

Abstract
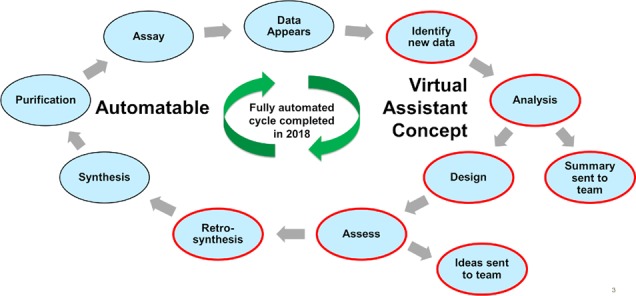
The virtual assistant concept is one that many technology companies have taken on despite having other well-developed and popular user interfaces. We wondered whether it would be possible to create an effective virtual assistant for a medicinal chemistry organization, the key being delivering the information the user would want to see, directly to them, at the right time. We introduce Kernel, an early prototype virtual assistant created at Lilly, and a number of examples of the scenarios that have been implemented to try to demonstrate the concept. A biochemical assay summary email is described that brings together new results and some basic analysis, delivered within an hour of new data appearing for that assay, and an email delivering new compound design ideas directly to the original submitter of a compound shortly after their compound was tested for the first time. We conclude with a high level description of the first example of a Design-Make-Test-Analyze cycle completed in the absence of any human intellectual input at Lilly. We believe that this concept has much potential in changing the way that computational results and analysis are delivered and consumed within a medicinal chemistry group, and we hope to inspire others to implement their own similar solutions.
Keywords: Virtual assistant, automation, data analysis
Within mainstream technology companies the virtual assistant concept has been heavily adopted, despite these companies having existing well-developed user interfaces. These include the likes of Google Assistant, Apple’s Siri, Microsoft’s Cortana, Amazon’s Alexa, and Samsung’s Bixby, each providing these companies different ways of interacting with users. One of the keys of the virtual assistant concept is providing the user with the information they want at the exact time that they want it. An example would be the way Google delivers information on journey time and traffic to Android phones just before the time people may begin their commute to and from their place of work.
Within the medicinal chemistry organization of a pharmaceutical company such as Lilly, the application of data analysis and other computational methods is inconsistent between teams. Many tools are desktop or web based and rely on the user knowing what kind of approach they want to take and then requesting it (possibly running it themselves). An alternate delivery of such analysis would be to recognize when the user might want to perform such analysis, compute the result in an automated way, and deliver that directly to the user. The first approach results in inconsistency and is fairly time-consuming for each individual user applying the desired approach. Whereas the second, if some approaches can be standardized and run in an automated way, should be far more efficient and much more scalable and potentially enable new solutions to be delivered.
Thinking about the way that the mainstream technology industry has embraced the virtual assistant concept, we wondered whether it would be possible to implement aspects of that approach within a medicinal chemistry environment. If we were to do this we would need to think about what drives medicinal chemistry cycles (primarily in the lead optimization phase).
Lilly, like many other groups doing medicinal chemistry, follows a Design-Make-Test-Analyze (DMTA) cycle1 which has been standard practice for a long time (although timelines for each cycle have come down with improvements in technology). Basically, a course of action is set given the current knowledge and understanding, and that direction is not perturbed until the emergence of new data. Decision making revolves around this appearance of new data. If we are thinking about how to implement a virtual assistant and want to be most impactful, we need to mimic this behavior and drive automated approaches off of the appearance of new information. With many assays being run by different people at different sites on a project, simply knowing that new data exists can be a challenge and is low hanging fruit a virtual assistant can help with. Couple this with additional automated analysis, and the virtual assistant can achieve significant impact.
If we now know when we want to deliver information to users to help their decision making, we need to think about what information and analysis is important to each project. Each project will have a number of assays within its flow scheme, and each one will have an amount of standard analysis that is performed (often duplicated by different users), plus some more custom, context dependent analysis. With a virtual assistant we would hope to be able to automate that standard analysis and present the data in such a way that the user could then apply anything more context dependent subsequently.
