

Abstract
Diabetes mellitus has been recognised as one of the four major non-communicable diseases that demands urgent attention from all key shareholders globally in an effort to address its prevalence and associated complications. It is considered as a top 10 cause of death globally, killing about 1.6 million people worldwide and is seen as the third highest risk factor for worldwide premature mortality due to hyperglycaemia and hyperglycaemic-induced oxidative stress and inflammation. There is a strong link between hyperglycaemia, hyperglycaemic-induced oxidative stress, inflammation and the development and progression of type 2 diabetes mellitus. Various reports have shown that chronic low-grade inflammation is associated with the risk of developing type 2 diabetes and that sub-clinical inflammation contributes to insulin resistance and is linked to the characteristics of metabolic syndrome which include hyperglycaemia. Oxidative stress stimulates the generation of inflammatory mediators and inflammation in turn enhances the production of reactive oxygen species. This interaction between diabetes, oxidative stress and inflammation is the primary motivation for the compilation of this review. Based on previous studies, the review examines the interaction between diabetes, oxidative stress and inflammation, factors promoting prevalence of diabetes mellitus, mechanisms involved in hyperglycaemia-induced oxidative stress with particular focus on type 2 diabetes and selected diabetic complications.
Keywords: Reactive oxygen species, progression, chronic inflammation, interplay, hyperglycaemia
Introduction
Scientists view diabetes mellitus from different angles. Some see it as an evolving disease with alterations in patterns as observed in both type 1 and type 2 diabetes with a broad variation in global incidence rates [1]. Badawi et al [2] view it as a significant worldwide health problem. Rehman & Akash [3] describe it as a complex and multifactorial metabolic syndrome with characteristic abnormal metabolism in carbohydrates, fats and proteins leading to hyperglycaemia and hyperlipidaemia. Apart from it been an evolving disease, Navarro and Mora [4] is more definitive on the kind of evolution that diabetes is undergoing. Specifically, the authors reported that it is evolving from metabolic disorder to an inflammatory condition. Their argument is based on the hypothesis proposed by Pickup & Crook [5] which suggest that long-term innate immune system activation, resulting in chronic inflammation brings about a disease instead of repair, potentially resulting in the development of type 2 diabetes. Interestingly, research reports have shown that low-grade inflammation is associated with the risk of developing type 2 diabetes and that sub-clinical inflammation contributes to insulin resistance and is linked to the characteristics of metabolic syndrome which include hyperglycaemia [6-9]. Oxidative stress has been reported as a known pathway in the pathogenesis of diabetic complications [10]. Hyperglycaemic-induced oxidative stress is believed to increase the levels of pro-inflammatory proteins with infiltrated macrophages secreting inflammatory cytokines which leads to local and systemic inflammation [11]. Increased secretion of tumour necrosis factor alpha (TNF-alpha) has been observed to be linked to obesity-related insulin resistance and obesity is a risk factor for the development of type 2 diabetes [12-14]. This interaction between diabetes, oxidative stress and inflammation is the motivation for this review. Therefore, this review looks at the interaction between diabetes, oxidative stress and inflammation with particular focus on type 2 diabetes and diabetic complications. The relationship between diabetes, oxidative stress and inflammation is shown in Figures 1, 2, 3, 4 and 5.
Figure 1.
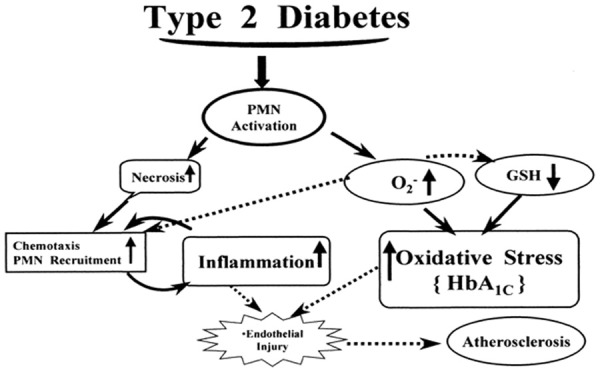
Relationship between type 2 diabetes, oxidative stress and inflammation (www.canacopegdl.com, accessed on 14 December 2018).
Figure 2.
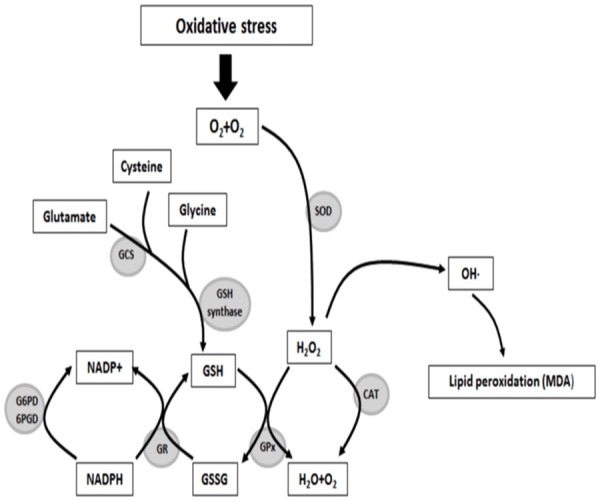
The role of oxidative stress in tissue injury/toxicity [144].
Figure 3.
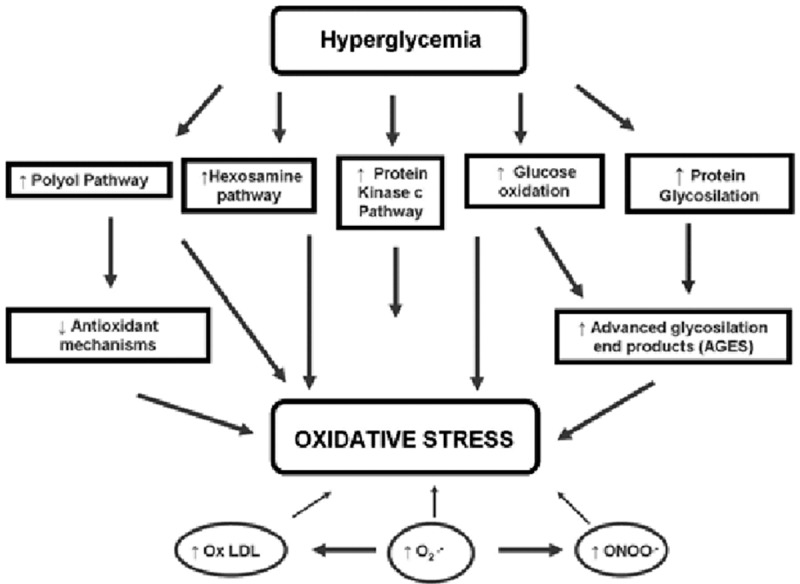
Hyperglycaemia-induced oxidative stress [145].
Figure 4.
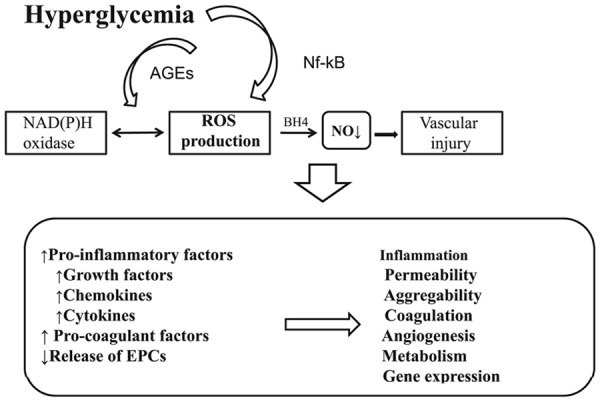
Hyperglycaemia-induced complications [146].
Figure 5.
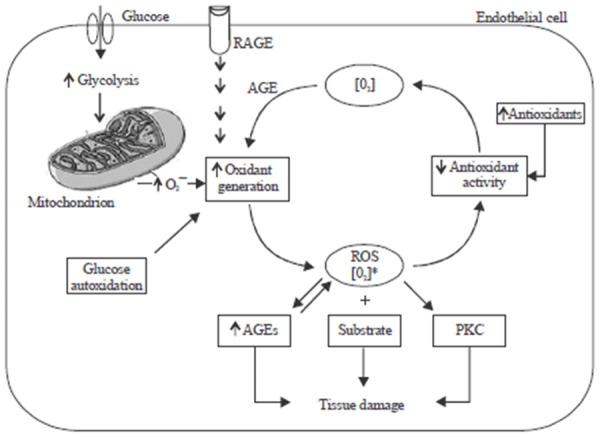
Oxidant generation, antioxidant activity, oxidative stress and damage in diabetes melltius [147].
Global perspective of diabetes
Diabetes mellitus has been recognised by the World Health Organization (WHO) as one of the four major non-communicable diseases that should attract urgent attention from all key shareholders; seen as the third highest risk factor for worldwide premature mortality due to hyperglycaemia [15,16]. In 2017, the number of diabetics worldwide by region is reported as follows: Western Pacific (159 million), Southeast Asia (82 million), Europe (58 million), North America and Caribbean (46 million), Middle East and North Africa (39 million), South and Central America (26 Million) and Africa (16 million) [17]. In Africa, the number of adults with impaired glucose tolerance (IGT) is expected to increase by 153% by 2045 and African region has the highest percentage of people with undiagnosed diabetes (that is about 70% of people with diabetes are unaware that they have diabetes) and 312000 were estimated to have died of diabetes in 2017 and about 73% deaths due to diabetes were people under the age of 60 years [17].
Data from the International Diabetes Federation (IDF) suggests that about 415 million people live with diabetes in the world with a prevalence rate of 8.8%. Of this, 75% live in low and middle income countries. It is estimated that by 2040, about 642 million people will be diabetic with type 2 diabetes mellitus as the major type of diabetes [18]. The increase in both type 1 and type 2 diabetes seem to cut across age, race and gender. In Finland, for example with the highest global incidence of type 1 diabetes and China with the lowest incidence of type 1 diabetes, it has increased by 2-5% per year. Type 2 diabetes is also reported to be increasing; sadly the increased incidence is occurring at a younger age in both adolescence and children. It is reported that in the USA, about one third of persons diagnosed with type 2 diabetes are adolescents [1]. The prevalence of type 2 diabetes in women has been reported to be on the increase globally which is more common in low-income countries with obesity and aging seen as the driving force [19]. The prevalence of gestational diabetes mellitus (GDM), a common pregnant complications has been observed to increase by over 30% in both developed and developing countries and has been linked to increased risk of developing cardio-metabolic disorders in women and their children [20,21].
Economic challenge of diabetes
Diabetic mellitus is a challenging health problem that poses serious economic burden to individuals and nations. In high income countries, reports have shown the increasing prevalence in low income countries [22,23]. Consequent to increased disability arising from complications that are associated with diabetes, the economy of nations is negatively affected. Diabetic patients’ ability to render maximum services to their respective organisations is also affected leading to reduced productivity. It has been reported in the USA that diabetic patients are 15 times more prone to lower limp amputation than non-diabetics [22,24]. Report indicates that global cost of diabetes is far above US$ 174 billion and for every $5, a $1 is spend on a diabetic patient [25]. In Sub Saharan Africa, over US$ 3.4 billion is spent annually while the USA spends about 50% of the total annual global expenditure in the treatment and management of diabetes mellitus. Lowest expenditure figures are reported in poorer developing countries where the treatment and management of diabetes mellitus is negatively affected [26].
Diabetes mellitus and risk factors
Type 2 diabetes is linked to impaired glucose tolerance due to insulin resistance; concomitant islet beta-cells injury may lead to insulin deficiency which impact on utilization of glucose by skeletal muscle, liver and adipose tissues [27]. Therefore, impaired glucose tolerance coupled with other factors such as genetic disposition, environmental factor, diet, physical inactivity and obesity do significantly contribute to the progression of insulin resistance and to the development of type 2 diabetes [2,28,29].
Obesity
The increase in the prevalence of obesity has led to increase in the incidence and prevalence of type 2 diabetes and epidemiological studies have reported on a strong relationship between obesity and risk of developing type 2 diabetes [29]. According to a report credited to Mokdad et al [30], for every kilogram increase in body weight, the risk of developing type 2 diabetes is believe to increase by 9%.
Reports showed that obesity is a principal cause of insulin resistance combined with metabolic dysregulation as well as hypertension and abnormal lipid metabolism predisposing individuals to the development of type 2 diabetes mellitus [31,2]. In obese individuals, insulin resistance has been shown to be linked to increased release of adipocyte-derived bioactive metabolites like lipids, free fatty acids, monocyte chemoattractant protein-1 and pro-inflammatory cytokines [31]. Research indicate that exposure of muscle cells to fatty acids impair insulin-mediated glucose uptake and consequently contribute to insulin resistance and insulin resistance in turn contribute to the development of type 2 diabetes [32,33]. To justify the possible contributory role of obesity to the development of insulin resistance and in turn in the development of type 2 diabetes, a study on young insulin-resistant lean children of type 2 diabetic individuals and insulin-sensitive controls of similar body mass index, revealed that in lean people, systemic inflammation may not play an important role in the development of insulin resistance [34]. However, human and animal models showed that tumour necrosis factor-alpha gene expression is up-regulated in adipose tissues, therefore linking it to pro-inflammatory cytokines released from adipose tissues to insulin resistance in type 2 diabetes mellitus [11,35].
Lifestyle
Food intake has been strongly related to obesity in terms of quantity of food, composition and quality [36]. High intake of red meat, sweets and fried food has been reported to contribute to increased risk for the development of insulin resistance and type 2 diabetes while intake of vegetables and fruits has been linked to reduced incidence of type 2 diabetes [37]. In 500 school children who consumed carbonated drinks for a selected period of time, the result indicated that serving additional carbonated drinks increased the incidence of obesity [38] and there is a report of a positive association between intake of sugars and the development of type 2 diabetes [39]. A study in Japan linked the consumption of white rice to increased risk of developing type 2 diabetes supporting the evidence of the association between diet and the risk of developing type 2 diabetes [40].
Physical inactivity is an important mortality risk factor in the development of type 2 diabetes with an increase of 20-30% of death compared with individuals who participate in at least 30 minutes of daily exercise [41]. Physical activity is reported to be linked to a significant decline in the risk of developing type 2 diabetes [42]. This is because physical activity is believed to increase insulin sensitivity and is reported to be more beneficial in preventing the progression of type 2 diabetes during the initial stage prior to the application of insulin therapy [43]. During physical activity, contracting skeletal muscle enhances glucose uptake into the cells; physical activity also increases blood flow in the muscle and promotes glucose transport into the muscle cells and physical activity has been reported to decrease intra-abdominal fat-a major risk factor for insulin resistance and in turn for the development of type 2 diabetes [44]. Report shows that there is a 20% increase risk of developing type 2 diabetes for daily increased watching of television for 2 hours [45].
Type 2 diabetes mellitus and oxidative stress
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the terms collectively used to describe free radicals and other non-radical reactive derivatives known as oxidants. Biological free radicals are highly unstable molecules which are products of normal cellular metabolism. They have electrons which can react with various organic substrates such as lipids, proteins and deoxyribonucleic acid (DNA). Free radicals are well recognized for playing a dual role as both deleterious and beneficial species, since they can be either harmful or beneficial to living systems [46]. At low or moderate levels free radicals (ROS and RNS) exert beneficial effects such as defence against infectious agents, induction of a mitogenic response and the maturation process of cellular structures [47]. ROS include superoxide anion (O2·-), hydroxyl (·OH), hydrogenperoxide (H2O2) and hypochlorous acid (HOCl) while RNS include nitric oxide (·NO), nitrogen dioxide (NO2·-) and peroxynitrite (OONO-). High concentrations of free radicals on the other hand result in deleterious processes that can damage cell structures due to oxidative stress [48].
Numerous experimental evidences have highlighted a direct link between oxidative stress and diabetes through the measurement of oxidative stress biomarkers in both diabetic patient and rodents. A hyperglycaemic state can lead to an increase in the levels of oxidative stress-induced DNA damage markers such as 8-hydroxy-2’-deoxyguanosine (8-OHdG) and 8-oxo-7, 8-dihydro-2’-deoxyguanosine; lipid-peroxidation products measured as thiobarbituric acid-reactive substances (TBARS); protein oxidation products such as nitrotyrosine and carbonyl levels and also lower the activity of antioxidant enzymes. Cell culture studies using pancreatic beta cells, aortic smooth muscle cells and endothelial cells have also provided evidence for an increase in ROS production in diabetes [49]. Exposure of β-cell line and isolated pancreatic islet cells to oxidative stress has been shown to inhibit the promoter activity and mRNA expression of the insulin gene therefore, decreasing insulin gene expression [50]. Oxidative stress is also strongly suspected to be involved in chronic hyperglycaemia-induced insulin resistance [51].
Experimental and clinical studies have shown that oxidative stress is involved in the pathogenesis of various diseases such as cardiovascular diseases and carcinogenesis. It is known to be playing a key role in the aetiology and pathophysiology of diabetes [48]. This is because prolonged exposure of both human and animal cells and tissues to hyperglycaemia is known to result in non-enzymatic glycation of proteins and the end products such as Schiff base and Amadori products, culminates in the production of reactive oxygen species (ROS) [3,4,52]. Chronic hyperglycaemia is seen as a principal factor in promoting the development of micro-vascular and macro-vascular complications in type 2 diabetes and hyperglycaemia is known to be responsible for the damage of DNA, lipids and proteins and the degree of damage has been linked to the degree of hyperglycaemic-induced production of reactive oxygen species and consequently oxidative stress [53]. Studies on the level of 8-hydroxy-deoxyguanosine modified proteins in GK-rats and Tucker diabetic rats by Akash et al [3] and Tanaka et al [54] respectively showed that hyperglycaemia is a main potential factor of oxidative stress in pancreatic beta-cells and that glucose-induced oxidative stress explains the mechanism behind glucotoxicity. To assess the effect of oxidative stress in type 2 diabetes mellitus, [53], recruited 309 diabetic individuals at the Diabetic Clinic at Charles Sturt University, Australia. The control group who were non-diabetic individuals were normoglycaemic, normotensive and had no history or evidence of cardiovascular disease. The investigators collected blood and urine specimens and measured blood glucose, lipids and oxidative stress biomarkers. The authors observed significant increase in the levels of glycosylated haemoglobin, lipids and oxidative stress biomarkers in diabetic group compared to non-diabetic and noted that the findings support the association between type 2 diabetes and hyperglycaemic-induced oxidative stress, chronic hyperglycaemia and progression to type 2 diabetes. The results also act as additional diagnostic tool in the screening and interpreting possible risk of developing diabetes in mild-to-moderate hyperglycaemic patients.
Oxidative stress leads to protein or enzyme inactivation such as SOD, GPX, CAT and reduced glutathione and reduction in these proteins promote oxidative stress [48]. In a study to investigate the role of oxidative stress in diabetes [55], selected diabetic patients on the basis of been diagnosed with oral glucose tolerance test (OGTT) and/or placed on anti-hyperglycaemic agents. Control group comprised individuals with no diabetes, CVD, renal or respiratory disease and all participants were comparable for age, sex, smoking, alcohol consumption, diet and physical activity. The authors reported that oxidative stress is apparent in the diabetics and in the development of associated complications.
Reduction of oxidative stress by blunting daily acute glucose fluctuations in patients with type 2 diabetes could be useful. Development of diabetic complications is very much related to dysglyceamia (chronic sustained hyperglycaemia and acute glycaemic fluctuations). It should be borne in mind that these types of dysglycaemia has been reported to lead to diabetic complications via two principal mechanisms which include activation of oxidative stress and increased activation of the innate immune system [56-58]. Oxidative stress has been positively linked to glycaemic variations over a daily period. To verify this further, [59] designed a randomised trial in which they evaluated the effects of daily glucose excursions on plasma oxidative stress parameters. All the participants had 48 hour continuous sub-cutaneous glucose monitoring at first and third visits. The authors concluded that activation of oxidative stress can be reduced by controlling acute glucose fluctuations over a daily period in type 2 diabetic patients.
Oxidative stress could fast-track the incidence of clinical manifestation of type 2 diabetes in patients and that oxidative stress is a key factor that enhances the development of atherosclerosis in type 2 diabetes [60]. To determine the relationship between oxidative stress and the development or progression of type 2 diabetes, [60] examined 45 non-smoking male and female participants aged 35-65 years old with type 2 diabetes and measured oxidative biomarkers. It was reported that oxidative stress as measured by the level of MDA did not significantly relate to fasting blood level. It is possible that the outcome of the study is related to the small sample size, therefore further study with larger sample size is suggested.
It has been shown that in diabetes, changes in the mitochondrial membrane could lead to activation of complexes in the electron transport chain thereby contributing to the production of oxygen radials. In addition, NADPH oxidase is documented to produce reactive oxygen species (ROS) and is viewed as the principal source of glucose-induced reactive oxygen species formation in the tissues and cells of diabetic models. It is important to note that xanthine oxidase plays a role in the generation of ROS which plays a role in the development of diabetes and diabetic complications. Glucose and its metabolites have been reported to react with hydrogen peroxide in the presence of iron and copper ions to form hydroxyl radical during auto-oxidation in diabetes thereby promoting the generation of ROS and development of diabetic complications [60].
Mechanism of diabetic-induced ros production and oxidative stress
In diabetes, ROS is produced through various routes such as increased polyol pathway, increased formation of advanced-glycation end-products (AGEs) and protein kinase C (PKC) activation [48,61].
Aldose reductase
This pathway which is dependent on nicotinamide adenine dinucleotide phosphate (NAD(P)H), catalyses the reduction of glucose to sorbitol accompanied by the oxidation of sorbitol to fructose by NAD+-dependent sorbitol dehydrogenase. It is believed that hyperglycaemia results in saturation of hexokinase to the extent that over 30% of glucose is moved into the polyol pathway [62,63]. The polyol pathway leads to a shortage of intracellular NAD(P)H and a surplus of NADH. Increased NADH generation is a source of substrate for NADH oxidase to generate ROS [64] causing DNA damage. The polyol pathway serves as a main source of ROS generation in the retina and sorbitol accumulation has been implicated in retinopathy in diabetic complication [61].
Advanced glycation end-products formation
Glucose can react spontaneously with free amino groups of protein to form Schiff bases. These Schiff bases through complex reactions can form advanced glycation end-products (AGEs) [65]. It is known that AGEs can cause tissue damage via formation of cross-links that alter protein structure and function and interaction of AGE with AGE-cell surface receptors on endothelial cells and macrophages resulting in activation of cell signalling and gene expression that induces oxidative stress and inflammation [10].
Protein kinase C
High glucose levels can stimulate ROS production via a PKC-dependent activation of NAD(P)H oxidase in cultured aortic endothelial cells, smooth muscle cells, and renal mesangial cells [66]. Laboratory evidence indicates that NAD(P)H oxidase-dependent production of ROS may cause DNA damage in diabetic renal tissues leading to the development of nephropathy [66]. Increased activity of the NAD(P)H oxidase has been reported in the retina of diabetic rats suggesting its involvement in the development of diabetic retinopathy [67].
The relationship between PKC, NADPH and ROS production
Protein kinase C (PKC) potentially regulates diabetic complications via various ways which includes the activation of endothelial nitric oxide synthase (eNOS), NAD(P)H oxidase, phospholipase A2 (PLA2), endothelin-1 (ET-1), vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), and by activating NF-KB [68]. Diacylglycerol activated PKC affects gene expression of key proteins and reported to lead to decrease blood flow, capillary occlusion, inflammation, free radicals generation and damage to cellular macromolecule [68,69]. High glucose levels can stimulate ROS production via a PKC-dependent activation of NAD(P)H oxidase in cultured aortic endothelial cells, smooth muscle cells, and renal mesangial cells [66]. NADPH oxidase-a primary enzyme found in phagocytic cells, is the main source of ROS generation in non-phagocytic cells such as endothelial cells, fibroblasts and smooth muscle cells. The expression of NAD(P)H oxidase components is up-regulated in vascular tissues from animal models of diabetes and in patients with diabetes and coronary artery disease [70]. Scientific report has shown that NAD(P)H oxidase-dependent production of ROS may cause DNA damage in diabetic renal tissues culminating in the development of nephropathy and increased activity of the NAD(P)H oxidase has also been reported in the retina of diabetic rats suggesting its involvement in the development of diabetic retinopathy [66,71].
Antioxidant enzymes and ROS production
High glucose concentrations causes oxidative stress. Red blood cells and other cells are vulnerable to oxidative stress consequent to high level of polyunsaturated fatty acids, ferrous ions and molecular oxygen [72,73]. Persistent hyperglycemia and increased oxidative stress play significant role in the development of secondary diabetic complications [70]. Cells have various defence systems to prevent or scavenge the over production of reactive oxygen species and oxidative stress. These include antioxidant enzymes such as superoxide dismutase, catalase and glutathione peroxidase. Superoxide dismutase scavenges superoxide radical by accelerating its conversion to hydrogen peroxide while glutathione peroxidase detoxifies hydrogen peroxides and lipid peroxides [74]. Catalase acts in the decomposition of hydrogen peroxide to water and oxygen. Hyperglycemia can interfere with the antioxidant defence system which could result into changes in the activities of antioxidant enzymes as reported in diabetic conditions. Changes in antioxidant defence system in a diabetic state as a significant reduction in SOD activity in red blood cells of diabetic animal model has been documented [73]. Such decrease in SOD activity in hyperglycemic state could be due to oxidative stress-induced inactivation. Increased hydrogen peroxide concentration for example is known to inactivate superoxide dismutase while glycosylation of superoxide dismutase and/or loss of Cu2+, a cofactor required for the enzyme activity can decrease its activity [75].
Interaction between oxidative stress and inflammation
Inflammation is an important physiological response of the body to various pathological processes such as pathogen invasion, tissue injury and irritants. This response involves infiltration and subsequent activation of the cells of the innate immune system and adaptive immune system to the site of injury and the production of inflammatory mediators such as cytokines. It is believed that the release of the inflammatory mediators is prompted by high glucose concentration and mediated by oxidative stress [76]. Chronic inflammation and oxidative stress have been implicated in the pathophysiology of diabetes mellitus. Inflammation and oxidative stress are inextricably linked in physiological and disease states [77]. Complex interactions between the oxidative stress and inflammatory pathways involve mechanisms for both mutual amplification (positive feedback or a “vicious cycle”). Inflammation is the primary immune system reaction to eliminate pathogens or other stimuli in order to restore the cells to normal state or replace destroyed tissue [78]. Following activation, innate immune system cells secrete pro-inflammatory cytokines and chemokines that stimulate the production of reactive oxygen species and/or reactive nitrogen species [78]. Pro-inflammatory cytokines can indirectly provoke oxidative stress by activating macrophages which is known to play a key function in removing the pathogen via the generation of reactive oxygen species [79]. It is important to note that chronic inflammation is a prolonged pathological condition recognised by tissue destruction and fibrosis, culminating in cell damage due to ROS overproduction from inflammatory cells.
Chronic inflammation elicits its cellular side-effects principally via continuous and excessive production of free radicals and depletion of antioxidants [80]. ROS also increases inflammation by activating certain stress-activated kinases. ROS can stimulate transcription factors such as nuclear factor-kappa B (NF-kB) and activator protein-1 (AP-1), to stimulate pro-inflammatory cytokine expression. Therefore, it could be reasonable to suggest that targeting oxidative stress-inflammatory cytokine signalling pathways could seem an attractive strategy for the treatment of diabetes. Under normal physiological conditions, oxidative stress and activation of the immune system are generally short-lived due to intrinsic negative feedback mechanisms like increased production of anti-oxidant compounds or anti-inflammatory cytokines. In certain chronic disease states, such as diabetes, both oxidative stress and inflammation remain activated and could form a positive influence in the control of diabetes or a negative impact depending on various factors [81].
The role of inflammation in the pathogenesis of type 2 diabetes mellitus
The idea that type 2 diabetes mellitus is an inflammatory diseases is seen as an important factor in the understanding of the factors that are involved in the development of type 2 diabetes. Early study by Schmidt et al [82] demonstrated that the presence of inflammatory mediators predicted the future incidence of type 2 diabetes in adults. Another study observed that increased plasma levels of sialic acid, interleukin-6 and C-reactive protein predicted the development of type 2 diabetes mellitus [16]. A study by Pradhan et al [83] shows that increased inflammatory biomarkers predicted insulin resistance and the development of type 2 diabetes. There is also a study that reported on the correlation between fasting insulin levels and C-reactive protein levels in plasma of diabetics showing that insulin resistance and inflammatory processes are linked [84].
Other studies have shown that low-grade inflammation is associated with the risk of developing type 2 diabetes mellitus and chronic sub-clinical inflammation is a factor in insulin resistance syndrome, a strong feature of metabolic syndrome [7,8]. Although the mechanism through which chronic inflammation stimulate the development of type 2 diabetes mellitus is not well understood, however, it has been observed that adipose tissue can synthesise main pro-inflammatory cytokines, tumour necrosis factor and interleukin-1 and -6 and that inflammatory biomarkers are linked with body fat mass, suggesting that activated innate immunity and inflammation are important biological factors in the pathogenesis of diabetes mellitus and in the complications of type 2 diabetes mellitus [85].
For instance, diabetic neuropathy is thought to develop as a consequence of hyperglycaemia-induced metabolic, enzymatic and micro-vascular alterations associated with the production of local and infiltrating pro-inflammatory cytokines with effects on neurons in the central, peripheral and autonomic nervous system and hyperglycaemia-induced production of cytokines have been reported [4]. Animal study has also shown that in diabetic retinopathy, mRNA expression for interleukin-1 and tumour necrosis factor-apha is significantly increased in the retina of diabetic animal whereas, inhibition of tumour necrosis factor-alpha showed improvement in the prevention of diabetic retinopathy [86].
