), by Fragment-Based Drug Design. J. Med. Chem. 2017, 60 (13), 5521–5542. 10.1021/acs.jmedchem.7b00231. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (15), 8554–9. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth J. S. Jr.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 2004, 126 (12), 3748–54. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–7. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell chemical biology 2018, 25 (1), 78–87. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Toure M.; Hellerschmied D.; Salami J.; Jaime-Figueroa S.; Ko E.; Hines J.; Crews C. M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–10. 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Hu J.; Xu F.; Chen Z.; Bai L.; Fernandez-Salas E.; Lin M.; Liu L.; Yang C. Y.; Zhao Y.; McEachern D.; Przybranowski S.; Wen B.; Sun D.; Wang S. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61 (2), 462–481. 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaction Biology Corp: http://www.reactionbiology.com/webapps/site/.
- Churcher I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?. J. Med. Chem. 2018, 61 (2), 444–452. 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- Patent WO 2015150995.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.