

Abstract
A highly chemoselective phenol cross-coupling reaction catalyzed by a Cr-salen catalyst was developed. Kinetic studies showed that the oxidation of Cr(III) to Cr(V) is the rate-determining step of the reaction. In addition, experimental stoichiometric analysis showed that a high valentCr(V) specie is the active catalyst for this process. The selectivity of the reaction was found to be determined by the cross-coupling carbon-carbon bond forming reaction, rather than any pre-coordination species. It appears that the lowest energy cross-coupling pathway requires a lesser degree of electronic reorganization in its transition state vs the lowest energy homo-coupling pathway. This result was supported by stoichiometric Cr(V) kinetics, 13C kinetic isotope effects, and DFT calculations. The understanding of the full landscape of this reaction, allowed us to develop a general analysis to predict the regioselectivity of the cross-coupling reaction.
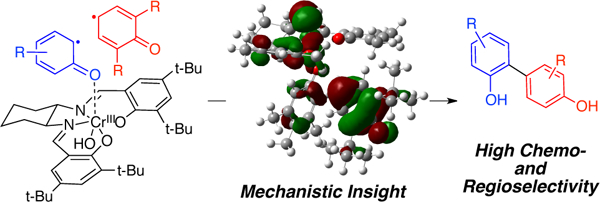
Graphical Abstract
INTRODUCTION
Synthesis of unsymmetrical biaryls is an important transformation in organic chemistry. The resultant products can be further utilized for the synthesis of natural products, pharmaceuticals, agrochemicals, polymers, and catalysis.1,2 Conventional syntheses of this important motif involve metal-catalyzed cross-couplings of activated arenes (Ar – X and Ar – M).3,4 These coupling reactions require several prefunctionalizaton steps that are intrinsically necessary to achieve reactivity and which also control regio- and chemoselectivity. In contrast, phenols can couple directly and this important transformation5 has been used to achieve the total synthesis of several natural products, particularly when symmetric or intramolecular couplings are involved. However, low site selectivity in phenol coupling reduces the utility of this approach in cross-coupling.
Several tactics to achieve cross-coupling of phenols have been developed over the years. Waldvogel elegantly demonstrated that the selective synthesis of partially protected unsymmetricbiphenols can be achieved by means of an metal-free anodic phenol cross-coupling approach.6 A disadvantage of this chemistry is the requirement for fluorinated solvents. In addition, in order for the cross-coupling to occur,6a one phenol must contain electron-donating groups (universally methoxy) and the other cannot. Further, a 1:3 ratio of the more oxidizable to the less oxidizable phenol is required to further suppress homo-coupling. Within this paradigm, remarkable results could be obtained especially where one partner was a monosubstituted phenol, which are notoriously difficult due to multiple sites available for coupling, little steric hindrance to suppress overcoupling, and typically high oxidation potentials. Overall, yields are modest with an average of 41% across 17 substrates and a high of 63%. In the 2014 report,6a only ortho-ortho couplings were described and a 2019 paper6f outlines only para-para couplings. Jeganmohan7 devised an innovative strategy using K2S2O8 as the oxidant together with an ammonium salt, wherein a more oxidizable phenol forms a radical cation and the ammonium salt assists in deprotonation of the other partner.7a However, this method requires the use of stoichiometric K2S2O8 (2 equiv) and an a 3:1 ratio of phenolic coupling partners. Again, one phenol must contain electron-donating groups (methoxy or dimethoxy) and yields are modest (47% average across 14 substrates with a high of 61%). Again, remarkable results can be obtained when one partner is phenol or a monosubstituted phenol. More recently, Pappo and coworkers8 developed two very successful different approaches to achieve highly selective oxidative cross-coupling of phenols catalyzed by an iron source. In a 2015 report,8a an iron catalyst was described where couplings were most successful when one substrate was more nucleophilic and the other was more oxidizable. In practice, one phenol contains electron-donating groups (methoxy or dimethoxy or trimethoxy) such that it is substantially more oxidizable. Within this framework, very high yields can be obtained (up to 96%) although 3:1 ratios of substrates are used in most such cases. A range of ortho-ortho, ortho-para, and para-para cross couplings were viable; in some cases, mixtures of isomers were obtained and the control elements for the regioselection with respect to the more oxidizable partner are unclear. Again, very good results can be obtained when one partner is monosubstituted phenol. In a 2017 report,8b an iron porphyrin catalyst was proposed to react via a radical-radical coupling where the more oxidizable phenol forms a transient free radical that preferentially reacts with a persistent radical formed upon ligation of the other phenol to the iron center. Selectivity is dictated by the greater binding affinity of one phenol to the iron center. Again, one phenol contains electron-donating groups (methoxy or dimethoxy) such that it is substantially more oxidizable. Yields are variable (15–77%), 2–3:1 ratios of substrates are used, and there is limited selectivity with certain subsets of phenols (especially mono ortho-substituted). Limitations of these iron catalyzed processes are the requirement for fluorinated solvents, the use of higher molecular weight oxidants (tert-butyl hydroperoxide or di-tert-butyl peroxide), and the need for an excess of one of the coupling partners to achieve favorable outcomes. Our group developed a catalytic cross-coupling method that allowed the synthesis of biphenols with high regio- and chemoselectivity by means of a Crsalen catalyst and dioxygen as terminal oxidant.9 A broader range of functionality was found to be compatible, notably high cross-selectivity was observed for partners with similar electronics and redox potentials, high regioselection was observed when multiple coupling sites were available on one partner, and 1.3–1.5:1 ratios of substrates were employed.
Few of these transformations are understood in detail. Waldvogel has shown that the cross-coupling of partially protected phenols (Scheme 1a) can be achieved via the oxidation of a phenol mediated by a boron-doped diamond (BDD) anode, which is trapped by an electron-rich arene to afford the final product.6a Pappo, in an approach involving FeCl3 as catalyst (Scheme 1b),9a suggests that the reaction proceeds via a chelated radical-anion coupling.
Scheme 1.
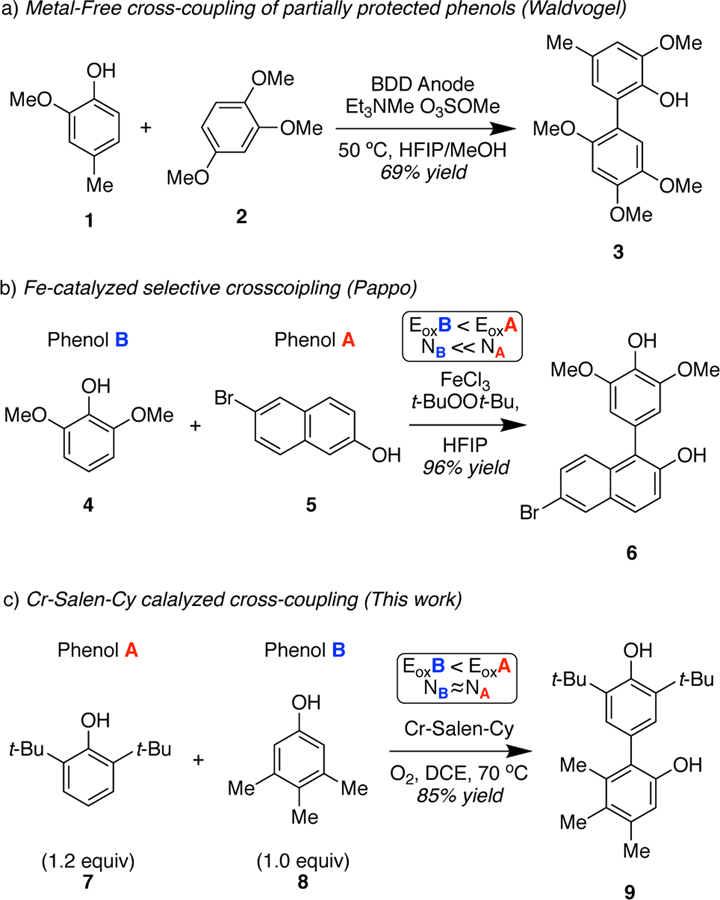
Examples of Cross-Coupling of Phenols by Employing Different Metal-Catalysts
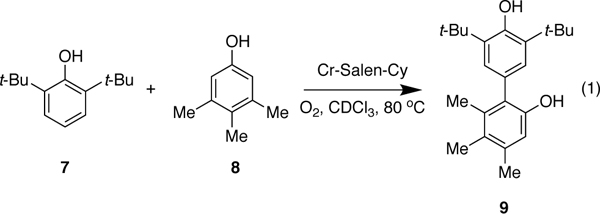
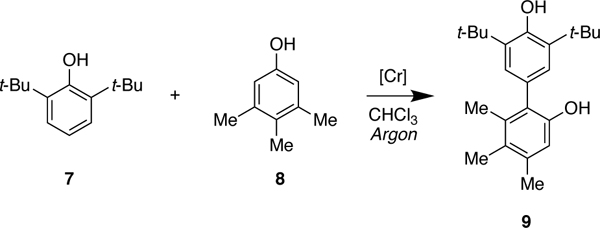
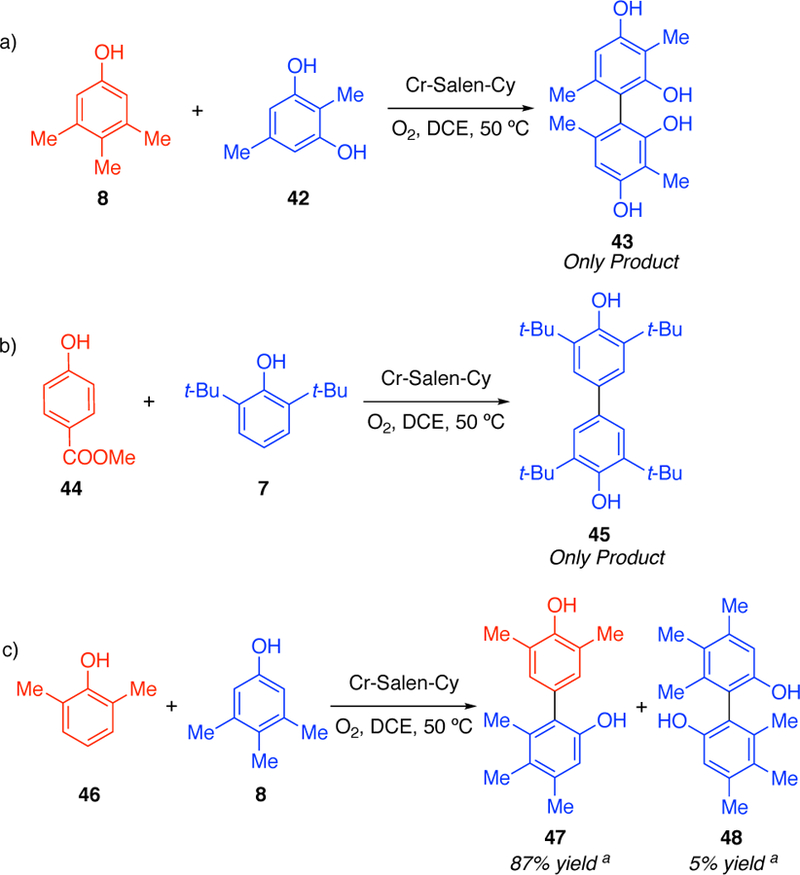
Pappo and coworkers have proposed that complementary electronic potential (Eox) and the theoretical global nucleophilicity (N) of the phenols govern these and related couplings.8, 10 A cross-coupling will be favored when there is a significant difference between the oxidation potential (EoxB < EoxA) and the global nucleophilicity (NB< NA) of the phenols. Even though their experimental results, and selected examples from other reports,7, 8, 10 are in good agreement with this model, we have discovered that there are substantial reactivity differences in the cross-coupling mediated by the Cr-salen catalyst. For example, when we attempted the reaction of 2,6-di-tert-butylphenol (7) and 3,4,5-trimethylphenol (8) where the latter substrate is more oxidizable, but their nucleophilicity is approximately the same (EB< EA, NB≈ NA) we obtain the cross-coupling product in good yield (85% isolated yield) without detectable homo-coupling (Scheme 1c).7
With goal of approaching biphenol natural products and other targets of interest (Chart 1),5c,11 we sought to understand the mechanism of the Cr-Salen-Cy catalyzed cross-coupling of phenols to be able to predict which substrate pairs couple well and to design further catalyst systems. Here, we present a full mechanistic picture of this transformation by a combination of experimental and computational studies. Catalytic and stoichiometric versions of the reaction were studied in order to understand the nature of the rate-determining and selectivity-determining steps.
Chart 1.

Examples of Unsymmetrical Biphenol Natural Products.
RESULTS AND DISCUSSION
Reaction Scope.
The Cr-Salen-Cy catalyst is effective in the coupling of a number of phenols containing alkyl, electron-donating, and weakly electron-withdrawing substituents (Figure 1). Cross-coupling between 2,6-substituted phenols and phenols with only unsubstituted ortho-positions available resulted in selective formation of ortho-para cross-coupling product (9–13). Both phenols of the products are hindered which prevents catalyst coordination. As such, further reaction to generate trimers is considerably more difficult. In addition, cross-coupling between 2,6-substituted phenols and phenols that had both ortho- and para-positions available resulted in the selective formation of para-para cross-coupled products (14–21). For these products, one phenol remains unhindered and can readily coordinate catalyst allowing formation of trimeric adducts at higher conversions or when greater amounts of the 2,6-disubstituted phenol is used.
Figure 1.

Scope and yields of oxidative cross-coupling of phenols using the Cr-Salen-Cy catalyst in DCE. aYield based on recovered starting material. bRemaining mass was starting material. cAround13%staring material observed by NMR and some decomposition. d Around 26% starting material observed by NMR, and 19 % homo-coupling of 6-methoxynaphthol (blue).
Finally, reactions with 2-naphthol, and its derivatives with electron-withdrawing, and electron-donating substituents, lead to cross-coupling products in good yields (22–30). The scope of the reaction did not extend to all phenols, particularly those with amino (oxidation of the nitrogen interferes) or electron-withdrawing substituents (no reaction due to higher oxidation potentials). The reaction conditions are straightforward requiring only catalyst and substrate with oxygen and a solvent at room temperature to 80 °C. A range of solvents are typically effective including dichloroethane, dichloromethane, benzene, toluene, and dichlorobenzene. In order to understand the high selectivity of this method, and potentially predict the formation of specific cross-coupling products, mechanistic studies were undertaken.
Catalytic Kinetic Studies.
To probe the reaction mechanism, kinetic studies of the Cr-Salencatalyzed cross-coupling of 2,6-di-tert-butylphenol(7) with 3,4,5-trimethylphenol (8) were initiated (eq 1). Overall, this process is well-behaved (Figure 2a) exhibiting linear behavior for the formation of product over time, and showing that the formation of water from the reduction of O2 does not affect the overall reaction.
Figure 2.
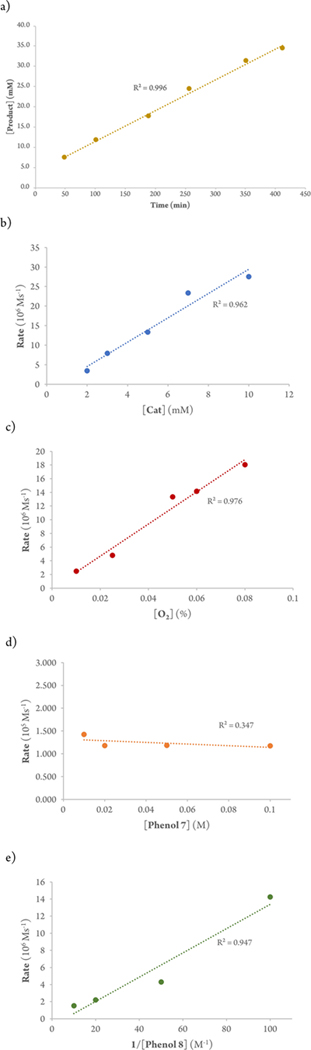
Catalytic kinetics for the cross-coupling reaction using Cr-Salen. (a) Formation of product over time for the catalytic reaction of 7 (0.060 M) with 8 (0.040 M) using Cr-Salen (0.004 M, 10 mol%) in dichloroethane at 60 °C under O2. (b) Correlation between initial rate and catalyst concentration. Both phenols = 0.1 M; [Cr-Salen] = 0.002, 0.003, 0.005, 0.007, 0.010 M; [CrSalen]]/[SM]o = 0.02, 0.03, 0.05, 0.07, 0.1, respectively, in CDCl3 under O2 at 80 °C. (c) Correlation between initial rate and oxygen concentration (10, 25, 50, 60, 80% O2/Ar) with [7] = [8] = 0.2 M and [catalyst] = 0.01 M (5 mol%) in CDCl3 at 80 °C. (d) Correlation between initial rate and [7] (0.01, 0.02, 0.05, 0.1 M) while [8] and [catalyst] were held constant at 0.1 M and 0.005 M (5 mol % with respect to 0.1 M phenol), respectively, in CDCl3 under O2 at 80 °C. (e) Correlation between initial rate and [8] (0.01, 0.02, 0.05, 0.1 M), while [7] and [catalyst] were held constant at 0.1 M and 0.005 M (5 mol % with respect to 0.1 M phenol), respectively, in CDCl3 under O2 at 80 °C.
The catalyst order was probed by holding the concentration of each of the phenols constant at 0.1 M, while the concentration of the Cr-Salen catalyst was varied (0.002–010 M). A double ln plot of initial rates vs catalyst concentration (see Supporting Information) revealed a first order dependence with respect to the catalyst which is also supported by a rate vs [cat]1 plot (Figure 2b). O2 concentration was probed by utilizing different ratios of [O2] and [Ar] gases at atmospheric pressure (10%, 25%, 50%, 60%, and 80% for oxygen; see SI). A first-order dependence on the concentration of oxygen was evident (Figure 2c).
Kinetic studies were next performed for each phenol. First, the concentration of 7 was varied over a 10-fold range while the concentration of 8 and the catalyst were held constant. A plot of rates vs concentration showed that the cross-coupling reaction is zeroorder with respect to 7 (Figure 2d). The kinetic profile of 8 was obtained utilizing the same approach. Notably, the reaction rate decreased as higher concentrations of 8 were used. A double ln plot revealed a slope of –0.94 consistent with negative first order dependence (See SI). In addition, a linear correlation for the rate vs 1/[8] (Figure 2e) was obtained. In line with this finding, higher concentrations of phenol 8 inhibit the cross-coupling reaction while promoting the homo-coupling of 8.
Together, these kinetic studies support an overall rate law of k[7]0 [8]−1[Cat]1 [O2]1. These data indicate that the rate deter-mining step of the catalytic reaction is the oxidation of the catalyst by interaction of the Cr(III) with dioxygen. Furthermore, Figure 2a indicates that reoxidation of Cr(III) is rate-limiting over the entire reaction course. The negative order in phenol 8 likely arises from facile binding of the unhindered electron-rich phenol to the Cr(III), which then prevents binding of oxygen and oxidation. The other phenol, 7, cannot engage in facile coordination with the Cr(III) due to the large ortho-tert-butyl groups, and thus exhibits a zero order.
Formation and Reactivity of Oxochromium(V) Salen and μ-Oxochromium Salen Dimer.
The kinetic data for the catalytic system indicate that the slow step is oxidation of the Cr(III) complex with dioxygen, which is in line with literature reports.12 Treatment of the Cr(III) complex at 80 °C under oxygen atmosphere caused a red shift in the UV-Vis spectrum, consistent with oxidation of Cr(III) to a Cr(V) species (Figure 3a vs 3b).13
Figure 3.

UV-Vis profiles of Cr-Salen upon treatment with oxygen at: (a) room temperature 4 h, (b) 80 °C, 4 h, (c) 80 °C, 4 h in the presence of 1 equiv TEMPO.
Dioxygen is a potent oxidant that can react via two electron pathways14a (peroxide byproduct) or four electron pathways14b (water byproduct).14 Peroxide was not observed in reaction mixtures when tested using starch/iodide. In addition, the chromium catalyst was found to convert hydrogen peroxide to dioxygen very quickly. As such, we assessed peroxide itself as an oxidant and found it to be effective (Table 1, entry 3 vs entry 5). tert-Butyl hydrogen peroxide was also effective (entry 2), but di-tert-butyl peroxide was not (entry 1). Other oxidants known to oxidize Cr(III) were also effective (entry 4). Thus, users can select the most appropriate oxidant depending on their application. For more sensitive substrates, it was found that peroxide gives rise to more byproduct formation and lower yields.
Table 1.
Impact of different oxidants on the formation of cross-coupling product.
 | ||
|---|---|---|
| entry | oxidant | conversion |
| 1 | (t-BuO)2 | NR |
| 2 | t-BuOOH | 75 % |
| 3 | H2O2 | 60 % |
| 4 | PhIO | 60 % |
| 5 | O2 | 38 % |
To further understand the nature of the active oxidant, we set out determine the oxidation state of the chromium species that causes the phenol oxidation. Literature reports indicate that Crsalen species undergoes oxidation with a range of oxidants to generate oxo-Cr(V)12a,15 and Cr(IV) equivalents. For the latter, reaction of oxo-Cr(V) salen with Cr(III) salen generates a μ-oxo-complex (Cr-O-Cr).15b, 15d Talsi and co-workers further defined the oxidation state of the binuclear salen complex as +5/+3 [i.e. Cr(V)O-Cr(III)]. They proposed that the binuclear μ-oxo-chromium complex interconverts with a mono-oxo-Cr(V) salen complex and a Cr(III)-salen complex (Scheme 2).15c In alkene oxidation, the mononuclear oxo-Cr(V) has been identified as the active oxidant with the binuclear μ-oxo complex serving as a reservoir of the active oxo-Cr(V) species.
Scheme 2.
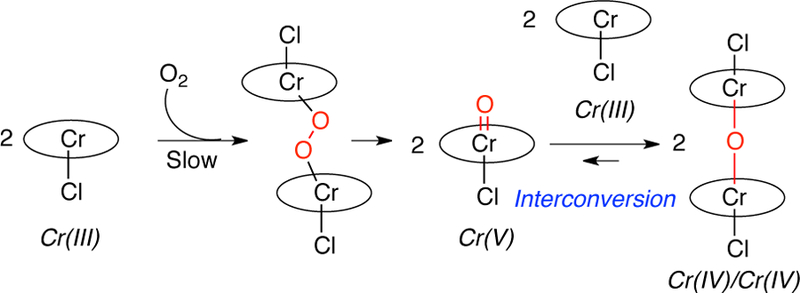
Formation of Oxo-chromium(V) and μ-Oxo-chromium Complexes
To determine which of the chromium species was involved in the cross-coupling reaction, stoichiometric experiments were performed under an argon atmosphere using phenols 7 and 8 (Table 2). Using only the oxo-Cr(V) complex, which was synthesized by treatment of the Cr(III)-salen with iodosobenzene,16 a proportional amount of cross-coupling product 9 was obtained (entries 1 and 2) at room temperature. This result indicates that the Cr(V) acts as a two-electron oxidant implicating a Cr(V) to Cr(III) redox couple. Combining the oxo-Cr(V) with the Cr(III) complex at room temperature caused no change in the characteristic Cr(V) color (green) and the reactivity was the same as with the oxo-Cr(V) alone. The Cr(IV) equivalent was generated by heating the oxo-Cr(V) with an excess of the Cr(III) complex to ensure that no residual oxo-Cr(V) remained (entry 3 – 4), which caused formation of a red colored solution with Cr(IV) spectral features. As shown in entry 3, this Cr(IV) equivalent is unreactive (c.f. entry 2). At higher temperature, however, the Cr(IV) equivalent did lead to a small amount of product. We hypothesize that a disfavorable equilibrium between the μ-oxo salen dimer and the oxo-Cr(V)/Cr(III) monomers would account for this observation.
Table 2.
Stoichiometric Experiments using Oxochromium(V) Salen and μ-Oxochromium-Salen Complexes.
 | ||||
|---|---|---|---|---|
| entry | [Cr] (equiv) | temp (°C) | time (h) | Conversionb |
| 1 | Cr [V] (0.1) | 25 | 3 | 10 % |
| 2 | Cr [V] (0.5) | 25 | 1 | 38 % |
| 3 | Cr [V] (0.5) + Cr [III] (1.0) | 25 | 24 | 0 % |
| 4a | Cr [V] (0.5) + Cr [III] (1.0) | 80 | 24 | 12 % |
The indicated amount of Cr [V] and Cr [III] were combined and heated at 80 °C for 2 h. This mixture was cooled to 25 °C and the phenol substrates were then added and stirred for the indicated time and temperature.
Determined by 1H NMR spectroscopy.
Hydrogen peroxide generates the Cr(V) via a different pathway as combination of Cr(III) and hydrogen peroxide quickly caused rapid formation (upon addition) of the characteristic green Cr(V) solution at room temperature whereas a slow formation of the Cr(V) was seen with dioxygen even at elevated temperature (Figure 3). However, depending on the substrate, hydrogen peroxide can also directly act on the substrates or products giving rise to byproducts.
Interaction of Phenols with Oxo-chromium(V) Salen.
In order to probe the mechanism of the reaction after the rate determining oxidation of the Cr(III) catalyst, single turnover reactions were undertaken with preformed oxo-Cr(V) complex.16 The onset oxidation potential of this complex was measured to be –1.64 eV which provides the upper limit of reactivity. Upon treatment of the oxo-Cr (V) complex with substrate 8, a rapid color change was observed. Thus, UV-Vis spectroscopy was used to monitor the reaction and the λmax of 8 at 633 nm was observed to decrease in the presence of phenols (Figure 4).
Figure 4.
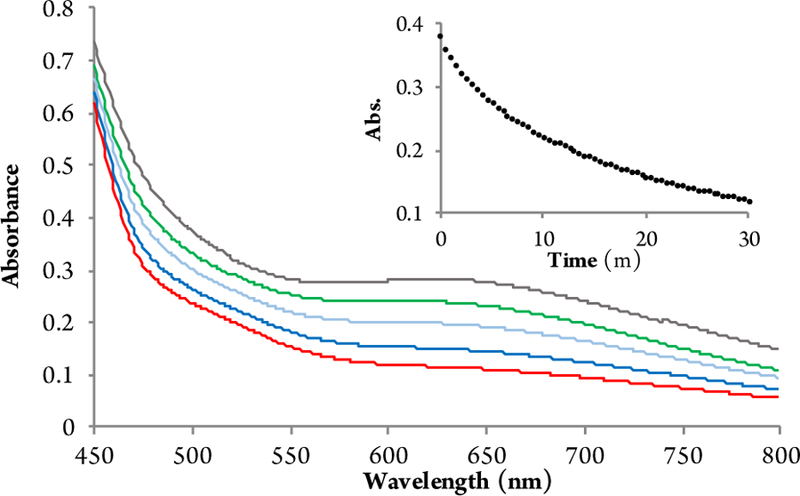
UV-Vis spectral changes observed for the reaction of 8 with Cr(V) complex in MeCN. Inset: decrease of the absorbance at 633 nm.
Time course experiments at 633 nm for the homo-coupling reactions of 7 and 8 were performed to measure rate of Cr(V) reduction in the presence of substrate (Figure 5). Notably, no change in the Cr(V) absorbance was noted in the absence of phenols under the reaction conditions (rt, MeCN). The reaction of 7 is much slower than the reaction of 8 [observed half-life t1/2 = 45.5 min (7), 8 min (8)].
Figure 5.

Exponential decay of the absorbance at 633 nm of (a) oxochromium(V) only (b) 7 (0.08 M) and oxo-chromium(V) complex (0.5 equiv) (c) 8 (0.08 M) and oxo-chromium(V) complex (0.5 equiv). All measurements were done in MeCN.
To understand the basis for this selectivity between phenols 7 and 8, several mechanisms were considered (Scheme 3) including outer sphere mechanisms involving hydrogen atom abstraction (path a), proton coupled electron transfer (path b PCET) or electron coupled proton transfer (path c ECPT). In addition, an inner sphere mechanism involving coordination of the phenol (path c) was evaluated.
Scheme 3.
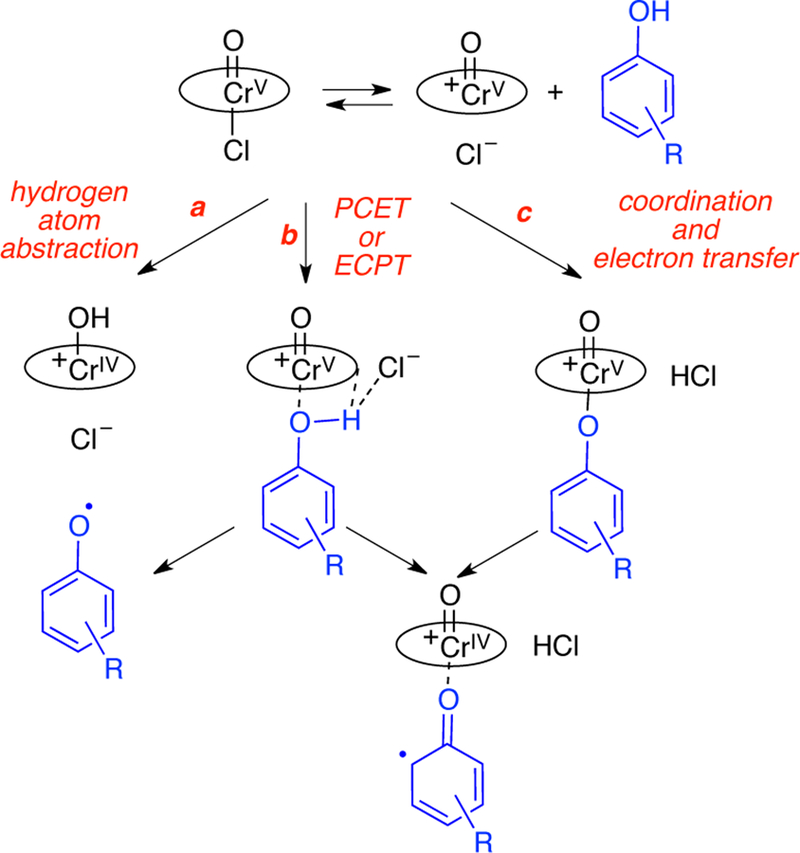
Possible pathways for initial oxidation of phenol by oxo-Cr(V) salen.
Deuterium Kinetic Isotope Effects (KIEs) for the HomoCoupling of 3,4,5-trimethylphenol.
To assess the hydrogen atom abstraction mechanism, deuterium kinetic isotope effects were measured. Parallel deuterium KIE studies were conducted for the homo-coupling reaction of proto- and deutero-8 (Scheme 4). The ortho positions of the phenol were deuterated along with the alcohol as a consequence of the deuterium incorporation protocol. For phenol reactions involving hydrogen atom abstraction mechanism, the KIE ranges from 1.2–5.9.17 The observed KIE of 1.14 thus rules out the possibility of the reaction proceeding via a rate-determining hydrogen atom abstraction from the phenol. This experiment is also inconsistent with alternative chromium oxidants that may form in the presence of oxygen, such as a Cr(IV) superoxide11c which engages in hydrogen atom abstraction.
Scheme 4.

Deuterium KIEs for the homo-coupling of 8 mediated by oxo-chromium(V).
Effect of Radical Inhibitors.
To further interrogate whether the phenoxy radical was forming, experiments were undertaken with radical inhibitors (Table 3). With diphenylethylene, BHT, and galvinoxyl, no change was seen in the conversion or the selectivity. On the other hand, TEMPO caused a shift in the selectivity. Based on the UV-Vis spectrum (Figure 3c above), TEMPO suppresses oxidation of the Cr(III). TEMPO is also sufficiently oxidizing to cause unselective coupling making it an inappropriate probe. Thus, it appears mechanisms that generate a free phenoxy radical are not operative, which includes hydrogen atom abstraction (path a, Scheme 3) or outer sphere electron transfer (path b, Scheme 3).
Table 3.
Effect of radical inhibitors on cross-coupling of 7 and 8 under catalytic conditions.
 | ||
|---|---|---|
| entry | radical inhibitor | NMR yield (%) |
| 1 | None | 78 |
| 2 | diphenylethylene | 80 |
| 3 | TEMPO | 35, 42 diphenoquinone |
| 4 | TEMPO (100 mol%) | 46, 75 diphenoquinone |
| 5 | BHT | 71 |
| 6 | galvinoxyl | 74 |
| ||
Oxidation Rates of Different Phenols.
To further the intermediacy of a phenoxide Cr(V) adduct (path c, Scheme 3) mass spectrometry experiments were undertaken after mixing phenol substrates for 1 h with the Cr(III) catalyst under oxygen (see SI). The Cr(III) salen, oxo-Cr(V) salen along with its acetonitrile complex, and μ-oxo chromium dimer were observed providing further evidence that oxygen can convert the Cr(III) into the oxo-Cr(V). However, no adducts with the phenols were found. Reasoning that such adducts are likely unstable due to a propensity to undergo electron transfer and react, indirect means were devised to gauge whether phenoxy coordination adducts were forming (path c, Scheme 3).
Specifically, the rates of oxidation of a series of 2,6-di-substituted phenols with the oxo-Cr(V) were measured to further probe this hypothesis (Figure 6). The results revealed that the reaction rate decreases as the steric bulk of the positions adjacent to the phenol increase: Me > iPr > tBu (half-life t1/2 = 23 min Me, 33.5 min i-Pr, 45.5 min t-Bu). Fitting of the data generated the rate constants shown in Figure 7.
Figure 6.
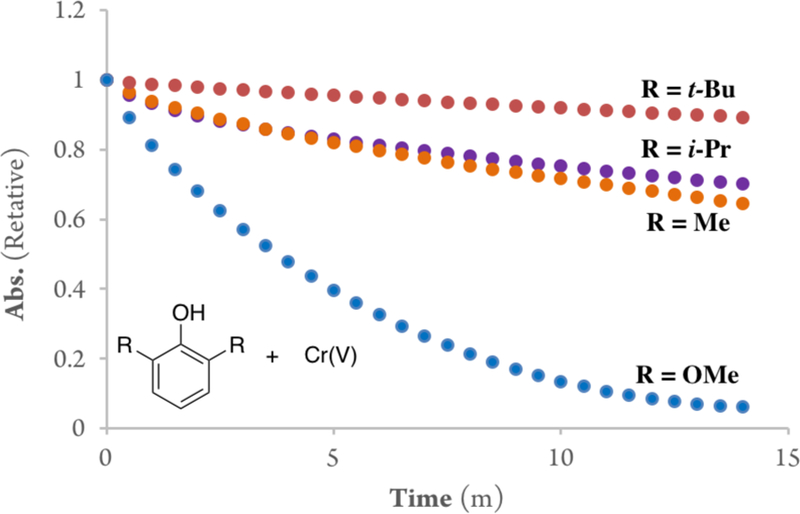
Absorbance decay profiles of Cr(V) to Cr(III) in MeCN at 633 nm in the presence of the 2,6-disubstituted phenols (0.5 mM, 2 equiv).
Figure 7.
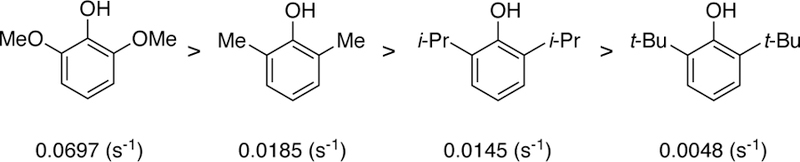
Reaction rate constants of 2,6-disubstituted phenols.
There was a good correlation between measured oxidation potentials and the reactions rates (see SI). Such a result is somewhat surprising when the electronic effects of the substituents are considered. The 2,6-dimethoxyphenol should be more reactive considering its electron rich character, but linear relationships were not seen between rate or oxidation potential using different Hammett parameters. It appears that the steric hindrance of the phenol plays a role in the relative reactivity both in this catalytic system and in oxidation at the electrode. Indeed, a good correlation was found between rate and the resonance Hammett parameter together with the B1 sterimol parameter (Figure 8). This scenario is consistent with a phenol coordinated species forming more readily from less sterically hindered phenols. Notably, this regression model allows reactivity to be estimated easily without having to undertake measurements of oxidation potentials.
Figure 8.
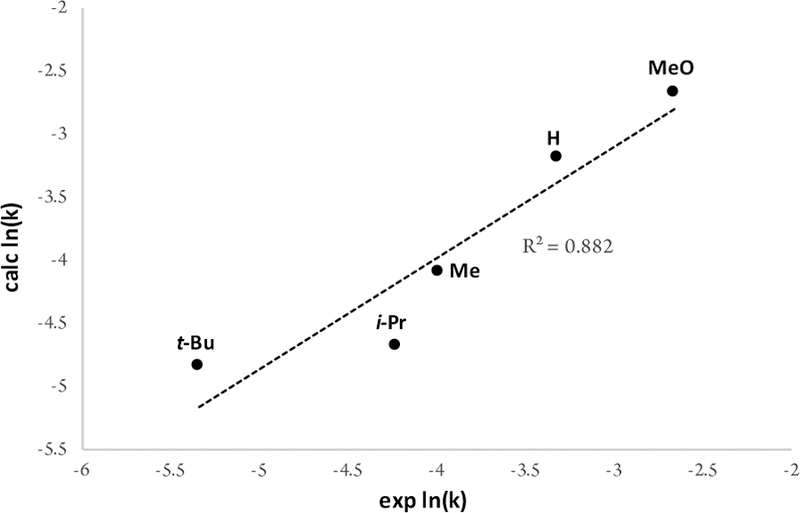
Multiple linear regression fit of ln(k) vs sterimol B1 parameter18 and the σR Hammett parameter19: ln(k) = –0.48(sterimolB)2 – 1.58(Hammett) – 3.58. Structures: 2,6-disubstituted phenols except for H = 3,4,5-trimethylphenol.
Oxo-chromium(V) Single Turnover Kinetics.
The above studies showed that the disappearance of oxochromium(V) could be readily monitored by UV-Vis spectroscopy, which provided an opportunity to gain insight into the kinetics after the rate determining step [oxidation of Cr(III)]. Single turnover experiments were, thus, undertaken to obtain the rate orders phenols 7 and 8 as well as oxochromium(V) (Figure 9).
Figure 9.
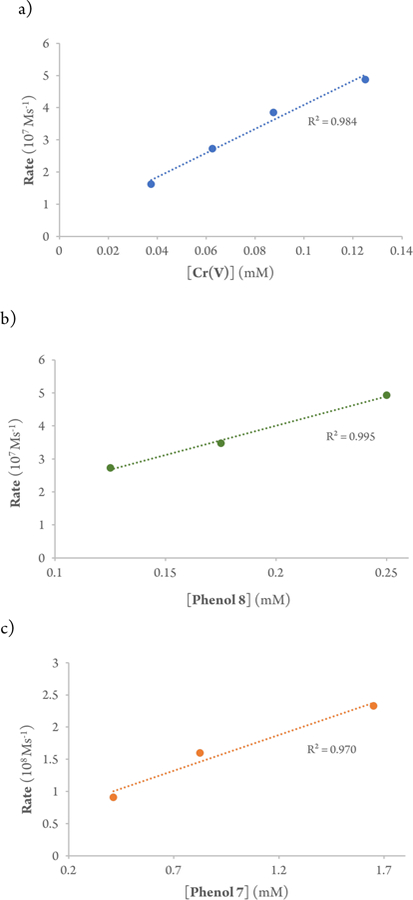
Stoichiometric kinetics for the cross-coupling reaction. (a) Correlation between reaction rate by monitoring loss of oxoCr(V) salen and concentration of Cr(V) (0.125, 0.0875, 0.0625, and 0.0375 mM) using an excess of 7 and 8 (2.5 mM). (b) Correlation between reaction rate by monitoring loss of oxo-Cr(V) salen and concentration of 8 (0.25, 0.175, and 0.125 mM) with excess 7 (2.5 mM) and Cr(V) (0.25 mM). (c) Correlation between reaction rate from GC measurements and concentration of 7 (0.41, 0.82, and 1.65 mM) using an excess of 8 (4.1 mM) and Cr(V) (0.41 mM).
Oxochromium(V).
Monitoring the consumption of the oxochromium(V) was undertaken at various concentrations (0.125, 0.0875, 0.0625, and 0.0375 mM) while holding the concentrations of 7 and 8 constant. A plot of ln rate vs ln concentration (See SI) shows that the reaction is first-order with respect to the oxoCr(V). This result is also supported by the linear correlation of the rate vs [Cr(V)]1 (Figure 9a).
Phenol 8.
A second set of kinetics was measured holding the oxo-Cr(V) and phenol 7 concentrations constant in order to determine the pseudo-rate order of phenol 8. The concentration of phenol 7 was varied (0.25, 0.175, and 0.125 mM) while the rate of loss of oxochromium(V) was measured. A rate vs [8]1 (Figure 9b) shows a linear correlation, which suggests that the reaction is first order with respect to 7 (results also supported by a double ln plot; see SI). However, a non-zero y-intercept was observed, which is attributed to simultaneous consumption of excess phenol 7 by the oxo-Cr(V) species.
Phenol 7.
A similar set of experiments was undertaken to probe phenol 7 using an excess of phenol 8. An apparent zero order consumption was observed. However, the oxidation profile of 7 and 8 (Figure 8) indicates that the latter oxidizes more quickly. Even though cross-coupling was predominantly observed, the UV/Vis method used is limited because the Cr(III) and Cr(IV) species have very similar UV absorption constants. Thus, only the Cr(V) to Cr(IV) transformation is observed which correlates to first one-electron oxidation rather than product formation.
Thus, a separate method, gas phase chromatography, was used in order to monitor formation of product. This approach allowed us to indirectly measure kinetics for phenol 7, by monitoring the formation of product over time. Pseudo-rate kinetics were determined by varying the concentration of phenol 7 (0.41, 0.82, and 1.65 mM) while the concentration of phenol 8 and oxochromium(V) were held constant. A plot of rate vs concentration showed a first-order dependence with respect to phenol 7 (Figure 9c).
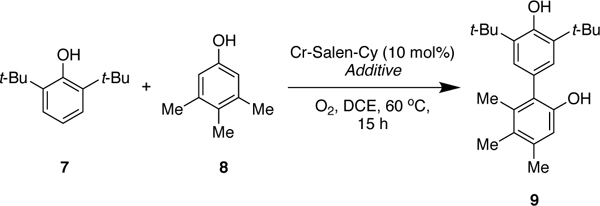
The above data is consistent with the catalytic cycle outlined in Figure 10. The catalytic kinetics show that the oxygen uptake by chromium is rate-determining (Figure 2). An oxo-Cr(V) was shown to be the active oxidant (Table 2, Figure 4). Further, reaction of oxo-Cr(V) with phenol was found to be kinetically faster than reaction of oxo-Cr(V) with Cr(III) to form the μ-oxo-Cr(IV) (Table 2). Under equilibrium conditions in the absence of phenol, the μ-oxo-Cr(IV) is the most stable species, and hence the catalyst resting state.
Figure 10.
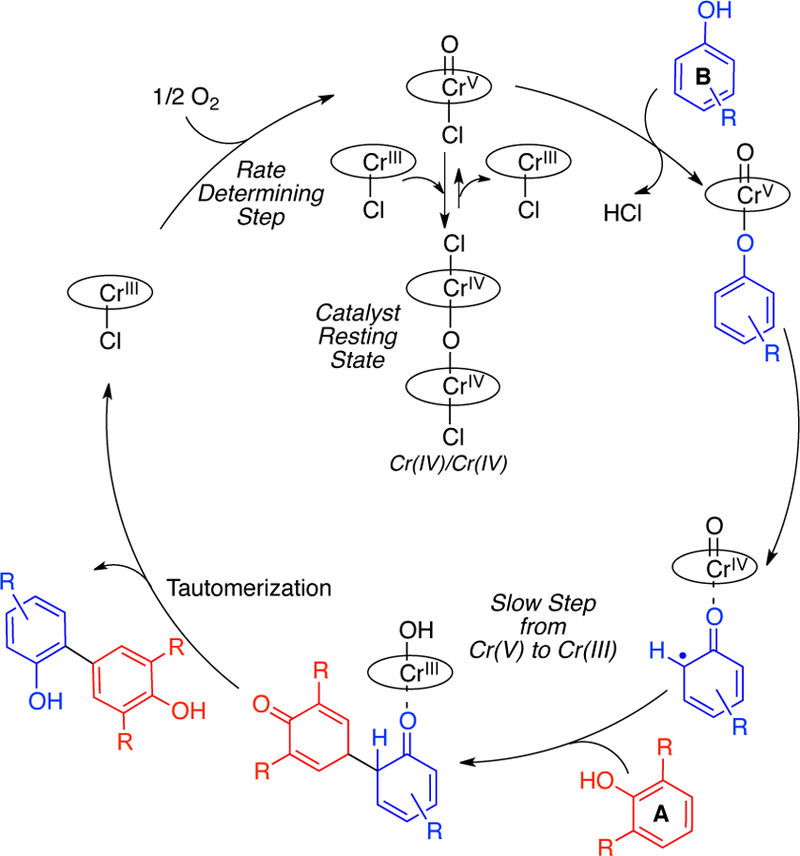
Preliminary catalytic cycle for cross-coupling phenols.
The slowest step from Cr(V) to Cr(III) is neither ligation or reaction of phenol B with the oxo-Cr(V), since such steps would be not be first order in phenol A (phenol 7 from Figure 9). These results indicate that a step involving both phenol A and phenol B is likely rate determining starting from the oxo-Cr(V). To gain further insight into this step that is likely also selectivity determining, the details of the proton and electron transfer events needed to be probed.
Role of the Phenol Hydroxyl Group.
For the mechanism in Figure 10, a free phenol is required for phenol B, but phenol A could be any nucleophilic arene. To probe this hypothesis, a comparison between the reactivity of TMS-phenols vs free-phenols was performed (Scheme 5).
Scheme 5.
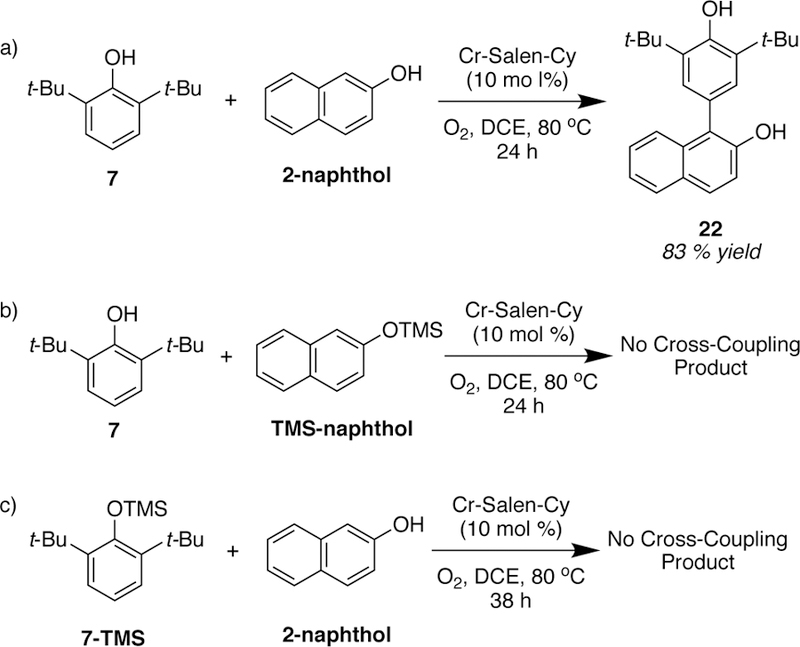
Reactions Performed to Probe the Importance of Free-Phenol.
The results showed that the cross-coupling of 7 with 2-naphthol afforded the desired cross-coupling product in 83% yield (Scheme 5a). However, when TMS-naphthol was submitted to the same reaction conditions, no detectable amount of the desired cross-coupling product was observed after 24 h (Scheme 5b). This result further supports that phenol B (TMS-naphthol) must possess a hydroxyl group in order to undergo ligand exchange and form the oxo-Cr(V)phenoxy adduct. A crystal structure showing coordination of pyridine N-oxide to the apical position of an oxo-Cr(V) salen20 also provides support for the proposed binding of phenol B.
Surprisingly, phenol A appears to be subject to the same requirement even though it is not proposed to ligate directly to the chromium. When 7-TMS was used in place of 7, no cross-coupling product was observed after 38 h (Scheme 5c). We postulate that phenol A must undergo a deprotonation prior to coupling, which modifies the catalytic cycle to involve coupling of the anion of phenol A with the chromium ligated phenol B.
To test this theory, experiments were performed with acid and base additives (Table 4). Three parallel reactions were conducted simultaneously over 15 h. Notably, the addition of acid (entry 2 vs entry 1) completely suppressed reactivity consistent with phenol deprotonation being integral. On the other hand, the results with a noncoordinating base, 2,6-di-tert-butyl-4-methylpyridine, (entry 3 vs entry 1) are consistent with generation of a phenoxide anion. The use of other bases, such as: Et3N, potassium tert-butoxide, potassium hydroxide, sodium hydroxide and potassium carbonate, led to decomposition.
Table 4.
Effect of acid or base.
 | ||
|---|---|---|
| entry | additive | NMR yield (%) |
| 1 | None | 78 |
| 2 | acetic acid (2 equiv) | 0 |
| 3 | 2,6-di-tert-butyl-4-methylpyridine (2 equiv) | 96 |
The higher yield with the hindered base vs without (entry 3 vs entry 1) was puzzling as the rate of the catalytic reaction is controlled by oxidation of the Cr(III) rather than the coupling of the phenoxide (see text below Figure 15). To assess whether phenoxide formation would accelerate the coupling, reactions with stoichometric Cr(V) was undertaken with and without base. The reaction with 2,6-di-tert-butyl-4-methylpyridine (2 equiv) and was found to be substantially faster (<1 min vs 30 min) providing further support for a deprotonation event.
Figure 15.
Proposed catalytic cycle for the cross-coupling of phenols.
Reaction Pathway Calculations.
Experimental data showed that the rate-determining step of the reaction was the formation of oxo-chromium(V), and that this specie was the active catalyst in the reaction. In addition, deuterium KIEs showed that the reaction does not proceed via a hydrogen atom abstraction mechanism. However, the nature of the cross-coupling selectivity was not fully understood. Based on these results, the reaction of 3,4,5-trimethylphenol (8) mediated by the oxo-chromium(V) was modeled using DFT calculations with 2,6-dimethylphenol as a model substrate vs the more computationally costly 2,6-di-tert-butylphenol (7). A simplified catalyst, containing methyl groups in place of tert-butyls, was also used in order to decrease the amount of computational time required to model the full system.
DFT calculations were performed using Gaussian 09. All geometry optimizations, and vibrational frequency calculations, of stationary points and transition states (TSs) were carried out at the UB3LYP level of theory with LANL2DZ basis set for Cr and 631G(d) basis set for all other atoms. Single point energies and solvent effects in DCE were computed at the UM06 level of theory with LANL2DZ basis set for Cr and 6–311+G(d,p) basis set for all other atoms, using the gas-phase optimized structures. Solvation energies were calculated by a self-consistent reaction field (SCRF) using the SMD solvation model. Both, homo- and cross-coupling of the phenols were calculated in order to explain the experimentally observed selectivity.
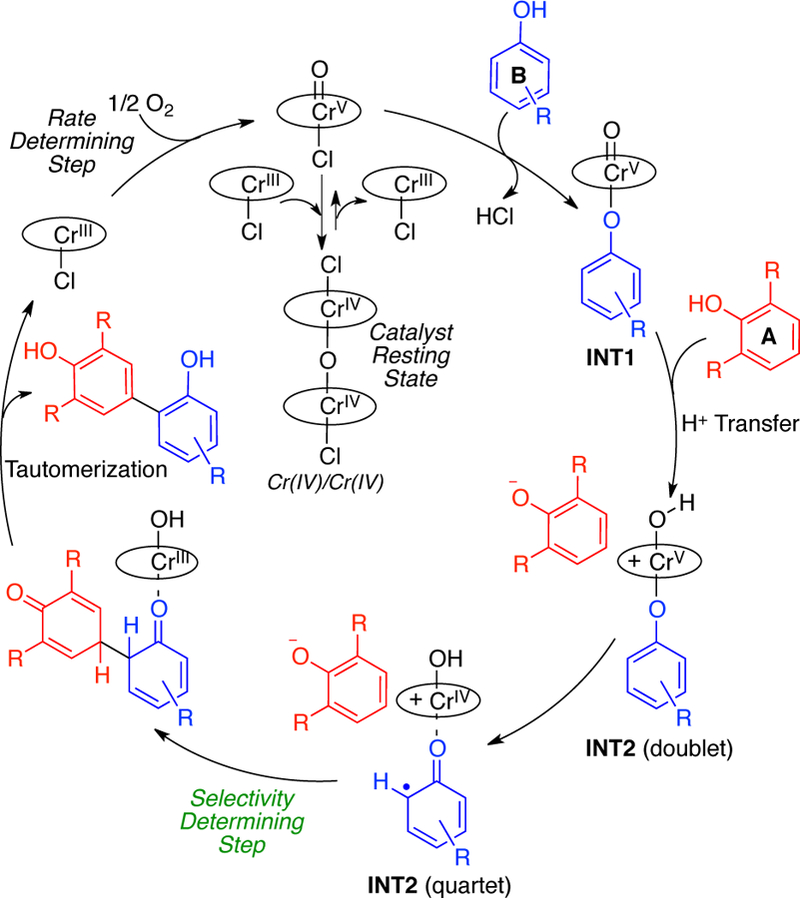
In addition, all possible spin states were assessed in order to evaluate the most favorable pathway (Figure 11). Starting from the active catalyst Cr(V), calculations suggest that the doublet spin state for this specie is considerably (7.6 kcal/mol) more stable. The next step of the process involves the coordination of the less hindered phenol 8 to afford INT1. A similar intermediate arising from the more hindered phenol (7 or 2,6-dimethylphenol) could not be located due to substantial steric interactions between the ortho-substituents of the phenol and the ligand of the catalyst. When comparing the energies of INT1, the quartet spin state was now found to be slightly lower (0.5 kcal/mol) in energy. The next step involves a proton transfer to the oxo-Cr(V) and subsequent oxidation of the coordinated phenol to afford INT2 via TSDeprot. Here, the doublet is slightly lower in energy for TSDeprot (0.6 kcal/mol) and the quartet for INT2 (0.5 kcal/mol). In the key cross-coupling of the two phenols, however, a large difference manifests in the barriers for the doublet (36.1 kcal/mol) vs the quartet (9.9 kcal/mol) with the former being essentially inaccessible. Overall, it appears that the doublet or quartet spin states are energetically similar after phenol coordination, but only the quartet leads to a reasonable barrier for the cross-coupling. Thus, an intersystem crossing occurs at some juncture. Addition of heavy atom additives such as iodobenzene did accelerate the reaction starting from the Cr(V) species consistent with such an event (see SI).21
Figure 11.
Doublet and quartet Gibbs free energy profile for the Cr-catalyzed cross-coupling reaction of phenols. Relative free energy values calculated with SMD-DCE-UM06/6–311+G(d,p); Cr: LANL2DZ//UB3LYP/6–31 G(d)
Notably, the calculations show that the slow step from the oxoCr(V), is deprotonation of the second phenol. However, there is charge separation in this transition state (TSDeprot) and such energies are notoriously difficult to compute even with solvation models.22 To determine if deprotonation, spin state crossover, or carbon-carbon bond formation was the slow and likely selectivity determining step, further experimental mechanistic studies were pursued.
13C Intermolecular Kinetic Isotope Effects.
13C KIE studies were performed for the catalytic cross-coupling of 7 with 8 (eq 2). The 13C KIEs for this homogeneous reaction were determined at natural abundance.23
For the analysis of samples of 7, two independent reactions at 70 °C were taken to 65 ± 2% and 78 ± 2% conversion. Reactions were carried out using an excess of 8 (2.5 equiv) to suppress formation of the bisphenoquinone of dimerized 7. The isolated unreacted phenol was analyzed by 13C NMR and compared to samples of the phenol that had not been submitted to the reaction conditions. The relative changes in 13C isotopic composition of the aromatic carbons were determined using the quaternary carbon of the tert-butyl groups as internal standard, assuming that their isotopic composition does not change during the course of the reaction. A similar approach was taken in order to analyze samples of 8, for which two independent reactions were taken to 78 ± 2% and 80 ± 2% conversion. Reactions were carried out using a slight excess of 7 (1.2 equiv) to suppress any possible homo-coupling of 8. From the changes in isotopic composition of the aromatic carbons, the 13C KIEs were calculated as previously described. 23
The results are summarized in Figure 12. From this intermolecular competition experiment, the slowest step involving the phenols can be probed even though the rate-determining step is oxidation of the catalyst. The para carbon of 7 exhibits a significant KIE. In addition, the ortho carbons of 8 also show significant and slightly larger KIEs.
Figure 12.

13C KIEs (k12/k13, 70 °C) for the reaction of 2,6-di-tert-butlyphenol (7) with 3,4,5-trimethylphenol (8).
Considering the three possibilities outlined above, selectivity being determined by a) deprotonation, b) spin state crossover, or c) the cross-coupling C-C bond formation, only the last is consistent with these isotope effects. In the first case the expected result will be a very small or negligible KIE; substrate 8 is not involved in the deprotonation and there is no change at the para carbon of 7. For the crossover, there is a change in the spin density on phenol 7 which might cause a very small secondary KIE and no change at all for phenol 8. However, a significant KIE is expected at the positions on both phenols involved in the C-C bond formation.24 With the observation of significant primary KIEs at the para carbon of 7 and the ortho carbons of 8, the results support the selectivity of the reaction being determined by the cross-coupling of the phenols.
These results in combination with the kinetics and DFT calculations suggest that the slow step from Cr(V) to Cr(III) is the phenol cross-coupling, which also controls the selectivity of the reaction.
Basis of Cross-Coupling Selectivity.
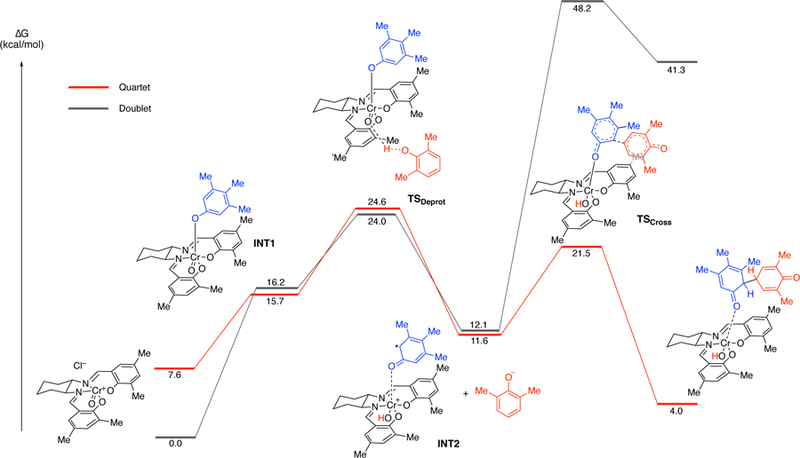
In order to understand why cross-coupling predominates over homo-coupling of the less hindered phenol (e.g., 8), further calculations were undertaken with 2,6-dimethylphenol (see SI) and 2,6-di-tert-butyl-phenol (Figure 13). Again, the barrier for the quartet was found to be substantially lower in energy and Figure 13 illustrates the free energy surface of the quartet spin state for both cross-coupling and homocoupling of 2,6-di-tert-butyl-phenol (7) with 3,4,5-trimethylphenol (8). Starting from INT1 where the less hindered phenol 8 is coordinated, either phenol 7 or phenol 8 can be deprotonated by the oxo-Cr(V) to form ion pair INT2. While two forms of INT2 with either phenoxide are close in energy, the barrier to the cross-coupling reaction via TSCROSS (6.8 kcal/mol), is lower than the barrier to the homo-coupling reaction via TSHOMO (11.9 kcal/mol). This 5.1 kcal/mol difference accounts for the observed selectivity, namely the absence of the homocoupling product for this substrate pair at the temperature of the reaction (60–80 °C). Subsequent release of the adduct and tautomerization leads to the product and regenerates the catalyst. A similar trend was observed for the reaction of 3,4,5-trimethylphenol (8) with 2,6-dimethylphenol (44), with the difference in the barrier for homo- vs cross-coupling being 1.3 kcal/mol, favoring the cross-coupling product. Experimental results showed that for this reaction a small amount of the homocoupling product is obtained (5% homo-coupling vs 87% cross-coupling, see Figure 18 and discussion below).
Figure 13.
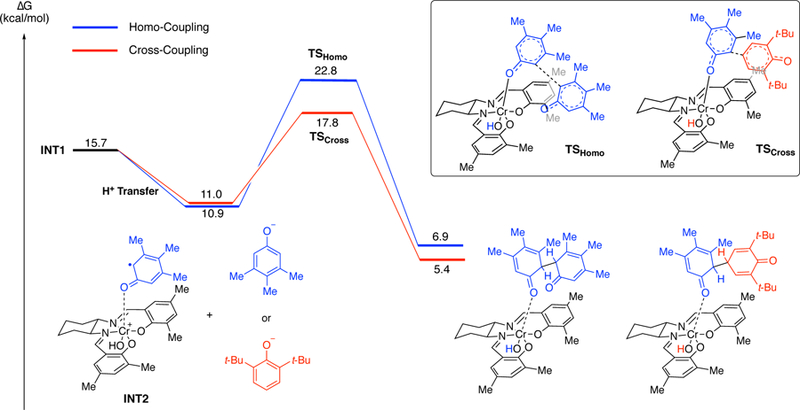
Gibbs free energy profile for the Cr-catalyzed cross- and homo-coupling reaction of phenol (quartet surface shown). Relative free energy values calculated with SMD-DCE- UM06/6–311+G(d,p); Cr: LANL2DZ//UB3LYP/6–31 G(d);Cr:LANL2DZ.
Figure 18.
Examples of reactions that did not selectively afford the desired cross-coupling product. a Isolated yield.
With the carbon-carbon bond forming event established as key to selectivity, the reasons behind the difference in energy of the transition states controlling the selectivity, TSCROSS and TSHOMO, were probed. The charges and spin densities were almost identical for each comparable center in the two transition states. To gauge the orbitals involved, the singly occupied orbitals of the quartet forms of INT2 were examined for the corresponding cross-coupling and homo-coupling (Figure 14 for 7 and 8, see SI for orbitals from 44 and 8). For the homo-coupling INT2, there is significant single electron character on the catalyst portion in all three of the singly occupied orbitals. For the HOMO and HOMO−2, there is also substantial single electron character on the phenol B bound to the chromium metal center. A similar distribution is seen in the HOMO and HOMO−1 orbitals of cross-coupling INT2. However, the HOMO−2 differs greatly between the INT2 precursors. In the homo-coupling, there is no single electron density on the phenoxide. On the other hand, there is a large amount of single electron character at phenol A. As such, a radical-anion best represents the homo-coupling INT2 whereas a radical-radical form best represents the cross-coupling INT2. The barrier for carbon-carbon bond formation from the radical-radical form is lower in energy as less electronic reorganization is required in the transition state. As a result, cross-coupling is more facile. The same orbital features were observed for the cross-coupling reaction with 2,6-dimethylphenol (See SI).
Figure 14.
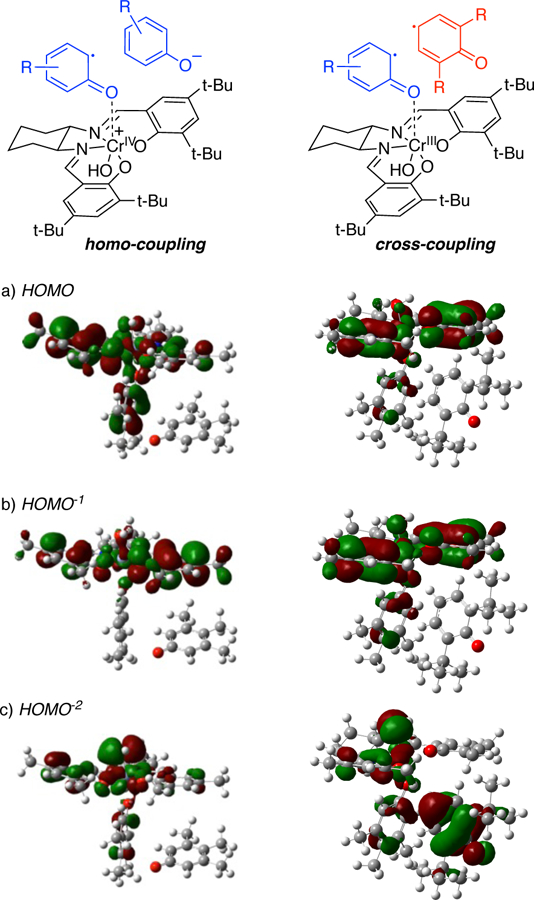
Singly occupied molecular orbitals (MOs) of INT2 for homo-coupling and cross-coupling.
Overall Mechanism.
The mechanism outlined in Figure 15 is consistent with the data described above. The rate-determining step of the reaction is the oxidation of the Cr(III) catalyst to the active Cr(V) oxidant. Single turnover rate orders with Cr(V), DFT calculations, and natural abundance KIE data show that the highest energy step involving the phenols is the C-C bond formation.
The selectivity of the reaction is determined at two stages. During the first stage, one of the phenols is better able to bind to the chromium catalyst and undergo oxidation forming INT1 (phenol B). A proton transfer from the second phenol (phenol A) to the catalyst leads to INT2, which constitutes the second decision point. More acidic substrates (i.e. phenol A) deprotonate more readily forming INT2 which results in carbon-carbon bond formation. After carbon-carbon bond formation, release of the adduct regenerates the catalyst. Subsequent tautomerization yields the final biphenol.
This mechanism can be used to assess the likelihood that any given pair of phenols will undergo an effective cross-coupling. For a phenol to participate, it must be oxidizable by the oxoCr(V) (–1.64 eV). For phenols in this range, the ability to bind the catalyst and oxidize first can be estimated with Hammett and sterimol parameters (see Figure 8 above).
Readily calculated gas phase acidities (Figure 16) provide a good measure of which phenol will most readily deprotonate. For example, the results of Scheme 1c are aptly explained. 3,4,5-rimethylphenol (Scheme 1c) (8) is clearly less hindered and more oxidizable that 2,6-di-tert-butylphenol (7), so it is taken up selectively by the chromium catalyst. Given the very similar global nucleophilicities of the neutral species (3.43 and 3.44, respectively, see SI) per Pappo’s analysis,8a it was unclear why the reaction was so high yielding and so selective; <2% of 3,4,5-trimethylphenol dimer was observed even though the global nucleophilicities would predict formation of this byproduct. Examination of the global nucleophilicities of the anions (Figure 16) also anticipate homo-coupling. On the other hand, the pKa values predict that 7 (127.5) would form the ion pair INT2 more readily than 8 (133.8). Further, addition of 2,6-di-tert-butyl-4-methylpyridine, gave rise to a higher yield of cross-coupled product (see Table 4, entry 3 vs 1) due greater selectivity (no 3,4,5-trimethylphenol dimer was observed) and, in this particular case, less decomposition. Similarly, the results from 9–19 and 21 in Figure 1 are in accord with this model. For 22–30, the naphthols are expected to be more oxidizable and more acidic. However, it appears that 2-naphthols do not deprotonate and homo-couple due to disfavorable sterics in formation of a hindered C–C biaryl bond.
Figure 16.
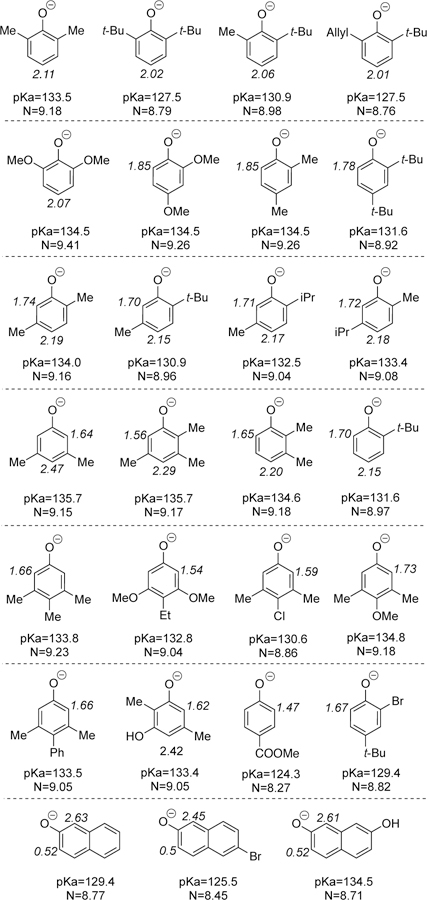
Calculated overall and site-nucleophilicities for several phenols. (N refers to overall nucleophilicity; site-nucleophilicitiesare labeled for selected positions of the phenols along with the charge in brackets. All nucleophilicities are in eV.)
Regioselectivity and Site Nucleophilicity.
In cases where more than one reaction site exists on the phenols, different regioisomers are possible. In all cases, only one isomer was observed in these processes. If two sites are available on phenol B, then reaction occurs at the less hindered site which explains the outcomes for 14–19 and 21 in Figure 1. Again, naphthols appear to be an exception (22–30) and reaction occurs at the site that causes the least loss of aromaticity in the keto-radical intermediate.
If two sites are available on phenol A, the most nucleophilic site reacts. While global nucleophilicity scales derived from HOMO energies9a provide a useful estimation of the nucleophilicity of the entire molecule, they do not permit multiple sites within a single molecule to be differentiated. In order to do so, we developed a measure of local kinetic nucleophilicities which is simple to implement using only the orbital coefficient at a given atom along with the HOMO energy level.25 The thus calculated site-specific nucleophilicities for a series of phenoxides are shown in Figure 16 along with the overall nucleophilicities.
For compound 20 from Figure 1, several anomalies arose in applying our model to this high yielding selective coupling. It was unclear which substrate was undergoing oxidation first. Cyclic voltammetry measurements revealed that 2-tert-butyl-6-methylphenol was slightly more oxidizable and functions as phenol B (see SI). The pKa values for the two monomers were, however, identical begging the question of the cross-selectivity. Theorizing that both anions could form, but that the more nucleophilic would react, we turned to global nucleophilicities. However, these values would anticipate a homo-coupling. Site nucleophilicities proved more predictive indicating that 2-tert-butyl-5-methylphenol would be more reactive. In addition, these parameters indicated that the para-position (2.15) within the molecule was substantially more nucleophilic than the ortho-position (1.70), thus accounting for the regioselection observed. Ortho- and para-positions both obtain electron density through resonance, but ortho-positions are more strongly affected by the inductive withdrawing effect of the OH, which reduces nucleophilicity.
Predictions from the Model.
Finally, our proposed mechanism for this reaction involves oxidation of a phenol that can readily bind the catalyst, followed by selective deprotonation of a second phenol to form an ion-pair. Intersystem crossing from this adduct then allows an intramolecular radical-radical recombination. By taking into consideration the electronics and binding-ability of phenols to the Cr-catalyst, along with pKa values and site nucleophilicities, we tested the ability of this mechanistic construct to predict if given pairs would react and if we could anticipate the product isomers.
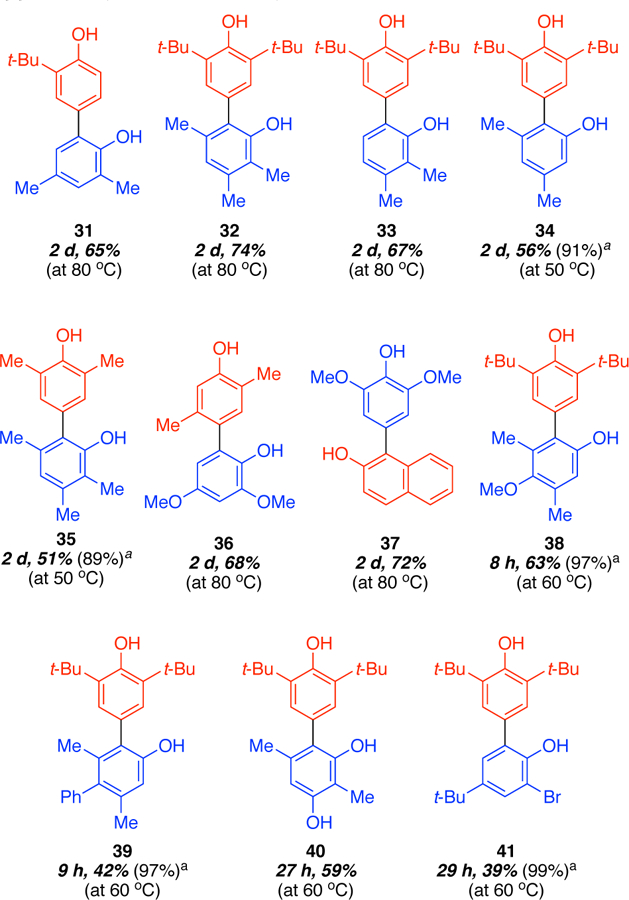
Our discovery efforts (Figure 1) had identified 2,6-disubstituted phenols as reaction partners that readily underwent selective cross-coupling with a range of phenol partners with the chromium catalysts. To determine whether it was possible to move away from having one partner being a 2,6-disubstituted phenol, 2-tert-butylphenol was assessed. We reasoned that this reaction partner would still be sufficiently hindered to disfavor direct binding to the chromium catalyst. For the other partner, 2,4-dimethylphenol was utilized. The molecular nucleophilicity scale rates the 2,4-dimethylphenol (neutral: 3.46 vs 3.21; anionic 9.26 vs 8.97) as more nucleophilic which predicts homo-coupling to predominate. On the other hand, the pKa values indicate that the 2-tert-butylphenol should deprotonate more readily (131.6 vs 134.5). In addition, the site nucleophilicity scale predicts that the most nucleophilic site is the para position of 2-tert-butylphenol (ortho 1.70, para 2.15). Together, these elements predict that a selective para-ortho cross-coupling should occur which was verified experimentally (31, Figure 17). Notably, good yields in coupling of mono-ortho substituted phenols like 2-tert-butylphenol are difficult to obtain.9
Figure 17.
Examples of cross-coupling products for which the region selectivity could be predicted by our model. a Yield based on recovered starting material.
Additional couplings were attempted (32–35, Figure 17) where our model, utilizing sterics/electronics and pKa, predicted that cross-coupling would occur. In all cases, good yields and selectivities were observed. Notably, a model based on oxidizability and global nucleophilicity anticipated homocoupling of phenol B as the major product.
For the phenol bound to the chromium catalyst (phenol B), the cross-coupling was hypothesized to occur at the least sterically hindered site. This hypothesis explains the para-para coupled products we obtained in the reaction scope. To shift the outcome, additional steric hindrance was introduced around the para- position to obtain ortho-para coupled products, such as in the use of 2,3,5trimethylphenol (32, 35), 2,3-dimethylphenol (33), and 3,5-dimethylphenol (34). In each case, coupling was observed at the least sterically hindered ortho- position.
For compound 36, 2,4-dimethoxyphenol is expected to be less sterically hindered with respect to binding the Cr catalysts as well as more oxidizable electronically. The 2,5-dimethylphenol is 0.5 pKa units more acidic predicting a cross-coupling. The site nucleophilicities further reinforce 2,5-dimethylphenoxide as more reactive than 2,4-dimethoxyphenoxide (1.85) and also corroborate the coupling at the para-position (2.19 vs 1.74).
In coupling of 2,6-dimethoxyphenol with 2-naphthol, the former is more readily oxidized (phenol B). Using global nucleophilicities alone would suggest that it is also the most nucleophilic, which would lead to the homo-coupled 2,6-dimethoxyphenol product. However, 2-naphthol is more acidic (129.4 vs 134.5) which correlates well with the observed outcome in formation of 37. The 1-position of 2-naphthoxide possessed a much higher site nucleophilicity (2.61 vs 0.52) than the 3-position in accord with general trends observed in 2-naphthol couplings.
Compounds 38 and 39 show that the method tolerates well variations in the electronic properties of the phenols. For example, in the case of 38, we expect the methoxy substituent to decrease the oxidation potential of the 3,5-dimethyl-4-methoxy phenol by donation of electron density into the ring. When pKa values are taken in consideration, we find that 2,6-di-tert-butylphenol (7) is significantly more acidic than 3,5-dimethyl-4-methoxy phenol (127.5 vs 134.8) allowing it to act as the phenol A partner, leading to the formation of the desired cross-coupling product. Compound 39 shows that the presence of slightly electron-withdrawing groups allows the formation of the desired cross-coupling products. In this case, the presence of a phenyl substituent on 3,5-dimethyl-4-phenyl phenol is expected to slightly increase the oxidation potential, but still allow it to act as the more oxidizable partner. When the pKa values of 7 and 3,5-dimethyl-4-phenyl phenol are compared we find that 7 is still more acidic (127.5 vs 133.5), allowing 7 to act as the nucleophilic partner, leading to the formation of the cross-coupling product.
In order to explore how the incorporation of additional hydroxyl groups would affect the cross-coupling, 2,5-dimethylresorcinol was coupled with 2,6-di-tert-butylphenol to afford 40. The 2,5-dimethylresorcinol acts as the more oxidizable partner, and the pKa values suggest that 2,6-di-tert-butylphenol reacts as the phenol A partner (127.5 vs 133.4), leading to formation of the desired compound 40.
Compound 41was obtained from the cross-coupling of 2,6-ditert-butylphenol with 2-bromo-4-tert-butyl phenol. The 2-bromo4-tert-butyl phenol acts as the more oxidizable partner, while 2,6di-tert-butylphenol is phenol A. The observed reactivity can be explained since 2,6-di-tert-butylphenol is more acidic (127.5 vs 129.4). The formation of desired compound 41 shows a broader applicability of this chemistry.
Limitations in Reactivity Anticipated from the Model.
The developed model has been shown to successfully predict the selective formation of cross-coupling products, based on the electronis, sterics, acidity, and site nucleophilicities of the reacting phenols. The model also correctly accounts for the selectivity of reactants that led to either homo-coupling of one of the phenolic species or to a mixture of homo- and cross-coupling products. A few examples are outlined in Figure 18.
For the reaction of 3,4,5-trimethylphenol (8) with 2,5-dimethylresorcinol (42) we only observed the homo-coupling product 43. When analyzing this result using our model we find that 42 is both more acidic and nucleophilic (pKa= 133.4, site N = 2.42) than its coupling partner 8 (pKa = 133.8, site N = 1.66) supporting the observed homo-coupling product.
Moreover, the reaction of methyl p-hydroxybenzoate (44) with 2,6-di-tert-butylphenol (7) showed the importance of the oxidation potential and the nucleophilicity of the phenols. For this reaction, the only product observed was 45, which corresponds to the homo-coupling of phenol 7. In this case, 44 has a high onset oxidation potential (1.47 eV) due to the electron-withdrawing nature of the ester group, rendering phenol 7 more oxidizable. In addition, the electron-withdrawing nature of the ester group will reduce the nucleophilicity of 44 (site N = 1.47 vs 2.02), decreasing the reactivity of the substrate after deprotonation and leading to the formation of homo-coupling product 45.
The reaction of 2,6-dimethylphenol (46) with 3,4,5-trimethylphenol (8) led to the formation of both homo- and cross-coupling products, in isolated yields of 5% and 87%, respectively. This reactivity is consistent with our model; the acidity of these two phenols is very similar (pKa=133.4 and 133.3 respectively) which will lead to a similar amount of deprotonated phenols in solution. However, the site nucleophilicity of 46 is significantly higher than that of 8 (2.11 vs 1.66) leading to the formation of the cross-coupling product predominantly.
CONCLUDING REMARKS
In summary, the mechanism of the selective Cr-catalyzed cross-coupling of phenols was investigated via a combination of experimental and computational studies. The results showed that the rate-determining step of the reaction is the oxidation of the Cr(III) to the active Cr(V). In addition, pseudo-rate orders, DFT calculations, and KIE data showed that the selectivity of the cross-coupling reaction is determined during carbon-carbon bond formation. The preponderance of the evidence supports the catalytic cycle outlined in Figure 15. The first step is rate-determining oxidation of the Cr(III) pre-catalyst to the active Cr(V) catalyst. In contrast to past work in epoxidation with chromium oxo species, oxygen was found to be an effective terminal oxidant.14d Even so, this step could be accelerated by use of different oxidants, so long as direct oxidation of the substrates could be avoided. Future catalyst design will focus on further accelerating this step, while also suppressing formation of the less reactive Cr(IV) complex. From here, the phenol that can more readily coordinate and oxidize (phenol B) generates Cr(V) adduct INT1 (Scheme 6, item 2). The oxo-Cr(V) causes deprotonation of a the more acidic phenol (phenol A) to generate ion pair INT2 (Scheme 6, item 3), which upon intersystem crossing would generate the quartet version that is best characterized as containing two phenoxy radicals. Carbon-carbon bond formation from this intermediate is lower in energy for the cross-coupling than the homo-coupling due to a lesser degree of electronic reorganization being needed. Regioselection is determined by steric hindrance for phenol B (Scheme 6, item 4) or site nucleophilicity for phenol A (Scheme 6, item 5). Finally, release of the adduct and tautomerization generates the product and regenerates the catalyst.
Scheme 6.
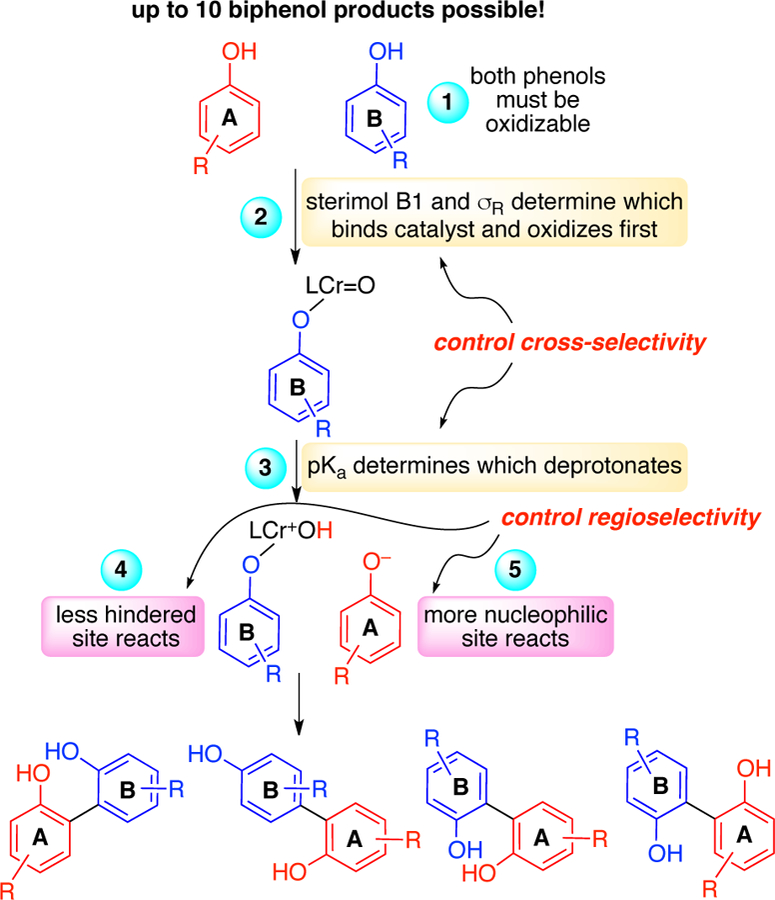
Flowchart to determine product outcomes.
The mechanistic construct outlined allows prediction of outcomes for different phenol pairs. Using steric and electronic parameters to assess catalyst binding/oxidation in combination with pKa values and site nucleophilicites shows good fidelity in anticipating reactivity, cross-selectivity, and regioselectivity. A greater degree of mechanistic understanding will allow the design of further catalyst systems and oxidants to access these important adducts.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NIH (GM-112684, GM-08765) and the NSF (CHE1464778, CHE1764298) for financial support of this research. Computational support was provided by XSEDE (TG-CHE120052). Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, 1S10RR022442, CHE-0840438, CHE-0848460, 1S10OD011980). Drs. Rakesh Kohli and Charles W. Ross III (UPenn) are acknowledged for obtaining HRMS data. We are grateful to Prof. David Chenoweth (UPenn) for use of a UV-vis spectrometer. We thank Prof. Joseph Subotnik (UPenn) and Prof. Daniel Singleton (Texas A&M University) for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental protocols and data, calculations, and NMR spectra. The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Quideau S; Feldman KS, Ellagitannin Chemistry. Chem. Rev 1996, 96, 475–504. [DOI] [PubMed] [Google Scholar]
- (2).Kočovský P; Vyskočil Š; Smrčina M, Non-Symmetrically Substituted 1,1’-Binaphthyls in Enantioselective Catalysis. Chem. Rev 2003, 103, 3213–3246. [DOI] [PubMed] [Google Scholar]
- (3).(a) Beletskaya IP; