

Abstract
BACKGROUND & AIMS:
Interleukin 6 (IL6) and tumor necrosis factor contribute to the development of colitis-associated cancer (CAC). We investigated these signaling pathways and the involvement of G protein subunit alpha i1 (GNAI1), GNAI2, and GNAI3 in the development of CAC in mice and humans.
METHODS:
B6;129 wild-type (control) or mice with disruption of Gnai1, Gnai2, and/or Gnai3 or conditional disruption of Gnai2 in CD11c+ or epithelial cells were given dextran sulfate sodium (DSS) to induce colitis followed by azoxymethane (AOM) to induce carcinogenesis; some mice were given an antibody against IL6. Feces were collected from mice, and the compositions of microbiomes were analyzed by polymerase chain reactions. Dendritic cells (DCs) and myeloid-derived suppressor cells (MDSCs) isolated from spleen and colon tissues were analyzed by flow cytometry. We performed immunoprecipitation and immunoblot analyses of colon tumor tissues, MDSCs, and mouse embryonic fibroblasts to study the expression levels of GNAI1, GNAI2, and GNAI3 and the interactions of GNAI1 and GNAI3 with proteins in the IL6 signaling pathway. We analyzed the expression of Gnai2 messenger RNA by CD11c+ cells in the colonic lamina propria by PrimeFlow, expression of IL6 in DCs by flow cytometry, and secretion of cytokines in sera and colon tissues by enzyme-linked immunosorbent assay. We obtained colon tumor and matched nontumor tissues from 83 patients with colorectal cancer having surgery in China and 35 patients with CAC in the United States. Mouse and human colon tissues were analyzed by histology, immunoblot, immunohistochemistry, and/or RNA-sequencing analyses.
RESULTS:
GNAI1 and GNAI3 (GNAI1;3) double-knockout (DKO) mice developed more severe colitis after administration of DSS and significantly more colonic tumors than control mice after administration of AOM plus DSS. Development of increased tumors in DKO mice was not associated with changes in fecal microbiomes but was associated with activation of nuclear factor (NF) ĸB and signal transducer and activator of transcription (STAT) 3; increased levels of GNAI2, nitric oxide synthase 2, and IL6; increased numbers of CD4+ DCs and MDSCs; and decreased numbers of CD8+ DCs. IL6 was mainly produced by CD4+/CD11b+, but not CD8+, DCs in DKO mice. Injection of DKO mice with a blocking antibody against IL6 reduced the expansion of MDSCs and the number of tumors that developed after CAC induction. Incubation of MDSCs or mouse embryonic fibroblasts with IL6 induced activation of either NF-ĸB by a JAK2-TRAF6-TAK1-CHUK/IKKB signaling pathway or STAT3 by JAK2. This activation resulted in expression of GNAI2, IL6 signal transducer (IL6ST, also called GP130) and nitric oxide synthase 2, and expansion of MDSCs; the expression levels of these proteins and expansion of MDSCs were further increased by the absence of GNAI1;3 in cells and mice. Conditional disruption of Gnai2 in CD11c+ cells of DKO mice prevented activation of NF-ĸB and STAT3 and changes in numbers of DCs and MDSCs. Colon tumor tissues from patients with CAC had reduced levels of GNAI1 and GNAI3 and increased levels of GNAI2 compared with normal tissues. Further analysis of a public human colorectal tumor DNA microarray database (GSE39582) showed that low Gani1 and Gnai3 messenger RNA expression and high Gnai2 messenger RNA expression were significantly associated with decreased relapse-free survival.
CONCLUSIONS:
GNAI1;3 suppresses DSS-plus-AOM–induced colon tumor development in mice, whereas expression of GNAI2 in CD11c+ cells and IL6 in CD4+/CD11b+ DCs appears to promote these effects. Strategies to induce GNAI1;3, or block GNAI2 and IL6, might be developed for the prevention or therapy of CAC in patients.
Keywords: CAC, Mouse Model, IBD, Transcription Factor
Graphical Abstract
About 20% of patients with ulcerative colitis develop colitis-associated cancer CAC over the course of 30 years.1 Genetic and pharmacologic evidence shows that proinflammatory cytokines (interleukin [IL] 6 and tumor necrosis factor [TNF]), transcription factors (nuclear factor kappa B (NF-ĸB) and signal transducer and activator of transcription 3 [STAT3]), and immune cells (dendritic cells [DCs] and MDSCs) play a critical role in CAC.2,3 IL6 and TNF are elevated in CAC and inflammatory bowel disease (IBD) patients,4,5 and their blockade diminishes CAC tumorigenesis in an azoxymethane (AOM)–dextran sulfate sodium (DSS)-induced CAC mouse model.3,5,6 IL6 and TNF are mainly produced by myeloid cells, including CD11c+ DCs in the tumor microenvironment. DCs, including CD8+ and CD4+/CD11b+ DCs, are primary antigen-presenting cells but have distinct functions. CD4+ DCs mainly produce IL6, whereas CD8+ DCs mainly produce IL12, preferentially activate CD8+ T cells, and are closely associated with the prognosis of multiple cancers.7–9 MDSCs are defined as Gr-1+CD11b+ cells and mainly include granulocytic MDSCs (CD11b+Ly6G+Ly6Clow/−) and monocytic MDSCs (CD11b+Ly6C+Ly6G−) and contribute to tumorigenesis.10–12
IL6 and TNF promote CAC through activation of the NF-ĸB–IL6–STAT3 axis.3,5 TNF sequentially activates TNF receptor associated factor 2 (TRAF2), transforming growth factor activated protein kinase-1 (TAK1), and the Iĸbα kinases α (CHUK) and β (IKĸB) that phosphorylate IĸBα [serine(S)32], resulting in NF-ĸB nuclear translocation and transcriptional activation.13–17 This activation can also be induced by NF-ĸBp65(S536) phosphorylation.18 Thus, IĸBα(S32) or NF-ĸBp65(S536) phosphorylation is widely used for measuring NF-ĸB activation. NF-ĸB triggers expression of IL6, which binds to IL6 receptor (IL6R) α, leading to activation of GP130 and Janus kinases 1 and 2 (JAK1 and JAK2) that phosphorylate STAT3[tyrosine(Y)705], resulting in STAT3 transcriptional activation. Thus, STAT3(Y705) phosphorylation is generally used for measuring STAT3 transcriptional activity. Deletion of STAT3 in the epithelial cells markedly protects mice from CAC upon AOM–DSS challenge.3,19 IL6Rα blockade or soluble GP130-Fc also suppresses CAC.3,19,20 Likewise, IKkB deletion in the epithelial cells or myeloid cells decreases IL6 production and CAC tumorigenesis.2,21,22
GNAI1, GNAI2, and GNAI3 are abundantly expressed in immune cells and participate in G protein-coupled receptor (GPCR) and non-GPCR signaling pathways.23,24 GNAI2 couples to CXCR2 governing neutrophil trafficking; GNAI2 ablation leads to spontaneous colitis and CAC in aged mice under certain environments.8,25–28 GNAI1 and GNAI3 (GNAI1;3) regulate cytokine responses to bacterial products.29,30 We previously showed that GNIA2 negatively regulated GNAI1;3, which were critical for epiderma growth factor receptor–mediated activation of Akt and extracellular signal regulated kinase 1/2, which are pivotal for cell growth, survival, and tumorigenesis.23,24,31 We hypothesized that GNAI1;3 mediate CAC tumorigenesis promoted by GNAI2 deficiency. Thus, we generated a combination of GNAI1, 2, and 3 individual, double-knockout (DKO) or triple conditional knockout (TKO) mice because total TKO mice are embryonically lethal.31 Despite a minor bone defect, GNAI1;3DKO and TKO mice were normal and reproductive. Although GNAI2KO mice (n = 40) developed spontaneous colitis, they did not develop colonic tumors during 9 months of observation under our environmental conditions (Guo and Chu, unpublished data 2013). Therefore, whether GNAI proteins by themselves are implicated in CAC tumorigenesis remains unclear.