Dalla-Vestra et al [87] reported that patients with type 2 diabetes with nephropathy indicated high concentrations of C-reactive protein, serum amyloid A, interleukin-6 and fibrinogen. It was noted in another study that db//db mice, a model of type 2 diabetes and diabetic nephropathy showed increased expression of intracellular adhesion molecule-1, known to enhance inflammation by increasing leucocyte and macrophage infiltration and adherence in glomeruli and tubules [88].
Studies by Nakamura et al [89] and Navarro et al [7] reported that mRNA expression for tumour necrosis factor-alpha is significantly high in the kidneys of diabetic rats when compared with the kidneys of non-diabetic rats. It is believed that cytokine is cytotoxic to glomerular, mesangial and epithelial cells and could potentially induce significant renal damage.
The identification of a novel gene-tanis whose expression is markedly affected by glucose and is dysregulated after fasting in the diabetic state is believed to be a receptor that binds to serum amyloid A-an acute-phase inflammatory response protein linked to the development of type 2 diabetes mellitus suggesting again that inflammation plays a role in the development of type 2 diabetes mellitus [91].
According to Bruun et al [92] and Belalcazar et al [93], fibrinogen, white blood cell count, C-reactive protein, serum amyloid A and pro-inflammatory cytokines such as interleukin-1 beta, interleukin-6 and chemokines such as MCP-1 are increased in the blood levels of individuals with type 2 diabetes mellitus. However, these parameters are decreased when individuals with type 2 diabetes mellitus are involved in robust lifestyle changes that significantly reduced the body weight. There seem to be an agreement among scientists that adipose tissue, muscle and pancreatic cells are locations of inflammation in the presence of obesity and type 2 diabetes and that these cells are very important in the production of pro-inflammatory cytokines. The cells act in autocrine and paracrine pathway to enhance insulin resistance by disrupting insulin signalling in peripheral tissues by means of activation of the C-JUN N-terminal kinase and nuclear factor-kappa B pathways are activated in more tissues in type 2 diabetes thereby playing a key function in enhancing tissue inflammation [31,94].
It is important to note that emerging evidence has shown that inflammasome NLRP3 (inflammasome is a multi-protein oligomer responsible for the activation of inflammatory responses. It promotes the maturation and secretion of pro-inflammatory cytokines Interleukin-1β and Interleukin-18) play a role in the pathogenesis of metabolic syndrome and type 2 diabetes mellitus. The activation of NLRP3 inflammasome in the pancreas by increased level of glucose and fatty acid followed by release of interleukin-1 beta led to beta cells dysfunction, apoptosis, insulin deficiency and progression to type 2 diabetes [95].
Obesity, physical inactivity, smoking, dietary diets, psychological stress and infections are seen as activating factors of the innate immune system which induced chronic low-grade inflammation which in turn via the secretion of pro-inflammatory cytokines enhance insulin resistance and insulin resistance leads to type 2 diabetes mellitus [96]. People with sedentary lifestyles have been reported to have higher glucose and insulin levels than active people from the same BMI group [97] while smoking has been positively associated with various inflammatory biomarkers [98]. On the other hand, dietary habits have been associated with the management and prevention of insulin resistance and diabetes [99]. For instance, higher consumption of red meat could increase insulin resistance in non-diabetic subjects [100]. To investigate the influence of lifestyle on inflammatory biomarkers in type 2 diabetes, Pitsavos et al [96] recruited 3042 (1514 males and 1528 females) in randomised controlled study. The authors reported an association between low-grade inflammation markers and glycaemic control parameters irrespective of demographic, clinical and lifestyle indices such as dietary factor. It was found that diabetic individuals demonstrated increased C-reactive protein, interleukin-6 and tumour necrosis factor than non-diabetic individuals. Following multi-adjusted analysis, the results also showed positive correlation of blood glucose and insulin levels with C-reactive protein and interleukin-6 levels confirming the hypothesis that low-grade inflammation is involved in the pathogenesis of type 2 diabetes mellitus.
Insulin resistance could increase the concentration of inflammatory biomarkers [94]. This association may reflects the effects of tumour necrosis factor and leptin (pro-inflammatory cytokines) on insulin sensitivity or secretion. Tumour necrosis factor could produce insulin resistance via decreased auto-phosphorylation of the insulin receptor, conversion of insulin-receptor substrate-1 into an inhibitor of insulin receptor tyrosine kinase activity, decrease GLUT transporter in muscle cells and increases in circulating free fatty acids [83,100,101]. It is believed that tumour necrosis factor and interleukin-6 could alter beta-cells function via direct action or stimulation of free fatty acid production. These processes enhance or promote the development of type 2 diabetes confirming the relationship between insulin resistance and inflammation and explains how resistance and inflammation promote the development of type 2 diabetes mellitus. From this interaction, we can gain a deeper understanding of the role of inflammation and its interaction with other factors in the pathogenesis of type 2 diabetes mellitus. Increased pro-inflammatory cytokines and activation of the inflammatory processes are key factors in the development of insulin resistance and type 2 diabetes. This knowledge and understanding should motivate scientists to develop an approach that can inhibit inflammation as a preventive means in the control of diabetes mellitus.
The interaction between hyperglycaemia, inflammation and oxidative stress in type 2 diabetes mellitus
Disturbances in glucose metabolism and resultant hyperglycaemia do play key roles in the development of type 2 diabetes. Chronic sustained hyperglycaemia has been reported as playing a role in the development of micro- and macro-vascular complications known to occur via series of mechanisms involving oxidative stress and inflammation [53,102]. Impaired fasting glucose could potentially lead to the development of type 2 diabetes mellitus which is related to increased oxidative stress and chronic sub-clinical inflammation [93]. As a consequence of hyperglycaemic-induced oxidative stress, ROS are known to damage DNA, lipids and proteins and the degree of damage or injury is associated with the duration of hyperglycaemia [103]. The oxidative stress which is initiated by hyperglycaemic is due to the increased intracellular and extracellular free radical concentrations. Decreased levels or activities of antioxidant protein (glutathione), enzymes (catalase, glutathione peroxidase, superoxide dismutase) and micronutrients (selenium and zinc) and vitamins such as vitamin C, E and A, as a result of hyperglycaemia may stimulate increase generation of reactive oxygen species, consequently oxidative stress may in turn stimulate the production of inflammatory cytokines and chemokines [104].
In a study by Soleiman et al [105] on the relationship between inflammation, oxidative stress and type 2 diabetes, it was suggested that controlling inflammation and oxidative stress is necessary for accelerating the treatment process and prevention of diabetic complications. In order to investigate the relationship between hyperglycaemia, oxidative stress and inflammation/inflammation biomarkers, [53], recruited 309 participants from the diabetic Health Screening Clinic in Australia. The study highlights the potential value that inflammatory and oxidative stress makers could contribute to the screening of patients with hyperglycaemic state. The findings documented the contribution of inflammation and oxidative stress in the progression of type 2 diabetes mellitus and demonstrated an association between hyperglycaemia, oxidative stress and inflammatory biomarkers in a screening population. The authors also noted that the effect of oxidative stress and its relationship with inflammatory reaction was further supported by a review of clinical status of patients showing a higher prevalence of self-reported CVD due to the association between type 2 diabetes mellitus, inflammation and oxidative stress. It was reported that hyperglycaemia is associated with increased risk of developing type 2 diabetes and that diabetes is associated with oxidative stress and inflammation responses. They observed differences in white blood cell count, blood glucose level, triglyceride, HDC-cholesterol, glycosylated haemoglobin between normoglyceamic and hyperglycaemic groups. Research reports have shown that individuals with increased levels of interleukin-6 and interleukin-1 beta has a 3-fold increase in the risk of developing type 2 diabetes whilst low levels of interleukin-1 beta showed no increased risk of developing type 2 diabetes [105-107].
It is vital to know that during hyperglycaemia, cytokine production (inflammation response to hyperglycaemia) is accompanied by increased levels of monocyte chemo-attractant protein-1 and reduced levels of insulin-like growth factor-1 [60,94,106]. It has been reported that elevated monocyte chemoattractant protein-1 could activate macrophage infiltration into adipose tissue causing induced adipose de-differentiation which contribute to hyperinsulinaemia and type 2 diabetes through insulin resistance [108].
Chronic disease progression is believed to be linked to the interaction between inflammation and oxidative stress. Increased interleukin-6 is reported to promote superoxide radicals and oxidative stress which in turn negatively impact on effective metabolism of free fatty acids. Oxidative stress thus induce mitochondrial uncoupling and increase generation of reactive oxygen species. In the cascade of reactions, increased generation of reactive oxygen species leads to further oxidative stress which also elicits inflammatory processes [109].
The pathophysiology of type 2 diabetes mellitus reveals that oxidative stress is one of the factors that plays a role in the pathogenesis of insulin resistance, impaired insulin secretion, glucose utilization, impaired hepatic glucose metabolism coupled with activation of inflammation pro-inflammation cytokines, culminating in type 2 diabetes [3]. Oxidative stress in pancreatic beta cells is known to be induced by high glucose level, hyperlipidaemia and inflammatory responses [110]. When a state of oxidative stress is established, it enhances the production of pro-inflammatory cytokines such as tumour necrosis factor (TNF). Increased generation of reactive oxygen species has been reported to increase in adipocytes as well and inflammation-induced oxidative stress has been noted as one of the most important factors in the development of type 2 diabetes [107]. It has been observed that pancreatic beta cells exposed to elevated concentration of free fatty acids and hyperglycaemia undergo apoptosis and that oxidative stress is the major factor that induced apoptosis in pancreatic beta cells [111]. Also, a relationship between the measurement of oxidative stress biomarkers and presence of pancreatic beta cells lesions has been observed in volunteer type 2 diabetic patients and significant reduction in the activities of antioxidant enzymes in patients with increased oxidative stress has been documented. Oxidative stress-induced autophagy has been implicated in the pathogenesis of type 2 diabetes. Among the factors that induce autophagy, is free fatty acids via lipid peroxidation and is known to induce pancreatic beta cells oxidative stress-induced autophagy by causing peripheral insulin resistance in type 2 diabetes [112].