Here, we introduce Kernel, an early prototype virtual assistant created with the aim to enhance medicinal chemistry at Lilly. The name of such an assistant, although having no bearing on the science being delivered, does assist in uptake and understanding among users. Colonel Eli Lilly, as the company’s founder, is well-known by Lilly employees and the virtual assistant name is a pun on both that, and the fact kernel is a term used within the computer science and machine learning fields, which has generally been well received by users. Unlike the mainstream technology companies utilizing voice recognition to interact with users, Kernel is currently configured to interact with users via email, and a number of POC workflows have been implemented in KNIME 3.3, high level details of which are provided below.2
The first task we sought to make inroads with was getting Kernel to summarize and analyze the data generated (typically each week) by the main biochemical assay on a project. Most projects will have a biochemical assay that serves as the workhorse for the project and will often be the first measure of activity of a newly synthesized compound on that project. As such, most medicinal chemists will be most interested in the output of that assay to see how the compounds they had made have performed.
In Figure 1, there is an example of the main biochemical assay summary email which tries to put together many of the standard analyses a medicinal chemist would (or should) do, to save them running more manual queries to achieve the same result. This email appears in a project team member’s inbox within an hour of the new data appearing in the database.
Figure 1.

Example of main biochemical assay summary email.
The first paragraph is a high level summary of what has been tested in that run, including the project name, number of compounds, the run date, and an assessment of diversity of the structures assessed using an internal 2D structural fingerprint. Following this, we have a link to an HTML page showing the new structures that have been tested that week (Figure 2). Here, the user can see the new activities using a single click from the email, along with other calculated properties of interest and efficiency metrics. Beyond the raw activity values, most users would want to know how these new activities compare to close analogues previously tested. To achieve this we implemented each of the compound ID’s as clickable links which display a number of matched molecular pairs (MMPs) to the newly tested compounds, displayed in a similar HTML format with the fold changes in activity and other properties calculated.
Figure 2.
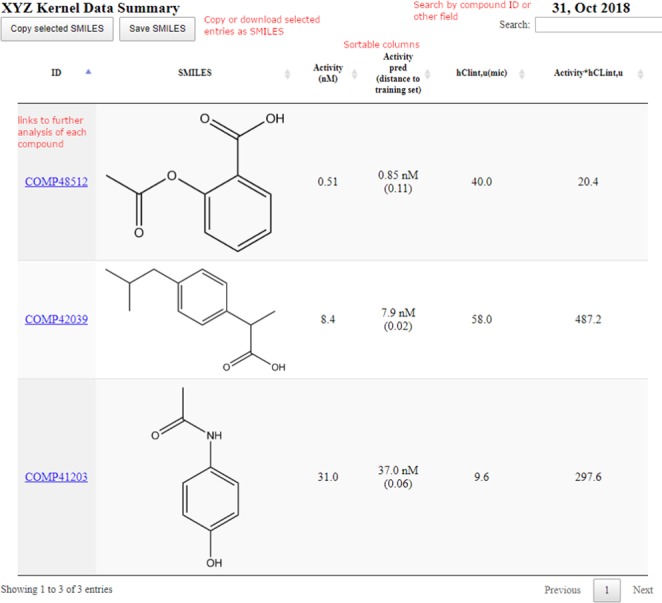
Example of newly tested structure activity data.
Something else that Kernel does in the main assay summary email is check potentially important things that users might not always do due to the inconvenience of doing it (i.e., running a number of database queries), and the low probability of it being an issue. These include checking the performance of the assay control compound and any repeated compounds. We have had examples whereby the control compound had been tested and found significantly different in activity to the historical performance, Kernel highlighted this and repeats were requested as a result. This is a good example of the benefit of the virtual assistant concept as there is significant time saving for the user, potential errors can be quickly flagged, and consistent behavior can be applied across projects.