Materials and Methods
Mice
Mice (female and male) on the B6;129 background were housed in a specific pathogen–free vivarium at the University of Hawaii (UH) according to National Institutes of Health guidelines. All experimental procedures were approved by the Institutional Animal Care and Use Committee. Mice with deletion of GNAI1, GNAI2, GNAI3, or GNAI1;3 and their genotyping have been described previously.32,33 To generate GNAI conditional TKO (GNAI123TKO) mice with deletion of GNAI2 in Itgax (CD11c) (GNAI123cTKO)–or Villin (GNAI123vilTKO)–expressing cells on the GNAI1;3DKO background, GNAI2flx/flx mice33 were crossed with GNAI1;3DKO mice and ItgaxCre or VillinCre mice (The Jackson Laboratory, Bar Harbor, ME). The genotypes of the mice were confirmed by polymerase chain reaction, and the absence of GNAI proteins was confirmed by immunoblotting (IB) (Supplementary Figure 1A–C).
Human Specimens
Eighty-three human specimens from the First Affiliated Hospital of Nanjing Medical University (FAHNMU) were used in this study. Fifty-seven fresh colon cancer tumor tissues and matched nontumorous colon tissues were collected with patients’ consent after surgical resections in 2016. Twenty-six human colon cancer tissues were retrospectively obtained from surgical resections that were fixed in buffered formalin, embedded in paraffin, and stored at the FAHNMU Tissue Repository. Ten normal, 9 colitis, 20 colitis-associated dysplasia (CAD), and 35 CAC colon tissue samples from Hawaii and Chicago were retrospectively obtained from surgical biopsies or resections and stored at the UH Cancer Center Tissue Repository and University of Chicago School of Medicine Tissue Repository. The corresponding clinical data were obtained from medical records and deidentified. The studies were approved by the institutional ethics committee of FAHNMU and the institutional review board and committee on human studies at UH and at the University of Chicago.
Results
GNAI1;3 Suppress Colitis and Associated Cancer Independent of the Microbiota
GNAI1;3 regulate proinflammatory cytokine responses to bacterial products.30 We determined if GNAI1;3 are implicated in colitis in a DSS-induced colitis mouse model. Upon DSS challenge, male GNAI1;3DKO mice exhibited more weight loss, shorter colon length, and a higher colitis severity score than wild-type (WT) controls (Figure 1A–C). GNAI1;3DKO colons showed more severe inflammation with more cryptitis/crypt abscess formation, erosion, or ulceration compared with WT controls (Figure 1D). GNAI1;3DKO colons produced more granulocyte and macrophage colony-stimulating factor (GM-CSF), IL6, and TNF than WT controls (Figure 1E).
Figure 1.

GNAI1;3 inhibit colitis, and their ablation promotes the initiation and progression of CAC, which is independent of microbiota. (A) Body weight changes during the course of acute colitis with DSS (n = 6). (B) The change of colon length. (C) Colitis severity score. (D) H&E-stained representative images of the colons of WT and GNAI1;3DKO mice with histology score (n = 6) on day 10. Scale bars: upper, 100 μm; lower, 25 μm. (E) The levels of GM-CSF, IL6, and TNF in the supernatant of colon culture (n = 3). (F) Representative images of colonic tumors from WT and GNAI1;3DKO mice. (G) Tumor number/colon of WT (n 15), GNAI1KO (n = 15), GNAI3KO (n = 13), or GNAI1;3DKO (n = 14) mice. (H) number/colon of WT (n = 5) and GNAI1;3DKO (n = 5) on days 0,13,49 and WT (n = 7) and GNAI3 (n = 7) on day 93. (I) Overall survival of challenged WT andGNAI1;3DKOmice (n = 12/group).(J) Tumor number/colon of WT (n = 21) and GNAI1;3 DKO mice (n = 20) under the CH condition or WT (n = 11) and GNAI1;3 DKO mice (n = 12) under the SH condition. (K) Polymerase chain reaction analysis of 16S ribosomal DNA of the indicated bacteria in the stool isolated from the indicated mice. All analyzed data are mean ± standard error of the mean (A, G, and H) or standard deviation (B–E, J). *P < .05, **P < .01, ***P < .001, ****P < .0001; 2-tailed Student t test. Bact B, Bacteroides species; Clostri B, Clostridiales species; Lacto B, Lactobacillaceae species; ns, not significant; SFB, segmented filamentous bacteria species.
In humans, patients with ulcerative colitis develop CAC by poorly understood mechanisms.1 We determined whether GNAI1 and GNAI3 are implicated in CAC in an AOM–DSS-induced CAC mouse model (Supplementary Figure 2A), which resembles many aspects of the pathogenic process of human ulcerative colitis and CAC.34 During the course of the challenge, the body weight of GNAI1;3DKO mice was significantly reduced compared with WT controls; the affected mutants had chronic colitis with progression to colitis-associated tumorigenesis (Figure 1F–H and Supplementary Figure 2B and C). GNAI3KO mice exhibited many more colonic tumors than GNAI1KO and WT mice (Figure 1F and G). GNAI1;3DKO mice exhibited even higher tumor multiplicity with shorter colons than single-KO mice (Figure 1F and G and Supplementary Figure 2D). The time-course experiment showed that up to 49 days, no apparent macroscopic tumors were noticed in the colon from WT mice; conversely, multiple tumors appeared in the colons of some of GNAI1;3DKO mice (Supplementary Figure 2E and F). By 93 days, tumor numbers were markedly increased in these mutants and only slightly increased in WT controls (Figure 1H and Supplementary Figure 2F). Pathologic analysis showed low-grade dysplasia tumors in the colonic mucosa of GNAI1;3DKO, but not WT, mice on day 49 (Supplementary Figure 2G). By 93 days, both WT and GNAI1;3DKO mice had high-grade dysplasia (HGD)/intramucosal carcinoma (Supplementary Figure 2G). During the 9 months of observation, GNAI1;3DKO mice exhibited a shorter lifespan (Figure 1I).
To determine if the tumorigenic phenotype in GNAI1;3DKO mice is dependent on the composition of the microbiota in the gut, we cohoused (CH) or separately housed (SH) WT and GNAI1;3DKO mice prior to and during AOM–DSS challenge. GNAI1;3DKO mice exhibited a significantly higher tumor burden than WT controls under either CH or SH conditions (Figure 1J). The numbers of colonic tumors in GNAI1;3DKO mice under the CH condition were not significantly different from those under the SH condition (Figure 1J). Moreover, although more severe colitis was observed in GNAI1;3DKO mice compared with WT controls, there were no significant differences in the colitis histology scores for GNAI1;3DKO mice under the CH or SH conditions (Supplementary Figure 2H). Furthermore, analysis of the compositions of the microbiota in the stools of the mice showed that both WT and GNAI1;3DKO mice under the CH and SH conditions had equal levels of Bacteroides, Clostridiales, Lactobacillaceae, and segmented filamentous bacteria species (Figure 1K). Therefore, these data suggest that enhanced colitis and tumorigenesis in GNAI1;3DKO mice are genotype specific but not microbiota specific.
GNAI1;3 Inhibit MDSC and CD4+ DC Expansion and Accumulation but Increase CD8+ DC Counts
To elucidate how GNAI1;3 deficiency promotes colitis and associated tumorigenesis, we examined gene expression profiles, which indicate the biological consequence of signaling pathways activated by GNAI1;3 loss, via an RNA-sequencing analysis with RNA isolated from the colonic tumor tissues of the AOM–DSS-challenged mice and the colon tissues of unchallenged mice. The expression levels of some inflammation- and cancer-associated genes (e.g., Ccl6, Ly6c1/Ly6c2, Mcl1, and TRAF6) were more induced, but Itgax was diminished in GNAI1;3DKO tumors compared with normal controls (Supplementary Figure 3A). Because Ly6C (Ly6c1/2) is a key marker of MDSCs and CD11c (Itgax) is a key marker of DCs, this result suggests that GNAI1;3 may modulate the expansion and accumulation of DCs and MDSCs.
We initially examined splenic DCs, including CD4+ and CD8+ DCs, and MDSCs in DSS-treated mice. Loss of GNAI1;3 led to significant increases in CD4+ DCs and CD11b+Ly6G+ cells but not MHCII+CD11c+ and CD8+ DCs compared with WT controls (Figure 2A). Next, we assessed splenic DCs and MDSCs in AOM–DSS-treated mice. Challenged GNAI1;3DKO mice exhibited splenomegaly (Supplementary Figure 3B) and 3–8-fold increases in splenic CD11b+Gr-1+-MDSCs, CD11b+Ly6C+–monocytic MDSCs, and CD11b+Ly6ClowLy6G+–granulocytic MDSCs compared with WT controls (Figure 2B). Temporal experiments further showed a significantly higher percentage of splenic MDSCs in GNAI1;3DKO mice than WT controls (Figure 2C) and showed a correlation of splenic MDSC expansion with the increased colonic tumor multiplicity (Figure 1H and Supplementary Figure 2F).
Figure 2.

GNAI1;3 deficiency stimulates MDSC expansion but reduces CD8+ DC counts. (A) Representative FACS plots of CD4+ DCs, CD8+ DCs, and CD11b+Ly6G+ MDSCs, together with quantitative analysis of major histocompatibility complex (MHC) II+CD11c+ DCs in the spleen of mice treated with DSS on day 10 (n = 4–6). (B) Representative FACS plots of splenic CD11b+Gr-1+ GNAI1;3DKO mice on day 100 (n = 5). (C) Representative FACS plots and quantitative analysis of splenic CD3–CD11b+Gr-1+ MDSCs from unchallenged (day 0, n = 5) or AOM–DSS-challenged WT and GNAI1;3DKO mice on day 49 (n = 5) or day 93 (n = 7). (D) Representative FACS plots of splenic CXCR2+ MDSCs of AOM–DSS-challenged WT and GNAI1;3DKO mice on day100 (n = 5). (E) IHC analysis of AOM–DSS-challenged mouse colonic tissues (n = 5) for granulocyte receptor-1 antigen (Gr-1) on day 100. Scale bars, 25 μm. (F) Representative FACS plots of CD11b+CD11c+ DCs and CD11b+Ly6G+ MDSCs in colonic LP of AOM–DSS-challenged WT and GNAI1;3DKO mice on day 63 (n = 5). (G) Percentages of CD3–MHCII+CD11b+CD11c+, CD4+CD11c+, and CD8+CD11c+ DCs (day 100) are shown (n = 5). All data shown are representative of 2 or 3 independent experiments. Data are mean ± standard deviation. *P < .05, 2-tailed Student t test. FACS, fluorescence-activated cell sorting.
Because NF-ĸB–regulated CXCR2 is critical for MDSC traf-ficking to the colon and subsequent CAC tumorigenesis,12 we examined its expression on MDSCs. CXCR2 expression was significantly higher in GNAI1;3DKO MDSCs than WT controls (Figure 2D). Consequently, a substantial infiltration of Gr-1+ MDSCs in the colon of AOM–DSS-challenged GNAI1;3DKO mice was shown by immunohistochemistry (IHC) (Figure 2E). Likewise, the high levels of GNAI1;3DKO CD11b+Ly6G+ MDSCs were found in the colonic lamina propria (LP) compared with WT controls (Figures 2F).
Furthermore, we assessed DCs in both the spleen and LP. In the spleen, both WT and GNAI1;3DKO mice showed similar numbers of MCHII+CD11b+CD11c+ DCs (Figure 2G). However, significantly lower CD8+ DC but higher CD4+ DC percentages were observed in GNAI1;3DKO mice (Figure 2G). In the LP, CD11b+ DCs were significantly accumulated in GNAI1;3DKO mice (Figure 2F). Because CD11c+ cells are major producers of IL6, which is important for CAC development,3 we assessed IL6 expression in DCs. GNAI1;3DKO CD11b+/CD4+ DCs, but not CD8+ DCs, produced high levels of IL6 (Supplementary Figure 3C), suggesting that IL6-producing CD4+ DCs play an important role in CAC promoted by GNAI1;3 deficiency.
Finally, we evaluated whether GNAI1;3 deficiency has an impact on frequencies of B cells and T cells in AOM–DSS-treated mice. There were no significant differences in these lymphocytes between treated WT and GNAI1;3DKO mice (Supplementary Figure 3D–F).
IL6 Activation of NF-ĸB and STAT3 Is Enhanced, and IL6 Is Critical for MDSC Expansion and CAC Tumorigenesis in GNAI1;3DKO Mice
These observations led us to test our hypothesis that IL6 levels are elevated in GNAI1;3DKO mice. IL6, but not TNF, showed an apparent increase in the blood of GNAI1;3DKO mice (Supplementary Figure 4A). The colonic secretion levels of IL6, together with TNF and GM-CSF, were significantly increased in GNAI1;3DKO mice compared with WT controls (Supplementary Figure 4B). These results are consistent with the high accumulation of CD4+/CD11b+ DCs and MDSCs, which are expanded by these cytokines,35 in the tumor microenvironment of GNAI1;3DKO mice (Figure 2E and F).
Because IL6 is essential for CAC development3, we tested our hypothesis that IL6 mediates MDSC expansion and CAC development in GNAI1;3DKO mice via an IL6 neutralization strategy (Figure 3A). IL6 blockage markedly diminished CAC tumor burden and MDSC expansion in the spleen and LP of GNAI1;3DKO mice (Figure 3B, D, and E). IL6 blockade largely abolished colonic bioavailability of IL6 but not GMCSF (Figure 3C). Moreover, in vitro IL6 expansion of bone marrow–derived MDSCs (BMDSCs) differentiated by GMCSF was significantly enhanced in the absence of GNAI1;3 (Figure 3F and G). Our data suggest that IL6 is up-regulated and mediates MDSC expansion and CAC tumorigenesis in GNAI1;3DKO mice.
Figure 3.

IL6 mediates colitis-associated tumorigenesis and MDSC expansion in GNAI1;3DKO mice, and its activation of NF-ĸB and expansion of MDSCs are enhanced by GNAI1;3 deficiency. (A) Schematic intraperitoneal injection schedule of IL6 monoclonal antibody (IL6mAb) and its isotype immunoglobulin G control (IgG). (B) Left panel shows tumor number/colon of AOM–DSS-challenged mice with injection on day 63 (n = 6–8). Right panel shows representative images of colonic tumors of the mice. (C) Levels of IL6 and GM-CSF in the supernatant of colon culture from indicated mice in (B) (n = 3). (D, E) Representative FACS plots of CD45+CD3−CD11b+Gr-1+ and CD45+CD3–CD11b+Ly6G+ MDSCs in (D) the LP or (E) the spleen of the mice in part B (n = 3–6). (F) WT BMDSCs were differentiated by GM-CSF in the presence or absence of IL6 and then assayed for CD11b+Gr-1+ MDSCs (n = 3). (G) Representative FACS plots of CD11b+Gr-1+ BMDSCs of WT and GNAI1;3DKO mice (n = 3) differentiated with GM-CSF plus IL6 in the presence or absence of BP-1–102 (5 μmol/L) (left). Quantitative analysis of CD11b+Gr-1+ BMDSCs shown in the left panel is graphed on the right. (H) IB analysis for the levels of p-STAT3(Y705), STAT3, INOS, GN1AI, GNAI3, and β-actin in indicated WT and G GNAI1;3DKO BMDSCs. (I) IB analysis for expression of INOS, p-STAT3, and β-actin in the indicated mouse colonic tissues. (J) IB analysis for the levels of INOS, p-NF-ĸBp65(S536), and bactin in WT and IKKBKO MEFs. (K) IB analysis for the levels of INOS, p-IĸBα(S32), and β-actin in WT and GNAI1;3DKO MEFs. (L) IB analysis for the levels of INOS, p-NF-ĸBp65(S536), NF-ĸB, and β-actin in GNAI1;3DKO MEFs in the presence or absence of ML-120. Data are mean ± standard deviation. **P < .01, ***P < .001, 2-tailed Student t test. Except in A–E, all data are representative of 2 or 3 independent experiments. mAb, monoclonal antibody; p-, phosphorylated.
To understand how IL6 promotes MDSC expansion and CAC in GNAI1;3DKO mice, we used both genetic and pharmacologic strategies to determine if IL6 directly activates NF-ĸB and whether GNAI1;3 block IL6-induced NF-ĸB and STAT3 activation. In BMDSCs, IL6-induced activation of STAT3 and up-regulation of nitric oxide synthase 2/inducible nitric oxide synthase (INOS), which is a target of NF-ĸB and is important for MDSC-induced protumor activity and initiation of colorectal cancer,35,36 were markedly enhanced in the absence of GNAI1;3 (Figure 3H). A specific NF-ĸB–STAT3 inhibitor, BP1–102,37 severely impaired IL6 expansion of BMDSCs and the subsequent enhancement by GNAI1;3 deficiency (Figure 3G). In the colon of AOM–DSS-challenged mice, GNAI1;3 loss led to significantly greater NF-ĸB and STAT3 activation and INOS up-regulation (Figure 3I and Supplementary Figure 4C). In mouse embryonic fibro-blasts (MEFs), deficiency of IKĸB almost abolished IL6 activation of NF-ĸB and up-regulation of INOS (Figure 3J), suggesting that IKĸB is the upstream kinase for NF-ĸB in IL6 signaling. In GNAI1;3DKO MEFs, IL6-induced activation of IKKB/NF-ĸB, as indicated by phosphorylation of IĸBa(S32) (p-IĸBa[S32]), and INOS up-regulation were markedly enhanced (Figure 3K); all of these were largely diminished by a specific IKĸB inhibitor, M-120B (Figure 3L). Likewise, NF-ĸB activation by TNF was enhanced in GNAI1;3DKO MEFs (Supplementary Figure 4D). Blockade of IKĸB and NF-ĸB activation by N-acetyl-L-cysteine38,39 largely abolished IL6 activation of NF-ĸB in GNAI1;3DKO MEFs (Supplementary Figure 4E).
Overall, these results suggest that IL6-induced NF-ĸB and STAT3 activation and this activation-dependent MDSC expansion are enhanced by GNAI1,3 deficiency and that IL6 is required for MDSC expansion and CAC tumorigenesis in GNAI1;3DKO mice.
GNAI1;3 Interrupt TAK1–TAB1 Interaction, and IL6 Induced JAK Activity-Dependent Complex Formation Among JAK2, TAK1, and TRAF6, Down-regulating NF-ĸB Activation and INOS Expression
To delineate how GNAI1;3 block IL6-induced NF-ĸB activation, we performed a bioinformatics analysis via a biological database and web resource of search tool for the retrieval of interacting genes/proteins (STRING); GNAI1 or GNAI3 was predicted to interact with TAK1 and TAK1 binding 1 (TAB1). Immunoprecipitation (IP)/IB assays showed that GNAI3 was preassociated with TAB1 and TAK1 and that TAK1’s association with GNAI3 was induced by IL6 (Figure 4A and Supplementary Figure 4F). GNAI1;3 loss significantly augmented TAK1’s association with TAB1 (Figure 4A). TAK1 loss led to severely impaired IL6 activation of NF-ĸB and INOS up-regulation but slightly enhanced STAT3 activation (Figure 4B). Specific TAK1 inhibition by 5z-7-oxozeaenol40 largely impaired augmentation of IL6-induced NF-ĸB activation and INOS up-regulation in WT and GNAI1;3DKO cells (Figure 4C and Supplementary Figure 4G). Moreover, IL6-induced TAK1 association with CHUK/IKĸB was further enhanced by GNAI1;3 deficiency (Supplementary Figure 4H).
Figure 4.

GNAI1;3 deficiency leads to enhanced TAK1–TAB1 interaction and IL6-induced JAK activity-dependent interactions among TAK1, JAK2, and TRAF6, which are required for IL6 activation of NF-ĸB, and GNAI2, GP130, and INOS up-regulation. (A) IP/IB and quantified analysis for interactions of TAK1 with TAB1 and GNAI3 (n = 3). (B) IB analysis for INOS, p-IĸBα(S32), p-STAT3(Y705), GNAI2, and β-actin in WT and TAK1KO MEFs. (C) IB analysis for INOS, p-NF-ĸBp65(S536), p-STAT3(Y705), and β-actin in WT MEFs treated with AZD or 5z. (D) IB analysis for INOS, p-IĸBα(S32), p-STAT3(Y705), and b-actin in WT and TRAF6KO MEFs, (E) IP/IB analysis for TAK1–TRAF6 interaction in WT and GNAI1;3DKO MEFs. (F) IP/IB analysis for TAK1–JAK2 interaction in WT and GNAI1;3DKO MEFs. (G) Representative FACS plots of GP130 on the surface of IL6-treated WT and GNAI1;3DKO MEFs. (H) IB for GP130, p-JAK2(Y1007/8), p-STAT3(Y705), and β-actin in treated WT and GNAI1;3DKO MEFs. (I) Representative FACS plots of CD11b+Gr-1+ WT BMDSCs differentiated with GM-CSF and IL6 in the presence or absence of AZD. (J) IB for GNAI2, p-IĸBα(S32), and β-actin in indicated WT and GNAI1;3DKO colonic tissues. (K) IB for GNAI2, p-IĸBα(S32), p-NF-ĸBp65(S536), and β-actin in WT and GNAI1;3DKO MEFs. (L) IB for GNAI2, p-IĸBa(S32), p-JAK2(Y1007/8), p-NF-ĸBp65(S536), p-STAT3(Y705), and β-actin in WT and TRAF6KO MEFs. (M) PrimeFlow analysis of Gnai2 mRNA expression in CD45+CD11c+ colonic LP isolated from indicated WT and GNAI1;3DKO mice on day 10 (n = 6). Data are mean ± standard deviation. *P < .05, **P < .01, ***P < .001; 2-tailed Student t test. All data in A–L are representative of 2 or 3 independent experiments. 5z, 5z-7-oxozeaenol; FACS, fluorescence-activated cell sorting; ns, not significant; p-, phosphorylated.
TRAF2/5 and TRAF6 are required for activation of TAK1 and NF-ĸB by TNF and IL1/toll-like receptors, respectively.16,17 The String program predicted that TRAF6 interacts with JAK2, indicating TRAF6 involvement in IL6 signaling. TRAF6 loss largely abolished NF-ĸB activation and INOS up-regulation but had no apparent effect on STAT3 activation by IL6 (Figure 4D). IL6 induced TRAF6’s association with TAK1 or JAK2 (Supplementary Figure 4I–K). Inhibition of JAKs by AZD1480 (AZD)41 markedly inhibited IL6-induced NF-ĸB activation and INOS up-regulation (Figure 4C). As expected, AZD abolished STAT3 activation (Figure 4C).
