

Abstract

Protein arginine methyltransferase 5 (PRMT5) is known to symmetrically dimethylate numerous cytosolic and nuclear proteins that are involved in a variety of cellular processes. Recent findings have revealed its potential as a cancer therapeutic target. PRMT5 possesses a cysteine (C449) in the active site, unique to PRMT5. Therefore, covalent PRMT5 inhibition is an attractive chemical approach. Herein, we report an exciting discovery of a series of novel hemiaminals that under physiological conditions can be converted to aldehydes and react with C449 to form covalent adducts, which presumably undergo an unprecedented elimination to form the thiol-vinyl ethers, as indicated by electron density in the co-crystal structure of the PRMT5/MEP50 complex.
Keywords: PRMT5, covalent inhibitor
Protein arginine methylation, catalyzed by a family of arginine methyltransferases, has emerged as one of the most common post-translational modifications (PTMs). As the major type II methyltransferase in mammals, protein arginine methyltransferase 5 (PRMT5) forms a hetero-octamer with MEP50,1 catalyzes the formation of symmetrical dimethylarginines (sDMA) on histones and non-histone substrates,2,3 and plays important roles in various cellular processes, including gene expression and RNA splicing.4,5 PRMT5 has been shown to be upregulated in many types of cancers.6−8 Loss of methylthioadenosine phosphorylase (MTAP) confers a selective dependence on PRMT5 and its binding partner, MEP50 (WDR77).9−11 MTAP is frequently lost due to its proximity to the commonly deleted tumor suppressor gene, CDKN2A. Together, these findings reveal PRMT5 as a potential therapeutic target for cancer. Indeed, PRMT5 selective inhibitors have recently been discovered and have demonstrated potent antitumor activity in preclinical models.12 Two molecules, GSK3326595, a substrate competitive inhibitor, and JNJ64619178, a SAM (S-adenosyl-l-methionine) mimetic/competitive inhibitor, have entered clinical trials for multiple cancer types.13,14 Most recently, two more small molecule PRMT5 inhibitors, PF-0693999915 and PRT54315 (structures not disclosed), entered clinical trials.
PRMT5 has been a drug target for many pharmaceutical and biotechnology companies, as well as academic research institutions, due to its critical function in cancer.16−22 Before the disclosure of LLY-283 and JNJ64619178,23−25 a major concern among scientists in the field was whether it was possible to obtain SAM mimetic nucleoside analogues with selectivity versus different PRMTs. Neither of the SAM analogues, SAH (S-adenosyl-l-homocysteine) or sinefungin, is a selective methyltransferase inhibitor. However, 5′-methylthioadenosine (MTA) has been shown to be a potent, selective PRMT5 inhibitor, thus providing an excellent starting point for a potential approach based on SAM mimetics.
Interestingly, PRMT5 possesses a cysteine (C449) in the SAM binding site, a unique feature for PRMT5 that is not present in the other PRMTs (which have a serine at the same position). The co-crystal structure of MTA in the PRMT5/MEP50 complex [Protein Data Bank (PDB) entry 5FA5] has revealed a small space that might accommodate a covalent warhead. The distances from C449 to atoms N6 and N7 of the SAM adenine ring were found to be 3.6 and 3.9 Å, respectively. Therefore, a covalent approach may offer potent and selective PRMT5 inhibitor analogues to SAM. On the other hand, C449 is not considered catalytic, so it may not be inherently reactive, which potentially makes this covalent approach challenging. However, there have been great successes in the discovery of covalent kinase inhibitors,26 such as the EGFR inhibitor Afatinib and the BTK inhibitor Ibrutinib. Also, a very exciting breakthrough was recently reported as the discovery of covalent inhibitors of KRAS,27 a target that has been considered “undruggable” for decades. All of these molecules modify the target proteins via noncatalytic cysteines.
Encouraged by these findings, we initiated a program to search for potent and selective covalent PRTM5 inhibitors. As described below, we report the discovery of a series of novel hemiaminal covalent PRMT5 inhibitors, their potency, characterization of covalent binding activity, and a co-crystal structure of the covalent adduct with the PRMT5/MEP50 complex.
A typical synthesis of hemiaminals is depicted in Scheme 1. Compound 1 was prepared following by the procedures previously described to make LLY-283.23 Treatment of acid 1 with NHMe(OMe) HCl salt and propylphosphonic anhydride (T3P) in the presence of Hünig’s base afforded Weinreb amide 2, which was then reacted with Grignard reagent to give ketone 3. Stereoselective hydride transfer reduction catalyzed by RuCl(p-cymene)[(R,R)-Ts-DPEN] provided alcohol 4 as a major diastereomer.28 Hydrolysis of acetonide 4 under acidic aqueous conditions afforded diol 5. Displacement of the pyrrolopyrimidine 6-Cl with 2,2- dimethoxyethanamine in the presence of Et3N afforded compound 6. Alternatively, the same displacement was carried out on compound 4 to provide acetal 7. Under acidic aqueous conditions, a cyclic hemiaminal ether 8 was obtained as a 1:1 mixture of diastereomers. Further hydrolysis of the methoxy group of 8 under 1 N HCl at reflux did not provide the corresponding aldehyde 10, yielding instead a hemiaminal 9. Compound 9 was finally converted to aldehyde 10 containing ∼30% 9 under basic conditions, which was confirmed by the peak in its 1H NMR spectrum at a δ of 9.61 ppm.
Scheme 1. Synthesis of PRMT5 Covalent Inhibitor Hemiaminal 9.

Reagents and conditions: (a) NHMe(OMe)·HCl, 50% T3P in EtOAc, Hünig’s base or Et3N, >90%; (b) 4-chlorophenylmagnesium bromide, THF, 0 °C, 80%; (c) 2 M aqueous sodium formate, RuCl(p-cymene)[(R,R)-Ts-DPEN], EtOAc, 16 h, 70%; (d) TFA, H2O; (e) 2,2-dimethoxy-ethan-1-amine, Et3N, iPrOH, reflux, 2–10 h, 66–72%; (f) 1 N aqueous HCl, reflux, 100%; (g) Amberlite basic.
Additionally, six-membered ring hemiaminals (such as compound 11 in Figure 1) and ketones (such as compound 12 in Figure 1) were prepared (see the synthetic routes in the Supporting Information). Like compound 9, 11 remains in its closed form as an HCl salt. On the other hand, ketone 12 only partially cyclized under acidic conditions, remaining as the keto form under neutral conditions.
Figure 1.

Structures of compounds 11 and 12.
Both hemiaminal 9 and aldehyde 10 were evaluated in a PRMT5/MEP50 biochemical assay to show comparable IC50 values of 11 nM (n = 3) and 19.5 nM (n = 1), respectively (see experimental details in the Supporting Information). Under the assay conditions (pH 8.0), 9 is likely converted to 10, which inhibits PRMT5 via formation of a covalent bond with C449. It is difficult to exclude the possibility that 9 might bind PRMT5 equally well to inhibit its enzymatic activities.
In addition, hemiaminal 9 was characterized for its covalent binding via jump dilution and time-dependent inactivation. The off rate was evaluated by the jump-dilution method comparing the wild type and C449S mutant of PRMT5. Hemiaminal 9 displayed an extremely slow off rate (koff) of 0.0022 ± 0.0002 min–1, which results in a residence time half-life (t1/2) of 315 ± 42 min. This is in contrast with the C449S mutant, which exhibited a faster koff of 0.045 ± 0.003 min–1, corresponding to a t1/2 of 15 ± 1 min. The significant koff shift clearly indicated that the covalent modification can happen only in the presence of C449. The time-dependent inactivation assay was carried out to estimate the apparent second-order rate constant (kinact/KI), which served as an index of irreversible inhibition potency. The apparent inactivation rate constant (kinact) and the initial inhibition constant (KI) were readily determined by a representative progress curve (Figure 2, top).29 Hemiaminal 9 inhibited the PRMT5/MEP50 complex [kinact/KI = (1.2 ± 0.2) × 105 M–1 min–1] very rapidly with a kinact of 0.068 ± 0.004 min–1 and a good binding affinity (KI = 55 ± 11 nM). The inhibitory efficiency was dependent on both the compound binding affinity and the nucleophilicity of the target cysteine (C449).
Figure 2.

Progress curve of inactivation of the PRMT5/MEP50 complex by hemiaminal 9.