Going beyond just showing the data and checking assay performance, we wanted to highlight which compounds are the most interesting from that run of the assay, which may not always be the ones that are the most potent. These should be interesting to the recipient of the email but may also show compounds that we can apply further computational approaches to (e.g., de novo compound design, described in more detail later in the manuscript), or they could be automatically submitted to downstream assays. Kernel currently tries to identify such compounds in two ways. One is to use efficiency metrics such as the product of the measured activity and the predicted human microsomal unbound intrinsic clearance (hCLint,u) which can be thought of as a measure of specific binding similar to a lipophilic ligand efficiency (LLE).3 The other is to use a ligand based QSAR model to compare predicted values to the measured ones. These sorts of machine learning models are very good at interpolation but much less so at extrapolation; therefore, predicted values provide a good baseline as to what might be expected to occur. Those compounds that show significant difference to the predicted values (typically > ∼7-fold) may have something interesting about them which is worth exploring further or may be an error in the assay and warrant repeat. Those falling within prediction, although they may be potent with good properties, are likely to fall within the existing SAR and may not be of significant interest. Details of the performance of the model are included in the email using statistics that are easily interpreted (e.g., median absolute error), along with a link to a plot of predicted vs measured pIC50 with each point able to be hovered over to reveal the compound structure so outliers can be easily visualized. Future work here will include automated binding mode prediction which could provide insight into QSAR model disconnects through the generation of new contacts with the receptor.
Finally, the identifiers of the newly tested molecules are included as a list which can be pasted directly into our other desktop tools for further custom analysis.
Alerting that an assay has been run, summarizing that data, and providing basic analysis that could potentially be coupled to pretty much every assay on a project is easily implemented and can save people time. Beyond that would be the ability to trigger additional computational approaches and deliver the results directly to those who would be interested. One such POC example we have implemented is the de novo design of derivatives of the newly tested molecules from the workhorse assay.
For active molecules identified by Kernel, we apply numerous MMP transformations to create novel progeny (e.g., atom perturbation or growing fragments at CH, NH, or OH, typically 50–150k per single molecule input), all of which are single point changes from the initially tested molecule (although any de novo design method could be used here). We restrict ourselves to molecules in the MMP space as this is typically how medicinal chemists operate as it helps understand the SAR. Currently we predict potency using an automatically generated and updated ligand based QSAR model,4,5 but other potency prediction approaches would be possible.6 We also predict a number of the ADME end points7 of interest, and those molecules that are most highly ranked by different criteria are then sent back to the original submitter of the compound for review via a similar email to the assay summary above.
The idea here is that ideas from the medicinal chemist are seeded with the more exhaustive sampling capable by the computer, and their additional insight adds value to the computational prediction to overall achieve higher quality output. Going forward for projects that are enabled with a protein structure bound with a relevant ligand, we envisage adding a structure-based assessment to the designed ligands and highlight those predicted to fill the pocket more optimally to drive potency, while balancing desired (predicted) properties.
As a pilot, we have deployed Kernel to four of our ongoing internal discovery projects. The aim here was not necessarily to make significant impact to the projects as what we have is an early unoptimized prototype, but to get a handle on how this change in paradigm could potentially impact medicinal chemistry and learn from this. Whenever compound predictions are sent to a chemist, the predictions and the date are saved to enable later analysis. As of this writing, 63 compounds have been suggested to a chemist by Kernel and later made by the same chemist (frequently not as a result of the suggestion from Kernel). We also looked for compounds that were predicted by Kernel but made by a different chemist on the project. These cases most likely correspond to independent compound design (i.e., the chemist came up with the design idea on their own). In these cases, we can look at the time between Kernel’s prediction and when the compound was made independently to see what kind of speedup could be possible if the chemist had seen the prediction and selected those compounds from the much larger number sent to them (typically ∼20-times more compounds than those made). On average, Kernel predictions were about 35 days (range of 4–72 days) ahead of the chemist’s. This represents the possibility of accelerating the progress of a discover project which could be a significant savings in both time and money.
Combining the two described scenarios with other computational tools and automated synthesis platforms at Lilly, in 2018 we were able to achieve a landmark moment of a fully automated DMTA cycle (Figure 3). On a test project, Kernel was able to identify that newly tested compounds had appeared in the database; from those found active, derivatives were designed as described above. The activity and properties of those were then predicted using ligand-based QSAR models. Those that met certain activity and property criteria, as well as being a certain fingerprint distance from the existing SAR, were then run through an internally developed automated retrosynthesis tool (which can be found as part of a larger open source release of internally developed cheminformatics methods: https://github.com/EliLillyCo/LillyMol).8 Molecules that met the criteria and could be made in one step using available reagents were then submitted for synthesis at our automated synthesis lab and made via the proposed route.9 Synthesized examples were then submitted for test completing a full DMTA cycle in the absence of any human intellectual input in 2018 for the first time at Lilly. Our implementation is still suboptimal, and in this case the synthesized compounds did not drive the project forward; however, as our ability to design and assess molecules computationally improves over time, this will likely only improve with future iterations. Achieving the first example of this is a significant step toward our ultimate goal of running this sort of approach alongside a medicinal chemistry team, allowing novel ideas to be generated and move the SAR in a different direction. This approach complements the automated hit finding approach we published recently.10
Figure 3.

Steps comprising fully automated DMTA cycle.
Others have also moved to create automated medicinal chemistry systems, such as the work from Stevan Djuric and his team at AbbVie shared in publication and oral presentation,11 as well as other microfluidic systems.12 These systems typically have the users define the seeds for expansion and the synthetic routes up front (some of which may be limited to being compatible with flow technologies) and may then iterate on compound designs. The system we define by comparison can choose any seed for expansion and can perform any chemistry that has been extracted from the historical electronic lab notebook or patents, and performed in batch mode, which is why we believe that there is significant potential here. As more automated synthetic chemistry systems appear,13 approaches such as this should become all the more relevant.
We believe that this virtual assistant concept has significant potential in changing the way the users receive computational results and analysis. Presenting results directly to users, at the right time, in an easily consumable format should increase uptake of the results and increase the effectiveness of those users. Beyond potency or ADME surrogate assays, examples of other assays we could apply this approach to could include compound stability assay, which for us is applied to all new submissions, and those found unstable could be highlighted to the original submitter. When new protein–ligand X-ray crystal structures are generated, the structure could be compared to what is known, and if novel the existing SAR could be docked to that structure and compared to proposed binding modes in other X-ray structures. The current interface via email will likely evolve too; we could imagine generating some sort of web interface providing a dashboard of the information, rather than the text heavy email. We believe there is lots of potential for novel automated approaches within this paradigm.
Overall, we describe a new concept of how to deliver information and results of computational approaches to scientists within a medicinal chemistry environment. The approach is generally applicable to most assays, the key being understanding what information the user would most like to see once alerted the new data is available. Tracking these assays can also then provide a trigger for subsequent more complex computational approaches and possibly automated downstream assay submissions. We describe our prototype summary of the results from a project workhorse biochemical assay, followed by de novo design of derivatives as an early POC. The concept is to automate the approaches and deliver the results directly to those that would want to see them. Finally, we highlight that we have achieved the first example at Lilly of a fully automated DMTA cycle and that this, along with other automated approaches, has the potential to complement and enhance medicinal chemistry at a pharmaceutical company such as Lilly. The POC we describe here has been well received internally (gathered through discussion with some users), and we are currently in the process of developing more optimized systems which are scalable across projects at Lilly, as well as coupling these systems to more assays and developing new approaches that may work well with the concept. We hope that others will be inspired by the concept we have shared to create similar systems within their own organizations.
Acknowledgments
The authors thank Christos Nicolaou, Jibo Wang, and others within the Computational Chemistry and Cheminformatics group at Lilly for generation of many of the tools we have used, Prashant Desai and his team for generation of ADME predictive models, James Lumley, Tom Wilkin, and their teams for development of internal KNIME infrastructure, Darryl Hilliard and others at the Automated Synthesis and Purification Laboratories at Lilly, Adam Sanderson and Andrew Williams for initial feedback on Kernel emails, Nelsen Lentz for discussion of IP issues, and all recipients of Kernel emails who have provided feedback.
Glossary
Abbreviations
- DMTA
Design-Make-Test-Analyze
- hCLint,u
human microsomal unbound intrinsic clearance
- HTML
Hypertext Markup Language
- IP
intellectual property
- LLE
lipophilic ligand efficiency
- MMP
Matched Molecular Pair
- POC
proof of concept
Author Contributions
All authors have given approval to the final version of the manuscript and contributed to the text and figures.