Oxidative stress and inflammation-enhanced diabetic complications
Diabetic hepatopathy
The liver plays important role in the regulation of carbohydrate metabolism both in physiological and pathological conditions. In type 2 diabetes mellitus, insulin resistance leads to hyperglycaemia in the liver and in turn further affect glucose uptake or utilization [113]. Growing evidence has linked a low-grade chronic inflammatory response to diabetic mellitus, its complications, especially liver-associated complications [114].
Reports have shown that diabetes mellitus is linked to a variety of liver alterations such as abnormal glycogen synthesis, fibrosis, cirrhosis, acute liver disease and liver hepatitis [115]. Over production of fatty acids in the liver and hyperglycaemia can negatively affect liver integrity and function of the liver thereby contributing to morbidity and mortality in people with diabetes mellitus [116]. It is known that the liver is one of the organs that is susceptible to hyperglycaemic-induced oxidative stress leading to hepatic damage. Oxidative stress and inflammatory cytokines contribute to the pathogenesis and progression of diabetes which affects the liver [117]. Diabetes, is now the most common cause of liver disease in the U.S.A and various types of liver diseases such as acute liver failure, cirrhosis, hepatocellular carcinoma can be seen in patients with type 2 diabetes [118]. Cryptogenic cirrhosis, of which diabetes is, by far, the most common cause, is now seen as the third leading indication for liver transplantation in the U.S. Caldwell et al, (1999) [119] accounting for 12.5% of deaths in patients with diabetes. Glycogenic hepatopathy is associated with abnormal glycogen accumulation in hepatocytes, a major cause of hepatomegaly in type 1 and type 2 [120].
Diabetic retinopathy
Diabetic retinopathy is a principal contributory factor to visual impairment among working adults especially in developed countries and with the increased prevalence of diabetes mellitus in developing countries, diabetic retinopathy is becoming of great concern among patients in developing countries [121]. The prevalence rate for diabetic retinopathy for all adults from 40 years is 28.5% (4.2 million people) and is expected to increase to 34.6% (93 million people) [13]. Research has documented that the retina has high levels of polyunsaturated fatty acids and coupled with the highest oxygen uptake and glucose oxidation compared to other tissues, making the retina to be susceptible to oxidative stress [122]. Oxidative stress by its activating activity, activates other metabolic pathways such as polyol pathway, advanced glycation end product pathway, protein kinase pathway, changes in the expression of vascular endothelial growth factor which further enhance the production of reactive oxygen species [119]. The role of inflammation in the development of diabetic retinopathy has been documented. For example, cyclooxygenase-2 that catalyses the formation of prostaglandin E2 is induced by interleukin-1, therefore COX-2 and prostaglandin E2 are important factors that contribute to the development of diabetic retinopathy and since inflammation is known to generate ROS, oxidative stress may directly or indirectly stimulate the release of inflammatory mediators [119].
Diabetic retinopathy results from damage to the small vasculature of the retina, multi cellular and the light sensitive tissue at the back of the eye. The retina capillaries are lined with endothelial cells responsible for maintaining the blood retinal barrier and are surrounded by smooth muscle cells, pericytes, which provide tone to the vessels [123]. The vascular lesions that are identified at the early stage of diabetic retinopathy include obliteration of capillaries and small arterioles, gradual thickening of vascular basement membrane, increased permeability of endothelial cells and formation of microaneurysms, vessel leakage and hemorrhage [124].
Diabetic nephropathy
With hyperglycaemia as the driving force, diabetic nephropathy is regarded as one of the most common complications of type 2 diabetes and the leading cause of end-stage renal disease [106,125]. It is a progressive disease with its pathogenesis influenced by factors such as duration of diabetes, oxidative stress-induced poor glycaemic control, hypertension, hypertriglyceridaemia, increased production of cytokines Il-6 and tumour necrosis factor from endothelial cells culminating in the induction of inflammation in type 2 diabetes [126,127]. It has been shown by Kafle et al [128], that prolonged duration of uncontrolled hyperglycaemia induces advanced glycation end-products which activates NFkB-factor. The NF-kB acts on the nucleus through transcription and release cytokines and that these cytokines are cytotoxic to glomerular and epithelial cells which induce direct renal damage. The cytokines may in turn have negative impact on protein permeability barrier of the glomerulus which may induce changes in haemodynamic factors of renal cells such as glomerular basement membrane thickening, renal mesangial cell (extra-glomerular or intra-glomerular cells) expansion and hyperplasia of extracellular matrix which represent an important lesion in diabetic nephropathy in people with long standing type 2 diabetes. Reduction in haemoglobin in patients with diabetic nephropathy is said to be related to reduction in the production of hormone erythropoietin [128]. Increased ROS production in diabetic condition has been associated with vasoconstriction, endothelial dys-function, modification of extracellular matrix proteins and increase renal sodium reabsorption [61]. The role of oxidative stress in diabetic nephropathy has been noted by the observation that inhibition of oxidative stress improves the featues linked to STZ-induced diabetic nephropathy [65]. Leucocytes, monocytes and macrophages have been implicated in the pathogenesis of diabetic nephropathy and inflammatory biomarkers have been linked with the risk of developing diabetic nephropathy [129]. Various reports support role of interleukin-1, 6 and 18 in the progression of diabetic nephropathy [4,130,131].
Diabetic neuropathy
Diabetic neuropathy is seen as the most common micro-vascular complications in diabetes mellitus. It is believed that 50% of patients with a 20-year history of diabetes develop neuropathy and that about 10% of diabetic neuropathy are linked to abnormal sensations and pain [132,133] and that the incidence of diabetic neuropathy increases with duration of diabetes and is fuelled by poor glycaemic control, hyperglycaemic-oxidative stress and production of inflammatory cytokines [134]. Oxidative stress is known to promote apoptosis in neurons and is seen as a strong factor that leads to damage to nervous system in diabetes [135]. In diabetic neuropathy, neurons are not only lost but their ability to regenerate is equally disturbed. In non-dividing cells such as the neurons, damage to proteins and other macromolecules hinder proteins and other macromolecules to perform their axonal transport and signaling [136]. Neuropathy is characterized by a progressive loss of nerve fibre function [137]. In the peripheral nervous system, diabetes causes a progressive decline of sensory nerves and damage to motor nerves [138].
Diabetic cardiomyopathy
It has been reported that various factors such as hyperglycaemia, insulin resistance, increased fatty acid metabolism work together to contribute to the development and progression of diabetic cardiomyopathy [139]. In spite of this recognition of the involvement of the above-mentioned factors, the interplay of oxidative stress combined with pro-inflammatory mediators do play important role in the development of biochemical and structural changes that are linked to diabetic cardiomyopathy [63,140]. Type 2 diabetes is a risk factor for coronary artery disease and stroke [141]. Diabetics have a 2- to 4-fold higher risk for cardiovascular events [142] and about 80% of diabetes-associated deaths are caused by cardiovascular disease [143].
Conclusion
Diabetes mellitus is a global public health problem affecting the poor, rich, educated, uneducated people as well as urban and rural dwellers in both developed and developing countries. Different factors are interplaying in the pathogenesis and progression of diabetes mellitus. The focus of this review is to examine the role of oxidative stress and inflammation in the pathogenesis and progression of diabetes mellitus and to critically examine the interplay based on scientific evidence that exist between diabetes mellitus, oxidative stress and inflammation. It also examines their interplay in the development of diabetic complications. The evidence from this review is very clear and shows that long-term innate immune activation that culminates in chronic inflammation plays a role in the development and progression of diabetes mellitus. It shows that inflammatory mediators such as interleukin-1-beta, 6, tumour necrosis factor-alpha are linked to type 2 diabetes mellitus and that these factors contribute to the generation of reactive oxygen species and together with hyperglycaemia induce further generation of reactive oxygen species. In the presence of defective antioxidant defence system either due to endogenous alteration or exogenous inadequacy that tilt the balance in favour of free radicals, oxidative stress develops. Interestingly, it can be observed from this review that various experimental and clinical studies support the direct link between oxidative stress and diabetes mellitus through the measurement of oxidative biomarkers in both diabetic and non-diabetic animals and humans and that oxidative stress is strongly involved in chronic hyperglycaemic-induced insulin resistance. It has been shown that the release of inflammatory mediators is prompted by hyperglycaemia and mediated by oxidative stress confirming the link between diabetes mellitus, oxidative stress and inflammation.
Acknowledgements
The author wish to acknowledge the financial support from the National Research Foundation (NRF) of South Africa and the Cape Peninsula University of Technology (CPUT).
Disclosure of conflict of interest
None.
References
- 1.Silink M. Childhood diabetes: a global perspective. Horm Res. 2002;57:1–5. doi: 10.1159/000053304. [DOI] [PubMed] [Google Scholar]
- 2.Badawi A, Klip A, Haddad P, Cole DE, Bailo BG, El-Sohemy A, Karmali M. Type 2 diabetes and inflammation: prospects for biomarkers of risk and nutritional intervention. Diabetes Metab Syndr Obes. 2010;3:173–86. doi: 10.2147/dmsott.s9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rehman K, Akash MSH. Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: how are they interlinked? J Cell Biochem. 2017;118:3577–3585. doi: 10.1002/jcb.26097. [DOI] [PubMed] [Google Scholar]
- 4.Navarro JF, Mora C. Role of inflammation in diabetic complications. Nephrol Dial Transplant. 2005;20:2601–4. doi: 10.1093/ndt/gfi155. [DOI] [PubMed] [Google Scholar]
- 5.Crook M. Is type 2 diabetes mellitus a disease of the innate immune system? Diabet Med. 2004;21:203–7. doi: 10.1046/j.1464-5491.2003.01030.x. [DOI] [PubMed] [Google Scholar]
- 6.Festa A, D’Agostino R Jr, Howard G, Mykkänen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the insulin resistance atherosclerosis study (IRAS) Circulation. 2000;102:42–7. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- 7.Frohlich M, Imhol A, Berg G. Association between C-reactive protein and features of the metabolic syndrome: a population based study. Diabetes Care. 2000;23:1835–9. doi: 10.2337/diacare.23.12.1835. [DOI] [PubMed] [Google Scholar]
- 8.Crook M. Type 2 diabetes mellitus: a disease of the innate immune system? An update. Diabet Med. 2004;21:203–207. doi: 10.1046/j.1464-5491.2003.01030.x. [DOI] [PubMed] [Google Scholar]
- 9.Hall V, Thomsen RW, Henriksen O, Lohse N. Diabetes in sub saharan africa 1999-2011: epidemiology and public health implications. A systematic review. BMC Public Health. 2011;11:564. doi: 10.1186/1471-2458-11-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Investig. 2005;115:1111–9. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westermann D, Van Linthout S, Dhayat S, Dhayat N, Schmidt A, Noutsias M, Song XY, Spillmann F, Riad A, Schultheiss HP, Tschöpe C. Tumor necrosis factor-alpha ntagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol. 2007;102:500–7. doi: 10.1007/s00395-007-0673-0. [DOI] [PubMed] [Google Scholar]
- 13.Zhang P, Zhang X, Brown J, Vistisen D, Sicree R, Shaw J, Nichols G. Global healthcare expenditure on diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:293–301. doi: 10.1016/j.diabres.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 14.Derosa G, D’Angelo A, Bonaventura A, Bianchi L, Romano D, Maffioli P. Effects of berberine on lipid profile in subjects with low cardiovascular risk. Expert Opin Biol Ther. 2013;13:475–82. doi: 10.1517/14712598.2013.776037. [DOI] [PubMed] [Google Scholar]
- 15.World Health Organization. Global Report on ddiabetes. Geneva, Switzerland: 2017. [Google Scholar]
- 16.Duncan BB. The burden of diabetes and hyperglycaemia in Brazil: past and present. Findings from the global burden of disease study 2015. Diabt Metab Syndr. 2017;9:1–18. doi: 10.1186/s13098-017-0216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.International Diabetes Federation (IDF) Diabetes Atlas. 3rd edition. Brussels, Belgium: International Diabetes Federation; 2017. [Google Scholar]
- 18.International Diabetes Federation (IDF) Diabetes Atlas. 3rd edition. Brussels, Belgium: International Diabetes Federation; 2015. [Google Scholar]
- 19.Roglic G. Diabetes in women: the global perspective. Int J Gynaecol Obstet. 2009;104(Suppl 1):S11–3. doi: 10.1016/j.ijgo.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara A. Increasing prevalence of gestational diabetes mellitus: a public health perspective. Diabetes Care. 2007;30:S141–S146. doi: 10.2337/dc07-s206. [DOI] [PubMed] [Google Scholar]
- 21.HAPO Study Cooperative Research Group. Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, Coustan DR, Hadden DR, McCance DR, Hod M, McIntyre HD, Oats JJ, Persson B, Rogers MS, Sacks DA. Hyperglycaemia and adverse pregnancyoutcomes. N Engl J Med. 2008;358:1991–2000. [Google Scholar]
- 22.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94:311–21. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997;14(Suppl 5):S1–85. [PubMed] [Google Scholar]
- 25.Freeman JS. The increasing epidemiology of diabetes and review of current treatment algorithms. J Am Osteopath Assoc. 2010;110(Suppl 7):eS2–6. [PubMed] [Google Scholar]
- 26.Oyagbemi AA, Salihu M, Oguntibeju OO, Esterhyse AJ, Farombi EO. Antioxidant-Antidiabetic Agents and Human Health. 2014. Some selected medicinal plants with antidiabetic potentials; pp. 95–113. [Google Scholar]
- 27.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–46. doi: 10.1016/S0140-6736(05)61032-X. [DOI] [PubMed] [Google Scholar]
- 28.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetic epidemic. Nat. 2001;414:782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 29.Alberti KG. Treating type 2 diabetes-today’s targets, tomorrow’s goals. Diabe Obes Metab. 2001;3:S3–S10. [PubMed] [Google Scholar]
- 30.Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the united, states, 1991-1998. JAMA. 1999;282:1519–1522. doi: 10.1001/jama.282.16.1519. [DOI] [PubMed] [Google Scholar]
- 31.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimopoulos N, Watson M, Sakamoto K, Hundal HS. Differential effects of palmitate and palmitoleate on insulin action and glucose utilization in raty L6 skeletal and muscle cells. Biochem J. 2006;399:473–481. doi: 10.1042/BJ20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bilan PJ, Samokhvalov V, Koshkina A, Schertzer JD, Samaan MC, Klip A. Direct and macrophage-mediated actions of fatty acids causing insulinresistance in muscle cells. Arch Physiol Biochem. 2009;115:176–190. doi: 10.1080/13813450903079314. [DOI] [PubMed] [Google Scholar]
- 34.King GL. The role of inflammatory cytokines in diabetes aand its complications. J Periodontol. 2008;79:1527–1534. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 35.Krogh-Madsen R, Plomgaard P, Moller K, Mittendorfer B, Pedersen BK. Incidence of TNF-α and IL-6 infusion on insulin sensitivity and expression of IL-18 inm humans. Am J Physiol Endocrinol Metab. 2006;291:E108–14. doi: 10.1152/ajpendo.00471.2005. [DOI] [PubMed] [Google Scholar]
- 36.Amin TT, Al-Sultan AI, Ali A. Overweight and obesity and their association with dietary habits and socio-demogtraphic characteristics among male primary school children in Al-Hassa, kingdom of saudi arabia. Eur J Nutr. 2008;47:310–8. doi: 10.1007/s00394-008-0727-6. [DOI] [PubMed] [Google Scholar]
- 37.Panagiotakos DB, Tzima N, Pitsavos C, Chrysohoou C, Papakonstantinou E, Zampelas A, Stefanadis C. The relationship between dietary habits, blood glucose and insulin levels among people without cardiovascular diseae and type 2 diabetes. Rev Diabet Stud. 2005;2:208–15. doi: 10.1900/RDS.2005.2.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khatib O. Non-communicable diseases: risk factors and regional strategies for prevention and care. East Mediterr Health J. 2004;10:778–788. [PubMed] [Google Scholar]
- 39.Villegas R, Shu XO, Gao YT, Yang G, Elasy T, Li H, Zheng W. Vegetable but fruit consumption reduces the risk of type 2 diabetes in Chinese women. J Nutr. 2008;138:574–80. doi: 10.1093/jn/138.3.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nanri A, Mizoue T, Noda M, Takahashi Y, Kato M, Inoue M, Tsugane S Japan Public Health Center-based Prospective Study Group. Rice intake and type 2 diabetes in Japanese men and women: the Japan public health centre-based prospective study. Am J Clin Nutr. 2010;92:1468–1477. doi: 10.3945/ajcn.2010.29512. [DOI] [PubMed] [Google Scholar]
- 41.Hosseini A, Abdollahi M. Diabetic neuropathy and oxidative stress: therapeutic perspectives. Oxid Med Cell Longev. 2013;2013:168039. doi: 10.1155/2013/168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugawara A, Kawai K, Motohashi S, Saito K, Kodama S, Yachi Y, Hirasawa R, Shimano H, Yamazaki K, Sone H. HbA(1c) variability and the development of microalbuminuria in type 2 diabetes: tsukuba kawai diabetes regist 2. Diabetologia. 2012;55:2128–31. doi: 10.1007/s00125-012-2572-7. [DOI] [PubMed] [Google Scholar]
- 43.Charokopou M, Sabater FJ, Townsend R, Roudaut M, McEwan P, Verheggen BG. Methods applied in cost-effectiveness models for treatment strategies in type 2 diabetes mellitus and their use in health technology assessments: a systematic review of the literature from 2008 to 2013. Curr Med Res Opin. 2016;32:207–18. doi: 10.1185/03007995.2015.1102722. [DOI] [PubMed] [Google Scholar]
- 44.Tucker DM, Palmer AJ. The cost-effectiveness of interventions in diabetes: a review of published economic evaluations in the UK setting with a eye on the future. Prim Care Diabetes. 2011;5:9–17. doi: 10.1016/j.pcd.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 45.ADVANCE Collaborative Group. Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 46.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Sachdev S, Davies KJ. Production, detection, and adaptive responses to free radicals in exercise. Free Radic Biol Med. 2008;44:215–23. doi: 10.1016/j.freeradbiomed.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 48.Halliwell B. The wanderings of a free radical. Free Radic Biol Med. 2009;46:531–42. doi: 10.1016/j.freeradbiomed.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 49.Lee YJ, Suh KS, Choi MC, Chon S, Oh S, Woo JT, Kim SW, Kim JW, Kim YS. Kaempferol protects HIT-15 pancreatic beta-cells from 2-deoxy-D-ribose-induced oxidative damage. Phytother Res. 2010;24:419–423. doi: 10.1002/ptr.2983. [DOI] [PubMed] [Google Scholar]
- 50.Hanazaki K. Problems associated with glucose toxicity: role of hyperglycemia-induced oxidative stress. World J Gastroenterol. 2009;15:4137–4142. doi: 10.3748/wjg.15.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eriksson JW. Metabolic stress in insulin’s target cells leads to ROS accumulation-a hypothetical common pathway causing insulin resistance. FEBS Lett. 2007;581:3734–3742. doi: 10.1016/j.febslet.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 52.Hojs R, Ekart R, Bevc S, Hojs N. Markers of inflammation and oxidative stress in the development and progression of renal disease in diabetic patients. Nephron. 2016;133:159–62. doi: 10.1159/000447434. [DOI] [PubMed] [Google Scholar]
- 53.Butkowski EG, Jelinek HF. Hyperglycaemia, oxidative stress and inflammation markers. Redox Rep. 2017;22:257–264. doi: 10.1080/13510002.2016.1215643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999;96:10857–62. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dayre A, Pouvreau C, Butkoswki EG, de Jong B, Jelinek HF. Diabesity increases inflammation and oxidative stress. Int J Pharm Sci Dev Res. 2016;2:012–018. [Google Scholar]
- 56.Brownlee M. Biochemistry and molecular cell biology of diabetic comp0lications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 57.Esposito K, Nappo F, Marfella R. Inflammatory cytokine concentrations are acutely increased by hyperglycaemia in humans: role of oxidative stress. Circ. 2002;106:2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae. [DOI] [PubMed] [Google Scholar]
- 58.Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycaemia of type 2 diabetic patinets: variations with increasing levels of HbA (1c) Diabetes Care. 2003;26:881–5. doi: 10.2337/diacare.26.3.881. [DOI] [PubMed] [Google Scholar]
- 59.Rizzo MR, Barbieri M, Marfella R, Paolisso G. Reduction of oxidative stress and inflammation by blunting daily acute glucose fluctuations in patients with type 2 diabetes: role of dipeptidyl peptidase-IV inhibition. Diabetes Care. 2012;35:2076–82. doi: 10.2337/dc12-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harani H, Otmane A, Makrelouf M, Ouadahi N, Abdi A, Berrah A, Zenati A, Alamir B, Koceir EA. The relationship between inflammation, oxidative stress and metabolic risc factors in type 2 diabetic patients. Ann Biol Clin (Paris) 2012;70:669–77. doi: 10.1684/abc.2012.0763. [DOI] [PubMed] [Google Scholar]
- 61.Ishill N, Patel KP, Lane PH. Nitric oxide synthesis and oxidative stress in the renal cortex of rats with diabetes mellitus. J Am Soc Nephrol. 2001;12:1630–9. doi: 10.1681/ASN.V1281630. [DOI] [PubMed] [Google Scholar]
- 62.González RG, Barnett P, Aguayo J, Cheng HM, Chylack LT Jr. Direct measurement of polyol pathway activity in the ocular lens. Diabetes. 1984;33:196–9. doi: 10.2337/diab.33.2.196. [DOI] [PubMed] [Google Scholar]
- 63.Funk SD, Yurdagul A Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. 2012;2012:569654. doi: 10.1155/2012/569654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morré DM, Lenaz G, Morré DJ. Surface oxidase and oxidative stress propagation in aging. J Exp Biol. 2000;203:1513–21. doi: 10.1242/jeb.203.10.1513. [DOI] [PubMed] [Google Scholar]
- 65.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM. Inhibition of NADPH oxidative prevents advanced glycation end product-mediated dama in diabetic nephropathy through a protein kinase C-apha-dependent pathway. Diabt. 2008;57:460–9. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- 66.Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, Nawata H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia. 2003;46:1428–37. doi: 10.1007/s00125-003-1205-6. [DOI] [PubMed] [Google Scholar]
- 67.Ellis D, Forrest KY, Erbey J, Orchard TJ. Urinary measurement of transforming growth factor-β and type IV collagen as new markers of renal injury: application in diabetic nephropathy. Am Assoc Clin Chem. 1998;44:21–27. [PubMed] [Google Scholar]
- 68.Noh H, King GL. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 2007;72:S49–S53. doi: 10.1038/sj.ki.5002386. [DOI] [PubMed] [Google Scholar]
- 69.Way KJ, Katai N, King GL. Protein kinase C and the development of diabetic vascular complications. Diabet Med. 2001;18:945–59. doi: 10.1046/j.0742-3071.2001.00638.x. [DOI] [PubMed] [Google Scholar]
- 70.Huang C, Kim Y, Caramori ML, Fish AJ, Rich SS, Miller ME, Russell GB, Mauer M. Cellular basis of diabetic nephropathy: II. The transforming growth factor-beta system and diabetic nephropathy lesions in type 1 diabetes. Diabetes. 2002;51:3577–81. doi: 10.2337/diabetes.51.12.3577. [DOI] [PubMed] [Google Scholar]
- 71.Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE. The voltage-gated proton channel, Hv1 enhances brain damage from ischemic stroke. Nat Neuro Sci. 2012;15:565–73. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yadav P, Sarkar S, Bhatnagar D. Lipid peroxidation and antioxidant enzymes in erythrocytes and tissues in aged diabetic rats. Indian J Exp Biol. 1997;35:389–392. [PubMed] [Google Scholar]
- 73.Hamden K, Carreau S, Jamoussi K, Miladi S, Lajmi S, Aloulou D, Ayadi F, Elfeki A. 1Alpha, 25 dihydroxyvitamin D3: therapeutic and preventive effects against oxidative stress, hepatic, pancreatic and renal injury in alloxan-induced diabetes in rats. J Nutr Sci Vitaminol (Tokyo) 2009;55:215–22. doi: 10.3177/jnsv.55.215. [DOI] [PubMed] [Google Scholar]
- 74.Jurkovič S, Osredkar J, Marc J. Molecular impact of glutathione peroxidases in antioxidant processes. Biochemia Medica. 2008;18:162–174. [Google Scholar]
- 75.Seven A, Guzel S, Seymen O, Civelek S, Bolayirli M, Uncu M, Burcak G. Effects of vitamin E supplementation on oxidative stress in streptozotocin induced diabetic rats: investigation of liver and plasma. Yonsei Med J. 2004;45:703–710. doi: 10.3349/ymj.2004.45.4.703. [DOI] [PubMed] [Google Scholar]
- 76.Gumieniczek A, Hopkała H, Roliński J, Bojarska-Junak A. Antioxidative and anti-inflammatory effects of repaglinide in plasma of diabetic animals. Pharmacol Res. 2005;52:162–166. doi: 10.1016/j.phrs.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 77.Ambade A, Mandrekar P. Oxidative stress and inflammation: essential partners in alcoholic liver disease. Int J Hepatol. 2012;2012:853175. doi: 10.1155/2012/853175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Emmendoerffer A, Hecht M, Boeker T, Mueller M, Heinrich U. Role of inflammation in chemical-induced lung cancer. Toxicol Lett. 2000;112-113:185–91. doi: 10.1016/s0378-4274(99)00285-4. [DOI] [PubMed] [Google Scholar]
- 79.Miliara X, Garnett JA, Tatsuta T, Abid Ali F, Baldie H, Pérez-Dorado I, Simpson P, Yague E, Langer T, Matthews S. Intra-mitochondrial signaling: interactions among mitoKATP, PKCε, ROS, and MPT. EMBO Rep. 2015;16:824–35. doi: 10.15252/embr.201540229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fialkow L, Wang Y, Downey GP. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic Biol Med. 2007;42:153–164. doi: 10.1016/j.freeradbiomed.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 81.Hold GL, El-Omar EM. Genetic aspects of inflammation and cancer. Biochem J. 2008;410:225–35. doi: 10.1042/BJ20071341. [DOI] [PubMed] [Google Scholar]
- 82.Jesmin J, Rashid MS, Jamil H, Hontecillas R, Bassaganya-Riera J. Gene regulatory network reveals oxidative stress as the underlying molecular mechanism of type 2 diabetes and hypertension. BMC Med Genomics. 2010;3:45. doi: 10.1186/1755-8794-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults: a cohort study. Lancet. 1999;353:1649–1652. doi: 10.1016/s0140-6736(99)01046-6. [DOI] [PubMed] [Google Scholar]
- 84.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6 and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 85.Lemieux I, Pascot A, Prud’homme D, Alméras N, Bogaty P, Nadeau A, Bergeron J, Després JP. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21:961–7. doi: 10.1161/01.atv.21.6.961. [DOI] [PubMed] [Google Scholar]
- 86.Pickup JC. Inflammation and activated immune system in the pathogenesis of type 2 diabetes. Diab Care. 2004;27:813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- 87.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Döhmen S, Adamis AP. Non-steroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-apha suppression. FASEB J. 2002;16:438–40. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 88.Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, Plebani M, Fioretto P. Acute-phase markers of inflammation and glomerular structure in patients with type-2 diabetes. J Am Soc Nephrol. 2005;16:S78–S82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 89.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesh GH. Intracellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–1722. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 90.Nakamura T, Fukui M, Ebihara I. mRNA expression of growth factors in glomeruli from diabetic rats. Diab. 1993;42:450–456. doi: 10.2337/diab.42.3.450. [DOI] [PubMed] [Google Scholar]
- 91.Wander GS, Khurana SB, Gulati R, Sachar RK, Gupta RK, Khurana S, Anand IS. Epidemiology of coronary heart disease and risk factors in a rural punjab population: prevalence and correlation with various risk factors. Indian Heart J. 1994;46:319–23. [PubMed] [Google Scholar]
- 92.Bruun JM, Helge JW, Richelsen B, Stallknecht B. Diet and exercise reduce low-grade inflammation and macrophage infiltration in adipose tissue but not in skeletal muscle in severely obese subjects. Am J Physiol Endocrinol Metab. 2006;290:E961–E967. doi: 10.1152/ajpendo.00506.2005. [DOI] [PubMed] [Google Scholar]
- 93.Belalcazar LM, Haffner SM, Lang W, Hoogeveen RC, Rushing J, Schwenke DC, Tracy RP, Pi-Sunyer FX, Kriska AM, Ballantyne CM Look AHEAD (Action for Health in Diabetes) Research Group. Lifestyle intervention and/or statins for the reduction of C-reactive protein in type 2 diabetes: from the look AHEAD study. Obesity (Silver Spring) 2013;21:944–950. doi: 10.1002/oby.20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105:141–50. doi: 10.1016/j.diabres.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 95.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammation activation. Nat Immunol. 2010;110:136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 96.Kavouras SA, Panagiotakos DB, Pitsavos C. Physical activity, obesity status, and glycemic control: the ATTICA study. Med Sci Sports Exerc. 2007;39:606–11. doi: 10.1249/mss.0b013e31803084eb. [DOI] [PubMed] [Google Scholar]