The nucleophilicity of C449 is pKa-dependent. Hence, we determined the pKa of C449 in the PRMT5 protein without MEP50 using iodoacetamide, an alkylation reagent known to rapidly modify deprotonated cysteine residues over the corresponding thiols. The PRMT5 protein alone was preincubated in buffers with pH values ranging from 3.0 to 9.4 with and without iodoacetamide. The pH-dependent inactivation of PRMT5 was characterized by comparing the methyltransferase activity in a flash-plate assay. As depicted in Figure 3, the observed pKa value of 7.4 ± 0.1 indicated that C449 partially exists as a thiolate ion under physiological or assay conditions.
Figure 3.

pH-dependent inactivation of PRMT5.
Aldehyde 10 was co-crystallized with the PRMT5/MEP50 complex to provide an X-ray co-crystal structure (PDB entry 6K1S) at 2.6 Å resolution (see the experimental details in the Supporting Information). As expected, on the basis of the electron density map, C449 is very close to the molecule, a distance within a bond length. To our surprise, the electron density does not support the existence of an OH group resulting from addition of -SH to the aldehyde (see Figure 4A). Instead, the S–C–C–N dihedral angle is 175.7°, with S–C–C and C–C–N bond angles of 116.0° and 125.9°, respectively (see Figure 4B), indicating a plausible trans double bond between the S and N atoms. As illustrated in Scheme 2, under physiological pH, an unprecedented and unexpected elimination of a H2O molecule appears to have taken place. This elimination could be driven by steric hindrance and inflexibility of the α-helix where C449 resides.
Figure 4.

Co-crystal structure of compound 10 with the PRMT5/MEP50 complex. (A) Compound 10 (stick view with an electron density map) in the SAM binding pocket of the PRMT5/MEP50 complex (cartoon view) showing the covalent bond formed between compound 10 and C449. (B) Polar contact of compound 10. (C) Overlay of co-crystal structures of EPZ015666, MTA, LLY-283, and compound 10 with the PRMT5/MEP50 complex, comparing the different conformations of the F327 side chain. The structures of EPZ015666 and LLY-283 are as shown.
Scheme 2. Proposed Mechanism of Vinyl-Thio Ether Formation.

Compound 10 forms a polar contact similar to that of MTA (PDB entry 5FA5) and LLY-283 (PDB entry 6CKC), with H-bond interactions between the aminopyrimidine ring and D419 and between the ribose hydroxyl groups and E392 and Y324, as shown in Figure 4B. The crystal structure also reveals the high flexibility of F327. As shown in Figure 4C, in the MTA structure, F327 is placed close to the substrate Arg but rotates out in 6CKC to accommodate the phenyl ring of LLY-283. In the case of compound 10, F327 moves farther from the SAM binding pocket, a movement induced by the bulkier p-Cl phenyl ring at the 5′ position of the ribose. F327 acts a crucial residue in the active site of PRMT5. It sits right between the cofactor and peptide substrate, thus acting as a gatekeeper, its side chain pivoting out of the way when needed. This flexibility has been also observed in the co-crystal structure with substrate competitive inhibitors such as EPZ015666 [PDB entry 4X61 (see Figure 4C)]. The F327 movement in PRMT5 and its significance have been recently reviewed.30
Compounds 6–12 were evaluated in a biochemical assay measuring inhibition of PRMT5/MEP50 complex-catalyzed histone methylation using an H4-based AcH4–23 peptide (FlashPlate) and in cellular assays measuring inhibition of sDMA and cell growth (see the experimental details in the Supporting Information). The potencies of each compound are summarized in Table 1. Neither compound 6 nor compound 8 was potent in any of these assays, which is consistent with their lack of chemical reactivity under assay conditions. Compound 11 showed potency against the PRMT5/MEP50 complex comparable to that of compound 9 but was less potent in cellular assays. This could be due to a more stable closed hemiaminal form or the fact that the corresponding aldehyde does not fit the active site as effectively as compound 10 does. Ketone 12 is not as potent as aldehyde 10 in either biochemical or cellular assay, perhaps because of its lower chemical reactivity versus cysteine or because of a lower affinity for the active site of the enzyme. Neither compound 11 nor compound 12 was a covalent modifier in jump-dilution experiments described above.
Table 1. Biochemical and Cellular Potency in Granta-519a.
| compound | PRMT5/MEP50 IC50 (μM) | sDMA IC50 (μM) | proliferation IC50 (μM) |
|---|---|---|---|
| I6 | 0.177 | >1 | >1 |
| 8 | 2.99 | >1 | >1 |
| 9 | 0.011 | 0.012 | 0.060 |
| 10 | 0.020 | 0.022 | 0.048 |
| 11 | 0.026 | 0.158 | 0.270 |
| 12 | 0.079 | 0.043 | 0.140 |
The values are average of at least two measurements or confirmed in an orthogonal assay with similar potency values.
Compound 9 was further profiled in cellular assays as shown in Figure 5. Figure 5A represents dose-responsive inhibition of cellular sDMA in Granta-519 cells, a mantle cell lymphoma cell line, after incubation for 3 days as determined in Western blots. Signals were quantified, normalized, and plotted to determine IC50 (see Figure 5B). Figure 5C represents time-dependent cell proliferation inhibition, in which cells died on day 10 at high concentrations (0.3 and 1 μM). Cell viabilities on day 10 were normalized to dimethyl sulfoxide and plotted to determine the proliferation IC50 as shown in Figure 5D (see details in the Supporting Information).
Figure 5.
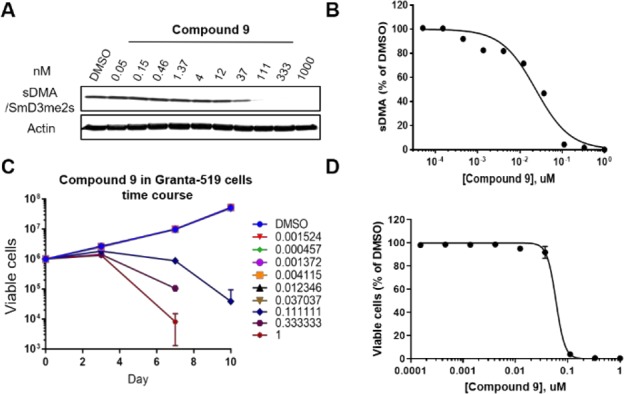
Data/curve and time course of compound 9 in cellular assays. (A) Representative image of the sDMA/SmD3me2s Western blot. (B) IC50 plot of panel A. (C) Time-dependent inhibition of cell proliferation. (D) IC50 plot of panel C on day 10.
Compound 9 was also tested against PRMT1 and PRMT4 (CARM1) in the HotSpot assay, using the PRMT5/MEP50 complex as a positive control.31 It proved to be potent versus PRMT5 in this assay, with an IC50 ot 31 nM (n = 3), a level of activity similar to that observed in our in-house FlashPlate assay. On the other hand, compound 9 was inactive against PRMT1 and PRMT4. Because compound 9 structurally resembles LLY-283, it is expected to be as selective as LLY-283 against a wider methyltransferase panel.
In conclusion, we have discovered a first-in-class PRMT5 covalent inhibitor hemiaminal 9. This compound will serve as an excellent tool molecule for studying the biological consequences of covalent inhibition of PRMT5 in vitro. Identifying covalent modifiers of methyltransferases could be as fruitful as identifying covalent kinase inhibitors, a mode of action with demonstrated success in addressing the potential for drug resistance. Other methyltransferases such as PRMT1 and EZH2 also have noncatalytic cysteine residues close to or at the SAM binding pockets, and indeed, covalent inactivation of PRMT1 with a peptide containing a reactive chloroacetamidine warhead has already been reported.32 In the case of PRMT5, evidence of covalent modification of PRMT5 at C449 is encouraging, because it shows that C449 has adequate reactivity. Most importantly, we observed an unprecedented covalent mechanism between aldehyde and cysteine, a possible elimination of a H2O molecule to form vinyl-thiol ether. This could offer a potential novel approach to identifying covalent modifiers of other important biological targets.
Acknowledgments
The authors acknowledge the chemists of Medicilon Inc. (Shanghai, China) for making some of the compounds described herein.
Biography
Hong Lin received her B.S. at Nanjing University, M.S. at Shanghai Institute of Organic Chemistry, and Ph.D. in Organic Chemistry at Brandeis University. Hong is Head of Early Discovery at Prelude Therapeutics, where she has led a discovery program that delivered three candidates within a two-year period, one of them currently in clinic. Structure-based drug discovery approach has been key to the success of this program. Prior to Prelude, Hong held multiple leadership roles in medicinal chemistry and virtual drug discovery groups within the therapeutic areas of oncology, neuroscience, and regenerative medicine during her 15 years of tenure at GlaxoSmithKline.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00074.
Biochemical and cellular assay conditions, protein purification and co-crystallization conditions, synthetic procedures, and characterization of compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y. W.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A. P.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Vishwanath S. N.; Erdjument-Bromage H.; Tempst P.; Sif S. Human SWI/SNFassociated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesiridis G. S.; Diamond E.; Van Duyne G. D. Role of pICLn in methylation of Sm proteins by PRMT5. J. Biol. Chem. 2009, 284, 21347–21359. 10.1074/jbc.M109.015578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. J.; Hoang S.; Mayo M. W.; Bekiranov S. Application of machine learning methods to histone methylation ChIP-Seq data reveals H4R3me2 globally represses gene expression. BMC Bioinf. 2010, 11, 396. 10.1186/1471-2105-11-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc R. S.; Richard S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65 (1), 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Bedford M. T. Protein arginine methyl-transferases and cancer. Nat. Rev. Cancer 2013, 13 (1), 37–50. 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Stopa N.; Krebs J. E.; Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A. Targeting protein arginine methyltransferase 5 in disease. Future Med. Chem. 2017, 9, 2081–2098. 10.4155/fmc-2017-0089. [DOI] [PubMed] [Google Scholar]
- Mavrakis K.; McDonald E. R.; Schlabach M. R.; Billy E.; Hoffman G. R.; deWeck A.; Ruddy D. A.; Venkatesan K.; McAllister G.; deBeaumont R.; Ho S.; Liu Y.; Yan-Neale Y.; Yang G. Z.; Lin F.; Yin H.; Gao H.; Kipp D. R.; Zhao S. P.; McNamara J. T.; Sprague E. R.; Cho Y. S.; Gu J.; Crawford K.; Capka V.; Hurov K.; Porter J. A.; Tallarico J.; Mickanin C.; Lees E.; Pagliarini R.; Keen N.; Schmelzle T.; Hofmann F.; Stegmeier F.; Sellers W. R.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to marked dependence on PRMT5. Science 2016, 351, 1208–1213. 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- Marjon K.; Cameron M. J.; Quang P.; Clasquin M. F.; Mandley E.; Kunii K.; Mcvay M.; Choe S.; Kernytsky A.; Gross S.; Konteatis Z.; Murtie J.; Blake M. L.; Travins J.; Dorsch M.; Biller S. A.; Marks K. M. MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell Rep. 2016, 15, 574–587. 10.1016/j.celrep.2016.03.043. [DOI] [PubMed] [Google Scholar]
- Kryukov G. V.; Wilson F. H.; Ruth J. R.; Paulk J.; Tsherniak A.; Marlow S. E.; Vazquez F.; Weir B. A.; Fitzgerald M. E.; Tanaka M.; Bielski C. M.; Scott J. M.; Dennis C.; Cowley G. S.; Boehm J. S.; Root D. E.; Golub T. R.; Clish C. B.; Bradner J. E.; Hahn W. C.; Garraway L. A. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E.; Kuplast K. G.; Majer C. R.; Boriack-Sjodin P. A.; Wigle T. J.; Johnston L. D.; Rioux N.; Munchhof M. J.; Jin L.; Jacques S. L.; West K. A.; Lin-garaj T.; Stickland K.; Ribich S. A.; Raimondi A.; Scott M. P.; Waters N. J.; Pollock R. M.; Smith J. J.; Barbash O.; Pappalardi M.; Ho T. F.; Nurse K.; Oza K. P.; Gallagher K. T.; Kruger R.; Moyer M. P.; Copeland R. A.; Chesworth R.; Duncan K. W. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11 (6), 432–7. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- NCT03614728 for GSK3326595 (https://clinicaltrials.gov/ct2/show/NCT03614728). NCT02783300 for GSK3326595 (https://clinicaltrials.gov/ct2/show/NCT02783300).
- NCT03573310 for JNJ64619178 (https://clinicaltrials.gov/ct2/show/NCT03573310).
- NCT03854227 for PF-06939999 (https://www.clinicaltrials.gov/ct2/show/NCT03854227). NCT03886831 for PRT543 (https://clinicaltrials.gov/ct2/show/NCT03886831).
- Luo M. K. Inhibitors of protein methyltransferases as chemical tools. Epigenomics 2015, 7, 1327–133. 10.2217/epi.15.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. U.; Konze K. D.; Jin J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Qian K.; Ho M. C.; Zheng Y. G. Small molecule inhibitors of protein arginin methyltransferases. Expert Opin. Invest. Drugs 2016, 25, 335–358. 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. U.; Jin J. Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol. 2017, 39, 100–108. 10.1016/j.cbpa.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. U.; Martini M. L.; Jin J. Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 2018, 118, 989–1068. 10.1021/acs.chemrev.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Hu W.; Yuan Y. Protein Arginine Methyltransferase 5 (PRMT5) as an Anti-Cancer Target and Its Inhibitor Discovery. J. Med. Chem. 2018, 61 (21), 9429–9441. 10.1021/acs.jmedchem.8b00598. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Li H. Novel Drugs Targeting the Epigenome. Curr. Pharmacol Rep. 2017, 3 (5), 268–285. 10.1007/s40495-017-0100-7. [DOI] [Google Scholar]
- Bonday Z. Q.; Cortez G. S.; Grogan M. J.; Anto-nysamy S.; Weichert K.; Bocchinfuso W. P.; Li F.; Kenne-dy S.; Li B.; Mader M. M.; Arrowsmith C. H.; Brown P. J.; Eram M. S.; Szewczyk M. M.; Barsyte-Lovejoy D.; Vedadi M.; Guccione E.; Campbell R. M. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9 (7), 612–617. 10.1021/acsmedchemlett.8b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehmer D.; Wu T.; Mannens G.; Beke L.; Vinken P.; Gaffney D.; Sun W.; Pande V.; Thuring J.-W.; Millar H.; Poggesi I.; Somers I.; Boeckx A.; Parade M.; van Heerde E.; Nys T.; Yanovich C.; Herkert B.; Verhulst T.; Jardin M. D.; Meerpoel L.; Moy C.; Diels G.; Viellevoye M.; Schepens W.; Poncelet A.; Linders J. T.; Lawson E. C.; Edwards J. P.; Chetty D.; Laquerre S.; Lorenzi M. V.. A novel PRMT5 inhibitor with potent in vitro and in vivo activity in preclinical lung cancer models [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2017; April 1–5, 2017, Washington, DC; American Association of Cancer Research: Philadelphia, 2017; Vol. 77 ( (Suppl. 13), ), Abstract DDT02-04. 10.1158/1538-7445.AM2017-DDT02-04 [DOI] [Google Scholar]
- Wu T.; Millar H.; Gaffney D.; Beke L.; Mannens G.; Vinken P.; Sommers I.; Thuring J.-W.; Sun W.; Moy C.; Pande V.; Zhou J.; Haddish-Berhane N.; Salvati M.; Laquerre S.; Lorenzi M. V.; Brehmer D.. JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivoactivity, demonstrated in several lung cancer models [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2018; April 14–18, 2018, Chicago; American Association of Cancer Research: Philadelphia, 2018; Vol. 78 ( (Suppl. 13), ), Abstract 4859. [Google Scholar]
- Bauer R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20, 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Janes M. R.; Li L.-S.; Hansen R.; Peters U.; Kessler L. V.; Chen Y.; Kucharski J. M.; Feng J.; Ely T.; Chen J. H.; Firdaus S. J.; Babbar A.; Ren P.; Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery 2016, 6, 316–329. 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- Tatlock J. H.; McAlpine I. J.; Tran-Dube M. B.; Wyeth M. J.; Rui E. Y.; Kumpf R. A.; McTigue M. A.. Substituted Nucleoside Derivatives Useful as Anticancer Agents. U.S. Patent 20160244475 A1, 2016.
- Tian W. X.; Tsou C. L. Determination of the rate constant of enzyme modification by measuring the substrate reaction in the presence of the modifier. Biochemistry 1982, 21 (5), 1028–32. 10.1021/bi00534a031. [DOI] [PubMed] [Google Scholar]
- Lin H.; Luengo J. I. Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorg. Med. Chem. Lett. 2019, 29 (11), 1264–1269. 10.1016/j.bmcl.2019.03.042. [DOI] [PubMed] [Google Scholar]
- PRMT1 and PRMT4 (CARM1). HotSpot assays at Reaction Biology Corp., Malvern, PA. [Google Scholar]
- Obianyo O.; Causey C. P.; Osborne T. C.; Jones J. E.; Lee Y. H.; Stallcup M. R.; Thompson P. R. A chloroacetamidine-based inactivator of protein arginine methyltransferase 1: design, synthesis, and in vitro and in vivo evaluation. ChemBioChem 2010, 11 (9), 1219–23. 10.1002/cbic.201000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.