The authors declare the following competing financial interest(s): L.R.V. and M.P.B. are employees and stockholders of Eli Lilly and Company.
References
- Andersson S.; Armstrong A.; Björe A.; Bowker S.; Chapman S.; Davies R.; Donald C.; Egner B.; Elebring T.; Holmqvist S.; Inghardt T.; Johannesson P.; Johansson M.; Johnstone C.; Kemmitt P.; Kihlberg J.; Korsgren P.; Lemurell M.; Moore J.; Pettersson J. A.; Pointon H.; Pontén F.; Schofield P.; Selmi N.; Whittamore P. Making medicinal chemistry more effective—application of Lean Sigma to improve processes, speed and quality. Drug Discovery Today 2009, 14 (11), 598–604. 10.1016/j.drudis.2009.03.005. [DOI] [PubMed] [Google Scholar]
- KNIME; KNIME AG: Zurich, Switzerland, https://www.knime.com/. [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Erickson J.; Wang J.; Webster Y.; Mader M.; Higgs R.; Watson I. Kinase Inhibitor Data Modeling and de Novo Inhibitor Design with Fragment Approaches. J. Med. Chem. 2009, 52, 6456–6466. 10.1021/jm901147e. [DOI] [PubMed] [Google Scholar]
- Baumgartner M. P.; Evans D. A. Lessons learned in induced fit docking and metadynamics in the Drug Design Data Resource Grand Challenge 2. J. Comput.-Aided Mol. Des. 2018, 32, 45–58. 10.1007/s10822-017-0081-y. [DOI] [PubMed] [Google Scholar]
- Gaieb Z.; Liu S.; Gathiaka S.; Chiu M.; Yang H.; Shao C.; Feher V. A.; Walters W. P.; Kuhn B.; Rudolph M. G.; Burley S. K.; Gilson M. K.; Amaro R. E. D3R Grand Challenge 2: blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput.-Aided Mol. Des. 2018, 32 (1), 1–20. 10.1007/s10822-017-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P. V.; Sawada G. A.; Watson I. A.; Raub T. J. Integration of in Silico and in Vitro Tools for Scaffold Optimization during Drug Discovery: Predicting P-Glycoprotein Efflux. Mol. Pharmaceutics 2013, 10, 1249–1261. 10.1021/mp300555n. [DOI] [PubMed] [Google Scholar]
- Watson I. A.; Wang J.; Nicolaou C. A. A retrosynthetic analysis algorithm implementation. J. Cheminform. 2019, 11, 1. 10.1186/s13321-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey A. G.; Masquelin T.; Hemmerle H. A remote-controlled adaptive medchem lab: an innovative approach to enable drug discovery in the 21st Century. Drug Discovery Today 2013, 18 (17–18), 795–802. 10.1016/j.drudis.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Nicolaou C. A.; Humblet C.; Hu H.; Martin E. M.; Dorsey F. C.; Castle T. M.; Burton K. I.; Hu H.; Hendle J.; Hickey M. J.; Duerksen J.; Wang J.; Erickson J. A. Idea2Data: Toward a New Paradigm for Drug Discovery. ACS Med. Chem. Lett. 2019, 10 (3), 278–286. 10.1021/acsmedchemlett.8b00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranczak A.; Tu N. P.; Marjanovic J.; Searle P. A.; Vasudevan A.; Djuric S. W. Integrated Platform for Expedited Synthesis–Purification–Testing of Small Molecule Libraries. ACS Med. Chem. Lett. 2017, 8, 461–465. 10.1021/acsmedchemlett.7b00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G. Automating drug discovery. Nat. Rev. Drug Discovery 2018, 17, 97–113. 10.1038/nrd.2017.232. [DOI] [PubMed] [Google Scholar]
- Steiner S.; Wolf J.; Glatzel S.; Andreou A.; Granda J. M.; Keenan G.; Hinkley T.; Aragon-Camarasa G.; Kitson P. J.; Angelone D.; Cronin L.. Organic synthesis in a modular robotic system driven by a chemical programming language Science 2019, 363, eaav2211. 10.1126/science.aav2211. [DOI] [PubMed] [Google Scholar]