- 97.Pannacciulli N, Cantatore FP, Minenna A, Bellacicco M, Giorgino R, De Pergola G. C-reactive protein is independently associated with total body fat, central fat and insulin resistance in adult women. Int J Obes Relat Metab Disord. 2001;25:1416–20. doi: 10.1038/sj.ijo.0801719. [DOI] [PubMed] [Google Scholar]
- 98.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–213. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 99.Cesari M, Penninx BW, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, Rubin SM, Ding J, Simonsick EM, Harris TB, Pahor M. Inflammatory markers and onset of cardiovascular events: results from the health ABC Study. Circulation. 2003;108:2317–2322. doi: 10.1161/01.CIR.0000097109.90783.FC. [DOI] [PubMed] [Google Scholar]
- 100.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 101.Van der Poll T, Romijn JA, Endert E, Borm JJ, Büller HR, Sauerwein HP. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol. 1998;24:E457–E465. doi: 10.1152/ajpendo.1991.261.4.E457. [DOI] [PubMed] [Google Scholar]
- 102.Collier B, Dosssett LA, May AK, Diaz JJ. Glucose control and the inflammatory response. Nutr Clin Pract. 2008;23:3–15. doi: 10.1177/011542650802300103. [DOI] [PubMed] [Google Scholar]
- 103.Tatsch E, Bochi GV, Piva SJ, De Carvalho JA, Kober H, Torbitz VD, Duarte T, Signor C, Coelho AC, Duarte MM, Montagner GF, Da Cruz IB, Moresco RN. Association between DNA strand breakage in type 2 diabetes. Mutat Res. 2012;732:16–20. doi: 10.1016/j.mrfmmm.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 104.Salazar J, Martínez MS, Chávez M, Toledo A, Añez R, Torres Y, Apruzzese V, Silva C, Rojas J, Bermúdez V. C-reactive protein: clinical and epidemiological perspectives. Cardiol Res Pract. 2014;2014:605810. doi: 10.1155/2014/605810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage: antioxidant response in postprandial hyperglycaemia in type 2 diabetes mellitus. Brit J Diab and Vasc Dis. 2011;11:53–54. [Google Scholar]
- 106.Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. 2012;30:49–59. doi: 10.1111/j.1755-5922.2010.00218.x. [DOI] [PubMed] [Google Scholar]
- 107.Donath MY, Dalmas É, Sauter NS, Böni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 2013;17:860–872. doi: 10.1016/j.cmet.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 108.Al Hannan F, Culligan KG. Human resistin and the RELM of inflammation in diabesity. Diab Metab Syndr. 2015;7:54. doi: 10.1186/s13098-015-0050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Codoñer-Franch P, Valls-Bellés V, Arilla-Codoñer A, Alonso-Iglesias E. Oxidant mechanisms in childhood obesity: the link between inflammation and oxidative stress. Trans Res. 2011;158:369–384. doi: 10.1016/j.trsl.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 110.Dinarello CA, Donath MY, Mandrup-Poulsen T. Role of Il-1 beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:314–21. doi: 10.1097/MED.0b013e32833bf6dc. [DOI] [PubMed] [Google Scholar]
- 111.Piro S, Anello M, Di Pietro C, Lizzio MN, Patanè G, Rabuazzo AM, Vigneri R, Purrello M, Purrello F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism. 2002;51:1340–7. doi: 10.1053/meta.2002.35200. [DOI] [PubMed] [Google Scholar]
- 112.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. HOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 114.Svistounov D, Smedsrød B. Hepatic clearance of advanced glycationend products myth or truth? J Hepatol. 2004;41:1038–40. doi: 10.1016/j.jhep.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 115.Guven A, Yavuz O, Cam M, Ercan F, Bukan N, Comunoglu C, Gokce F. Effects of melatonin on STZ-induced diabetic liver injury in rats. Acta Histochem. 2006;108:85–93. doi: 10.1016/j.acthis.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 116.Mohamed J, Nazratun Nafizah AH, Zariyantey AH, Budin SB. Mechanisms of diabetes-induced liver damage. Sultan Qaboos Univ Med J. 2016;16:e132–41. doi: 10.18295/squmj.2016.16.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Figgers CL. Dietary caloric restriction modifies inflammatory responses in the livers of STZ-induced diabetic rats. Nutr Res. 2006;26:483–490. [Google Scholar]
- 118.Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30:734–43. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 119.Caldwell RB, Bartoli M, Behzadian MA. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Tragets. 2005;6:511–524. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- 120.Jardim J, Trindade E, Carneiro F, Dias JA. Hepatomegaly in Type-1 diabetes mellitus: when to suspect of glycogenic hepatopathy? J Medical Cases. 2013;4:726–728. [Google Scholar]
- 121.Anne R, Sonia P, Patrice F. Role of inflammation in diabetic retinopathy. Int J Mol Sci. 2018;19:942–73. doi: 10.3390/ijms19040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Du Y, Miller CM, Kern TS. Hyperglycaemia increases mitochondrial superoxide in retina and retinal cells. Free Rad Biol Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 123.Santos JM, Mohammad G, Zhong Q, Kowluru RA. Diabetic retinopathy, superoxide damage and antioxidants. Curr Pharm Biotechnol. 2011;12:352–61. doi: 10.2174/138920111794480507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hammes H. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37:39–43. doi: 10.1055/s-2005-861361. [DOI] [PubMed] [Google Scholar]
- 125.Raine AF. Epidemiology, development and treatment of end-stage renal failure in type 2 (non-insulin dependent) diabetic patients in europe. Diabetologia. 1993;36:1099–1104. doi: 10.1007/BF02374505. [DOI] [PubMed] [Google Scholar]
- 126.Wolf G, Ziyadeh FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 2007;106:p26–31. doi: 10.1159/000101797. [DOI] [PubMed] [Google Scholar]
- 127.Diabetes Control and Complications Trial Research Group. Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, Rand L, Siebert C. The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 128.Kafle D, Signh N, Singh SK, Singh N, Islam N. Relationship between hyperglycaemia, inflammation and oxidative stress in type-2 diabetic nephropathy subjects. Int J Pharm and Biol Arch. 2012;3:1203–1206. [Google Scholar]
- 129.Kiritoshi S, Nishikawa T, Sonoda K, Kukidome D, Senokuchi T, Matsuo T, Matsumura T, Tokunaga H, Brownlee M, Araki E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes. 2003;52:2570–7. doi: 10.2337/diabetes.52.10.2570. [DOI] [PubMed] [Google Scholar]
- 130.Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, Endoh M, Nomoto Y, Sakai H. In situ hydridization of interleukin6 in diabetic nephropathy. Diabetes. 1995;44:1233–8. doi: 10.2337/diab.44.10.1233. [DOI] [PubMed] [Google Scholar]
- 131.Wong CK, Ho AW, Tong PC, Yeung CY, Kong AP, Lun SW, Chan JC, Lam CW. Aberant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetes with nephropathy. Clin Exp Immunol. 2007;149:123–31. doi: 10.1111/j.1365-2249.2007.03389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Apfel SC. Nerve regeneration in diabetic neuropathy. Diabetes Obes Metab. 1999;1:3–11. doi: 10.1046/j.1463-1326.1999.00006.x. [DOI] [PubMed] [Google Scholar]
- 133.Calcutt NA. Potential mechanisms of neuropathic pain in diabetes. Int Rev Neurobiol. 2002;50:205–228. doi: 10.1016/s0074-7742(02)50078-7. [DOI] [PubMed] [Google Scholar]
- 134.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004;25:612–28. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 135.Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis. 1999;6:347–63. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- 136.Metodiewa D, Kośka C. Reactive oxygen species and reactive nitrogen species: relevance to cyto-neuron toxic events and neurological disodera. An overview. Neurotox Res. 2000;1:197–233. doi: 10.1007/BF03033290. [DOI] [PubMed] [Google Scholar]
- 137.American Diabetes Association. Standards of medical care in diabetes-2007. Diabetes Care. 2007;30(Suppl 1):S4–S41. doi: 10.2337/dc07-S004. [DOI] [PubMed] [Google Scholar]
- 138.Pazdro R, Burgess JR. The role of vitamin E and oxidative stress in diabetes complications. Mech Ageing Dev. 2010;131:276–86. doi: 10.1016/j.mad.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 139.Varga ZV, Giricz Z, Liaudet L, Haskó G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cel;l death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:232–42. doi: 10.1016/j.bbadis.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Woodward M, Rumley A, Tunstall-Pedoe H, Lowe GD. Associations of blood rheology and interleuklin-6 with cardio-vascular risk factors and prevalent cardio-vascular disease. Br J Haematol. 1999;104:246–57. doi: 10.1046/j.1365-2141.1999.01158.x. [DOI] [PubMed] [Google Scholar]
- 141.Orchard TJ, Costacou T, Kretowski A, Nesto RW. Type 1 diabetes and coronary artery disease. Diabetes Care. 2006;29:2528–38. doi: 10.2337/dc06-1161. [DOI] [PubMed] [Google Scholar]
- 142.Ding H, Triggle CR. Endothelial cell dysfunction and the vascular complications associated with type 2 diabetes: assessing the health of the endothelium. Vasc Health Risk Manag. 2005;1:55–71. doi: 10.2147/vhrm.1.1.55.58939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Winer N, Sowers JR. Epidemiology of diabetes. J Clin Pharmacol. 2004;44:397–405. doi: 10.1177/0091270004263017. [DOI] [PubMed] [Google Scholar]
- 144.McDonnell-Dowling K, Kelly JP. The role of oxidative stress in methamphetamine-induced toxicity and sources of variation in the design of animal studies. Curr Neuropharmacol. 2017;15:300–314. doi: 10.2174/1570159X14666160428110329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Poblete-Aro C, Russell-Guzmán J, Parra P, Soto-Muñoz M, Villegas-González B, Cofré-Bolados C, Herrera-Valenzuela T. Oxidative stress in diabetes mellitus. Int J Biol Chem. 2015;9:92–109. doi: 10.4067/s0034-98872018000300362. [DOI] [PubMed] [Google Scholar]
- 146.Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol. 2013;62:667–76. doi: 10.1016/j.jacc.2013.03.089. [DOI] [PubMed] [Google Scholar]
- 147.Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. 2002;1:1. doi: 10.1186/1475-2840-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]