To further gain insight into how GNAI1;3 regulate NF-ĸB activation by IL6, we determined the protein–protein interactions among GNAI1;3, JAK2, TAK1, and TRAF6 by IP/IB assays. GNAI1;3 were in a protein complex containing JAK2, TAK1, and TRAF6 (Supplementary Figure 4I and J). GNAI1;3 absence potentiated TAK1’s interactions with JAK2 or TRAF6 (Figure 4E and F). Inactivation of JAK2 by AZD diminished IL6-induced interactions of TAK1 with JAK2 or TRAF6 in both WT and GNAI1;3DKO cells (Figure 4E and F). Taken together, these results suggest that GNAI1;3 block TAK1’s interaction with TAB1 and interfere with the complex formation among JAK2, TAK1, and TRAF6 leading, to diminished IL6 activation of IKĸB/NF-ĸB and INOS up-regulation.
GNAI2, GP130, and MDSCs Are Targets of the IL6–JAK–NF-ĸB Axis, Which Is Attenuated by GNAI1;3
The enhanced activation of IL6 signaling in GNAI1;3DKO cells prompted us to examine the expression of IL6 receptor GP130. GP130 basal level was higher in GNAI1;3DKO cells than in WT controls and was strikingly increased by IL6 in GNAI1;3DKO cells (Figure 4G and H). This up-regulation was markedly inhibited by AZD (Figure 4H and Supplementary Figure 5A), indicating that JAK2 is required for increased GP130 expression in GNAI1;3DKO cells. GeneCards database analysis indicated that the promoter of Il6st contains both NF-ĸB and STAT3 binding sites. Inhibition of both NF-ĸB and STAT3 by AZD or NF-ĸB by N-acetyl-L-cysteine markedly diminished IL6-induced GP130 up-regulation (Figure 4H and Supplementary 5B). AZD also inhibited IL6-induced BMDSC expansion in a dose-dependent manner (Figure 4I).
GNAI1;3 are up-regulated in GNAI2KO cells,23,31 and GNAI2 expression is NF-ĸB dependent.42 We surmised that GNAI2 is up-regulated in GNAI1;3DKO cells and mice. Indeed, a high level of GNAI2 was observed in GNAI1;3DKO colon tissues compared with WT controls (Figure 4J and Supplementary 5C). GNAI1;3 loss led to enhanced IL6-induced NF-ĸB activation and GNAI2 up-regulation in MEFs (Figure 4K), which was largely impaired by IKĸB/NF-ĸB inhibition (Supplementary Figure 5D). Inhibition of JAK2 or loss of TAK1 or TRAF6 led to markedly diminished GNAI2 up-regulation by IL6 (Figure 4B and L and Supplementary Figure 5E). Conversely, the GNAI–adenosine 3′5′-cyclic monophosphate–protein kinase A axis had no apparent effect on IL6-induced GNAI2 up-regulation (Supplementary Figure 5F).
Because high IL6 level was detected in GNAI1;3DKO CD11c+ cells (Supplementary Figure 3C), we determined whether Gnai2 messenger RNA (mRNA) is also up-regulated in CD11c+ cells of the LP via PrimeFlow RNA analysis. As shown, GNAI1;3 deficiency led to significant up-regulation of Gnai2 mRNA in CD11c+ but not CD11b+ cells, which was further induced by DSS treatment (Figure 4M, and Supplementary Figure 5G and H). Gnai1 mRNA or Gnai3 mRNA was not significantly up-regulated (Supplementary Figure 5I). Collectively, these results suggest that GNAI1;3 deficiency augments GNAI2 up-regulation, which requires JAK-TAK1-TRAF6–dependent NF-ĸB activation in IL6 signaling.
GNAI2 Promotes CAC, and Its Deletion in CD11c+ Cells, but not Epithelial Cells, Mitigates CAC Tumorigenesis, NF-ĸB/STAT3 Activation, MDSC, and CD4+ DC Expansion and Accumulation and Restores CD8+ DC Counts in GNAI1;3DKO Mice
To discover whether GNAI2 is implicated in CAC tumorigenesis, we challenged GNAI2KO mice with DSS and DSS–AOM. Similar to GNAI1;3DKO mice, upon DSS challenge, GNAI2KO mice exhibited significant weight loss, a higher colitis severity score, and more severe colitis, shortened colons, and higher levels of colonic secretion of IL6 and IL12 than WT controls (Figure 5A–D and Supplementary Figure 6A and B). Unexpectedly, upon AOM–DSS challenge, GNAI2KO mice exhibited significantly less CAC tumorigenesis with higher levels of colonic secretion of interferon gamma, IL12, IL17, and TNF than WT controls (Figure 5E and F and Supplementary Figure 6C–G).
Figure 5.

GNAI2 inhibits colitis but promotes CAC. (A) Body weight change of WT and GNAI2KO mice during the course of acute colitis with DSS (n = 12). (B–E) On day 10: (B) colitis severity score (n = 6), (C) change of the colon length, and (D) H&E representative images and histologic analysis of the colon of WT and GNAI2KO mice. Scale bars: upper, 100 μm; lower, 25 μm. (E–G) On day 63: (E) representative images of the colon and the change of the colon length, (F) tumor number/colon of indicated AOM–DSS-treated WT and GNAI2KO mice, and (G) H&E representative images of colonic tumors from WT and GNAI2KO mice. Scale bars: upper, 100 μm; lower, 25 μm. (H–J) On day 63: (H) representative images of the colon and the change of the colon length, (I) tumor number/colon of indicated AOM–DSS-treated WT and GNAI2cKO mice, and (J) H&E representative images of colonic tumors from number/colon of indicated AOM–DSS-treated WT and GNAI2cKO mice. Scale bars: upper, 100 μm; lower, 25 μm.
Because GNAI2 was up-regulated in CD11c+ cells (Figure 4M), we hypothesized that GNAI2 in CD11c+ cells promotes CAC. Thus, we generated mice with GNAI2 deletion in CD11c+ cells (GNAI2cKO) or Lck+ cells (GNAI2LckKO) as a control and challenged them with AOM–DSS. GNAI2cKO, but not GNAI2LckKO, mice exhibited significantly fewer CAC tumors than WT controls (Figure 5H and I and Supplementary Figure 6H). Pathology analysis showed that WT, GNAI2KO, and GNAI2cKO mice displayed HGD tumors in the colon at the end of AOM–DSS treatment (Figure 5G and J).
These observations led us to hypothesize that the GNAI2 in CD11c+ cells mediates CAC tumorigenesis in GNAI1;3DKO mice. To this end, we generated mice with GNAI2 deletion in CD11c+ cells (GNAI123cTKO) or epithelial cells (GNAI123vilTKO) as a control on a GNAI1;3DKO background. Intriguingly, GNAI123vilTKO mice exhibited a similar CAC tumorigenesis and similar levels of colonic secretion of IL6, TNF, and GM-CSF to GNAI1;3DKO mice (Figure 6A and Supplementary Figure 7A–G). Conversely, GNAI123cTKO mice had markedly reduced tumor multiplicity compared with GNAI1;3DKO mice under either CH or SH conditions (Figure 6B and C and Supplementary Figure 7H), indicating that GNAI2-expressing CD11c+ cells, but not epithelial cells, are important for CAC promoted by GNAI1;3 deficiency and that the observed tumorigenic phenotype is genotype specific rather than microbiota specific. Pathologic analysis showed that WT, GNAI123cTKO, and GNAI123vilTKO mice had HGD tumors in the colon (Supplementary Figure 7I).
Figure 6.

Deletion of GNAI2 in CD11c+ cells, but not the epithelial cells, of GNAI1;3DKO mice diminishes colonic tumor multiplicity, CD4+ DC and MDSC expansion, and NF-ĸB and STAT3 activation but restores CD8+ DC and CD11c+ MDSC counts. (A) Tumor number/colon of AOM–DSS-challenged WT (n = 22), GNAI1;3DKO (n = 11), or GNAI123vilTKO (n = 27) mice on day 72. (B) Tumor number/colon of AOM–DSS-challenged mice (WT, n = 10; GNAI1;3DKO, n = 28; GNAI123cTKO, n = 23) on day 100. (C) Tumor number/colon of AOM–DSS-challenged mice (GNAI1;3DKO, n = 9; GNAI123cTKO, n = 10) under the CH condition for 87 days. (D) IHC analysis for Gr-1, p-NF-ĸBp65(S536), and p-STAT3(Y705) in colonic tissues from AOM–DSS-challenged WT, GNAI1;3DKO, and GNAI123cTKO mice (n = 3). Scale bars: 25 μm. (E) Representative FACS plots of splenic CD3−CD11b+Gr-1+ and CD3−CD11b+Ly6C+ MDSCs from indicated AOM–DSS-challenged WT, GNAI1;3DKO, and GNAI123cTKO at day 120. n = 4. (F) Representative FACS plots of splenic CD3−CD11b+Gr-1+p-STAT3+ MDSCs of AOM–DSS-challenged WT, GNAI1;3DKO, and GNAI123cTKO mice. (G) Representative FACS plots of splenic CD11c+ CD3−CD11b+Gr-1+ and CD11c+ CD3−CD11b+Ly6C+ MDSCs of AOM–DSS-treated WT, GNAI1;3DKO, and GNAI123cTKO mice. (H) Representative FACS plots of splenic CD11b+, CD4+, and CD8+ MHCII+CD11c+ DCs from AOM–DSS-challenged WT, GNAI1;3DKO, and GNAI123cTKO mice. n = 4. Quantitative analysis of MDSCs and DCs is shown next to the plots or images. All data are mean ± standard deviation (C–H) or standard error of the mean (A–B). *P < .05, **P < .01, ***P < .001, ****P < .0001. FACS, fluorescence-activated cell sorting; ns, not significant.
IHC staining showed that MDSC infiltration and activation of NF-ĸB and STAT3 in the colon were significantly compromised in GNAI123cTKO mice compared with GNAI1;3DKO mice (Figure 6D). The numbers of splenic CD11b+Gr-1+ and CD11b+Ly6C+ MDSCs and activation of STAT3 in MDSCs were remarkably decreased in GNAI123cTKO mice (Figure 6E and F), suggesting that deletion of GNAI2 in CD11c+ cells leads to impaired MDSC expansion and STAT3 activation in GNAI1;3DKO mice.
To further elucidate how GNAI2 deletion in CD11c+ cells leads to compromised MDSC accumulation in GNAI1;3DKO mice, we determined whether MDSCs express CD11c. Approximately 21%–41% of WT MDSCs expressed CD11c, and this expression was significantly reduced in GNAI1;3DKO mice but restored in GNAI123cTKO mice (Figure 6G).
Our previous study showed that GNAI1;3DKO mice exhibited significantly increased CD4+ DCs but decreased CD8+ DCs upon AOM–DSS challenge (Figure 2G). This phenotype was corrected by GNAI2 deletion in CD11c+ DCs in GNAI1;3DKO mice (Figure 6H).
Collectively, these data show that GNAI2 in CD11c+ cells contributes to CAC formation, NF-ĸB–STAT3 activation, MDSC and CD4+ DC expansion and accumulation, and reduction of CD8+ DCs in GNAI1;3DKO mice.
Differential Expression of GNAI1;3 and GNAI2 Are Associated With Human CAC
Finally, we determined whether GNAI proteins are implicated in human CAC tumorigenesis. IHC analysis indicated that GNAI1 or GNAI3 expression levels were significantly lower in CAD, adjacent normal, or CAC colon tissues compared with normal controls (Figure 7A). In contrast, GNAI2 expression was significantly higher in CAD and CAC colon tissues than in normal controls (Figure 7A). These results suggest that the low levels of GNAI1 and GNAI3 and the high level of GNAI2 are significantly associated with CAC. We further assessed NF-ĸB activation and GP130 up-regulation in these specimens. We observed marked NF-ĸB activation and GP130 expression in CAC tumor tissues (Figure 7B and C and Supplementary Figure 8B), underscoring the importance of GP130 and NF-ĸB in CAC tumorigenesis.
Figure 7.

Low GNAI1;3 expression and high GNAI2 expression are associated with colorectal tumorigenesis and poor patient survival. (A–C) IHC staining for (A) GNAI1-, GNAI2-, and GNAI3-, (B) p-NF-ĸBp65(S536)–, and (C) GP130-positive cells in the normal, dysplastic (CAD), and adjacent nonneoplastic or neoplastic (CAC) epithelial cells and corresponding surrounding stromal cells. Quantitative analyses of GNAI1, GNAI2, GNAI3, and GP130 are shown under the images. All data are mean ± standard deviation. *P < .05, **P < .01, ***P < .001; 2-tailed Student t test. Scale bars: 25 mm. (D) Heatmap analysis of GSE4107 CRC mucosal tissue DNA microarray for expression of Gnai1, Gnai2, and Gnai3 mRNA. (E) IB analysis for GNAI1, GNAI2, or GNIA3 in CRC patients’ normal colon (N), peritumor (PT), or tumor (T) tissues. (F) Relapse-free survival of 4 subgroups of CRC patients classified by different combinations of Gnai1 and Gnai3 expression levels (left) and Gnai2 and Gnai3 expression levels (right).
To extend our findings in CAC to colorectal cancer (CRC), we first analyzed a public DNA microarray database, GSE4107, generated by using RNA from 10 healthy individuals and 12 CRC patients.43 The expression levels of Gnai1 and Gnai3 were lower, but Gnai2 was higher in the CRC patients than healthy control individuals (Figure 7D). In our CRC patient cohort, compared with normal control individuals, GNAI1, GNAI3, and GNAI1;3 were expressed at low levels in 46.6%, 62.0%, and 39.7% of patients, respectively; GNAI2 was up-regulated in 38.6%, and GNAI2 was up-regulated but GNAI3 was down-regulated in 21.2% of patients (Figure 7E and Supplementary Figure 8C). IHC analysis also indicated that GNAI1 and GNAI3 expression levels were low that but levels of GNAI2 were high in CRC tumor tissues (Supplementary Figure 8D). Analysis of Gnai1, Gnai2, and Gnai3 RNA expression in a public human CRC primary tumor DNA microarray database, GSE39582,44 showed that low Gnai1 and Gnai3 expression or high Gnai2 expression is significantly associated with decreased relapse-free survival (Figure 7F).
Discussion
In this study, we have shown that low expression of GNAI1;3 and high expression of GNAI2 are significantly associated with human CAC. We have further shown that GNAI1;3DKO exhibit markedly increased CAC tumorigenesis, whereas GNAI2KO mice show a significantly diminished CAC tumorigenesis upon challenge with AOM–DSS. However, the mechanisms by which GNAI proteins regulate CAC tumorigenesis remain elusive.
It is known that the microbiota in the gut regulate immune system components and play an important role in colitis and associated cancer.34 A study suggests that IL33KO mice are highly susceptible to colitis and CAC due to a dysbiotic microbiota.45 Other studies report that TRUC mice, which harbor deletion of T-bet in CD11c+ cells on a Rag2KO background, develop spontaneous colitis and CAC in an IL6- and TNF-dependent but gut microbiota–independent manner.46,47 Our CH and SH experiments show that the enhanced CAC tumorigenesis in GNAI1;3DKO mice is independent of microbiota and is associated with marked increases in the levels of IL6 and GNAI2 and activation of NF-ĸB and STAT3.
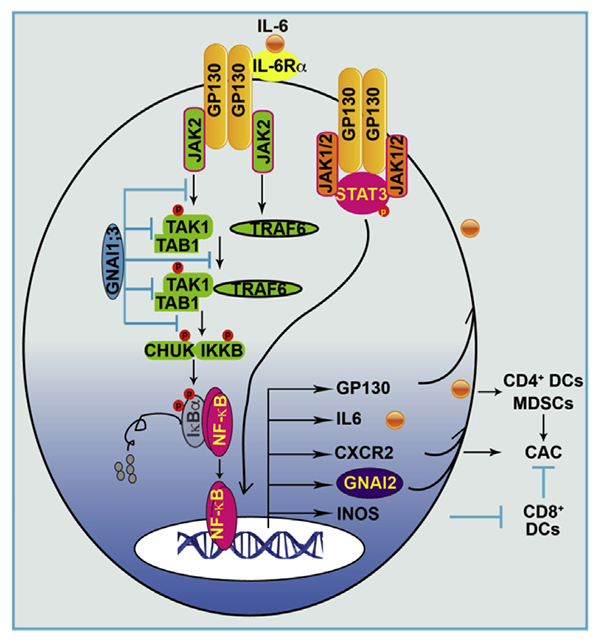
The current understanding of IL6 signaling during CAC development is that IL6 ligates to IL6R, which recognizes the assembly of the GP130-JAK1/2 complex, the main function of which is to generate phosphorylated STAT3, leading to CAC.5 The studies reported here further show that in addition to this complex in IL6 signaling, there is the assembly of a novel GP130-JAK2-TRAF6-TAK1/TAB1-IKKB complex, the main function of which is to trigger NF-ĸB activation, leading to expression of CXCR2, GNAI2, GP130, and INOS (Supplementary Figure 9). IL6 activation of NF-ĸB and subsequent GNAI2 up-regulation are augmented by GNAI1;3 deficiency. Blockade of IL6 or deletion of GNAI2 in CD11c-expressing cells greatly diminished MDSC expansion and CAC tumorigenesis promoted by GNAI1;3 deficiency.
We found that this IL6–NF-ĸB signaling cascade also contains a positive feedback loop, by which IL6 not only activates GP130 but also induces its expression through JAK, NF-ĸB, and STAT3 activation (Supplementary Figure 9). The deregulation of GP130 leads to numerous pathophysiologic consequences including colorectal tumorigenesis.19,48 Our IHC analysis shows that GP130 is significantly up-regulated and associated with elevated NF-ĸB activation in human CAC tumor tissues, in which GNAI1;3 expression levels are significantly low and GNAI2 expression level is significantly high. Unexpectedly, GNAI1;3 suppress the basal level of GP130 and its induction by IL6. GNAI1;3 also inhibit TAK1-TAB1 interaction, which is constitutive and required for NF-ĸB activation.49 Loss of GNAI1;3 leads to the high basal levels of GP130 and TAK1–TAB1 interaction, which are likely responsible for high basal activation of NF-ĸB and STAT3 and up-regulation of GNAI2 and INOS in GNAI1;3DKO cells.
TRAF6 is upstream to TAK1 in IL1/toll-like receptor signaling.16,17,50 This is also the case for IL6–NF-ĸB signaling. We discovered that GNAI1;3 form a complex with TAK1, TRAF6, and JAK2 and interrupt IL6-induced association of TAK1 with JAK2 and TRAF6. Inhibition of JAKs or ablation of either TRAF6 or TAK1 severely diminishes IL6 activation of NF-ĸB and GNAI2 and INOS up-regulation.
Previous studies reported that GNAI2 suppresses CD8+ DCs to produce IL-128 and that GNAI2KO mice develop spontaneous colitis and CAC under certain environments.28 However, although GNAI2KO mice develop spontaneous colitis, they did not develop colonic tumors in our vivarium. Surprisingly, GNAI2KO mice or mice with GNAI2 deletion in CD11c+ cells exhibit significantly less CAC tumorigenesis upon AOM–DSS challenge, suggesting that GNAI2 in CD11c+ cells is important for CAC development. In GNAI1;3DKO mice, GNAI2 is highly up-regulated in CD11c+ cells, and IL6 is greatly produced in CD11b+/CD4+ but not CD8+ DCs. Because GNAI2 and MDSCs are targets of IL6, it is expected that high IL6-producing CD11c+ cells also express high levels of GNAI2 and stimulate MDSC expansion in GNAI1;3DKO mice. GNAI2 is coupled to CXCR2,25,26 which is up-regulated in GNAI1;3DKO mice and is required for MDSC trafficking and CAC tumorigenesis.12 We found that MDSCs also express CD11c. Thus, IL6 neutralization or GNAI2 deletion in CD11c+ cells greatly diminishes CAC tumorigenesis and MDSC expansion in GNAI1;3DKO mice.
In summary, our data suggest that GNAI1;3 suppress GNAI2-mediated MDSC expansion and CAC tumorigenesis through negatively regulating IL6 signaling. GNAI1;3 act as potential tumor suppressors, and GNAI2 acts as a tumor-stimulating factor. Induction of GNAI1;3 or blockade of GNAI2 and IL6 could be preventive and therapeutic avenues for CAC.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Interleukin 6 (IL6) and tumor necrosis factor (TNF) contribute to the development of colitis-associated cancer (CAC).
NEW FINDINGS
A significant association was found between low expression of G protein subunit alpha i1 (GNAI1) and Gnai3, and high expression of Gnai2, with development of CAC. In mice, loss of GNAI1 and GNAI3 resulted in increased expression of IL6 and GNAI2 in CD11c+ cells, which promoted development of colitis and growth of colon tumors.
LIMITATIONS
These studies were only performed in mice or in tumor samples collected from patients.
IMPACT
GNAI1, GNAI2, and GNAI3 could be used as prognostic factors for patients with CAC. Strategies to induce expression of GNAI1 and GNAI3, or block GNAI2 and IL6, might be developed for prevention or therapy of CAC in patients.
Acknowledgements
We thank Drs Michele Carbone, Junfang Ji, Charles Rosser, and Haining Yang for critically reading the manuscript, Hugh Luck for expert help with H&E and IHC, and Drs Jeremie Oliver and Jun Zhang for technical support. We are grateful to the Chu-Yao family for their financial support. Wen-Ming Chu was a scholar of the Leukemia and Lymphoma Society.
Funding
This study was supported by the National Natural Science Foundation of China (NNSFC) 81100274 & 81570522 (to Xiqiao Zhou), the State Key Programs of the NNSFC 81430062 (to Beicheng Sun), the Key project of National Science and Technology of China 2016ZX08011005 (to Ailin Tao), Z01-ES-101643 of the Intramural Research Program of the National Institutes of Health (NIH) (to Lutz Birnbaumer), P30GM114737 (to Molecular and Cellular Immunology Core), P30CA071789–16 (to Microscopy, Imaging, and Flow Cytometry Shared Resource), 2P20GM103457 (to W. Steven Ward), P30CA071789 (to Randall F. Holcombe), NIH R01 GM118915 to Guangyu Wu, NIH U01CA188387–01 (to Wei Jia and Wen-Ming Chu), the Hawaii Community Foundation, the University of Hawaii (UH) Foundation Pilot Award, UH Start-Up and Bridged Funds, and NIH R01 AI054128 (to Wen-Ming Chu). Pathology slides were created at the Pathology Core of UH Cancer Center.
Abbreviations used in this paper:
- AOM
azoxymethane
- AZD
AZD1480
- BMDSC
bone marrow–derived myeloid-derived suppressor cell
- CAC
colitis-associated cancer
- CAD
colitis-associated dysplasia
- CH
cohoused
- CRC
colorectal cancer
- DC
dendritic cell
- DKO
double knockout
- DSS
dextran sulfate sodium
- FAHNMU
First Affiliated Hospital of Nanjing Medical University
- GM-CSF
granulocyte and colony-stimulating factor
- GNAI
G protein subunit alpha i
- GNAI1;3
GNAI1 and GNAI3
- Gr-1
granulocyte receptor-1 antigen
- HGD
high-grade dysplasia
- IB
immunoblot
- IBD
inflammatory bowel disease
- IHC
immunohistochemistry
- IL
interleukin
- IL6R
interleukin 6 receptor
- INOS
inducible nitric oxide synthase, or nitric oxide synthase 2
- IP
immunoprecipitation
- LP
lamina propria
- MDSC
myeloid-derived suppressor cell
- MEF
mouse embryonic fibroblast
- MHC
major histocompatibility complex
- mRNA
messenger RNA
- NF-ĸB
nuclear factor ĸB
- SH
separately housed
- STAT3
signal transducer and activator of transcription 3
- STRING
Search Tool for the Retrieval of Interacting Genes/Proteins
- TKO
triple conditional knockout
- TNF
tumor necrosis factor
- UH
University of Hawaii
- WT
wild type
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2019.02.040.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Foersch S, Neurath MF. Colitis-associated neoplasia: molecular basis and clinical translation. Cell Mol Life Sci 2014;71:3523–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greten FR, Eckmann L, Greten TF, et al. IKKBeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004;118:285–296. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009;15:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terzic J, Grivennikov S, Karin E, et al. Inflammation and colon cancer. Gastroenterology 2010;138:2101–2114. [DOI] [PubMed] [Google Scholar]
- 5.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis 2011;7(Suppl 1):i104–i108. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 2011; 12:715–723. [DOI] [PubMed] [Google Scholar]
- 7.Hansen M, Andersen MH. The role of dendritic cells in cancer. Semin Immunopathol 2017;39:307–316. [DOI] [PubMed] [Google Scholar]
- 8.He J, Gurunathan S, Iwasaki A, et al. Primary role for Gi protein signaling in the regulation of interleukin 12 production and the induction of T helper cell type 1 responses. J Exp Med 2000;191:1605–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003;3:133–146. [DOI] [PubMed] [Google Scholar]
- 10.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res 2017;5:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 2004;10:48–54. [DOI] [PubMed] [Google Scholar]
- 12.Katoh H, Wang D, Daikoku T, et al. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013;24:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu WM. Tumor necrosis factor. Cancer Lett 2013; 328:222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li ZW, Chu W, Hu Y, et al. The IKKBeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med 1999;189:1839–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu WM, Ostertag D, Li ZW, et al. JNK2 and IKKBeta are required for activating the innate response to viral infection. Immunity 1999;11:721–731. [DOI] [PubMed] [Google Scholar]
- 16.Zhang P, Chan J, Dragoi AM, et al. Activation of IKK by thymosin alpha1 requires the TRAF6 signalling pathway. EMBO Rep 2005;6:531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem 2009; 78:769–796. [DOI] [PubMed] [Google Scholar]
- 18.Christian F, Smith EL, Carmody RJ. The regulation of NF-kappaB subunits by phosphorylation. Cells 2016;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009;15:91–102. [DOI] [PubMed] [Google Scholar]
- 20.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004;21:491–501. [DOI] [PubMed] [Google Scholar]
- 21.Danese S, Grisham M, Hodge J, et al. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol 2016;310:G155–G162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Grivennikov SI, Karin M. Implications of anti-cytokine therapy in colorectal cancer and autoimmune diseases. Ann Rheum Dis 2013;72(Suppl 2):ii100–ii103. [DOI] [PubMed] [Google Scholar]
- 23.Cao C, Huang X, Han Y, et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal 2009; 2:ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Dela Cruz R, Ji F, et al. G(i)alpha proteins exhibit functional differences in the activation of ERK1/2, Akt and mTORC1 by growth factors in normal and breast cancer cells. Cell Commun Signal 2014;12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pero RS, Borchers MT, Spicher K, et al. Galphai2-mediated signaling events in the endothelium are involved in controlling leukocyte extravasation. Proc Natl Acad Sci U S A 2007;104:4371–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zarbock A, Deem TL, Burcin TL, et al. Galphai2 is required for chemokine-induced neutrophil arrest. Blood 2007;110:3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho H, Kamenyeva O, Yung S, et al. The loss of RGS protein-Galpha(i2) interactions results in markedly impaired mouse neutrophil trafficking to inflammatory sites. Mol Cell Biol 2012;32:4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudolph U, Finegold MJ, Rich SS, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet 1995;10:143–150. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Wang D, Chen Z, et al. Gαi1 and Gα3 regulate macrophage polarization by forming a complex containing CD14 and Gab1. Proc Natl Acad Sci U S A 2015; 112:4731–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan H, Williams DL, Zingarelli B, et al. Differential regulation of lipopolysaccharide and Gram-positive bacteria induced cytokine and chemokine production in splenocytes by Galphai proteins. Biochim Biophys Acta 2006; 1763:1051–1058. [DOI] [PubMed] [Google Scholar]
- 31.Gohla A, Klement K, Piekorz RP, et al. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A 2007;104:3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plummer NW, Spicher K, Malphurs J, et al. Development of the mammalian axial skeleton requires signaling through the Galpha(i) subfamily of heterotrimeric G proteins. Proc Natl Acad Sci U S A 2012;109:21366–21371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ustyugova IV, Zhi L, Abramowitz J, et al. IEX-1 deficiency protects against colonic cancer. Mol Cancer Res 2012; 10:760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Robertis M, Massi E, Poeta ML, et al. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog 2011;10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015;125:3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaked H, Hofseth LJ, Chumanevich A, et al. Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci U S A 2012;109:14007–14012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Yue P, Page BD, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A 2012;109:9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKĸBeta. Cell 2007;130:918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oka S, Kamata H, Kamata K, et al. N-acetylcysteine suppresses TNF-induced NF-kappaB activation through inhibition of IkappaB kinases. FEBS Lett 2000;472:196–202. [DOI] [PubMed] [Google Scholar]
- 40.Singh A, Sweeney MF, Yu M, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012;148:639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009;16:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arinze IJ, Kawai Y. Transcriptional activation of the human Galphai2 gene promoter through nuclear factor-kappaB and antioxidant response elements. J Biol Chem 2005;280:9786–9795. [DOI] [PubMed] [Google Scholar]
- 43.Cui M, Yuan J, Li J, et al. Gene expression analysis of colorectal cancer by bioinformatics strategy. Hepatogastroenterology 2014;61:1942–1945. [PubMed] [Google Scholar]
- 44.Marisa L, de Reynies A, Duval A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med 2013;10:e1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malik A, Sharma D, Zhu Q, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J Clin Invest 2016;126:4469–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powell N, Lo JW, Biancheri P, et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology 2015;149:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garrett WS, Punit S, Gallini CA, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell 2009;16:208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howlett M, Menheniott TR, Judd LM, et al. Cytokine signalling via gp130 in gastric cancer. Biochim Biophys Acta 2009;1793:1623–1633. [DOI] [PubMed] [Google Scholar]
- 49.Hirata Y, Takahashi M, Morishita T, et al. Post-translational modifications of the TAK1-TAB complex. Int J Mol Sci 2017;18:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng X, Zhang P, Wang X, et al. Signaling pathways leading to the activation of IKK and MAPK by thymosin alpha1. Ann N Y Acad Sci 2007;1112:339–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.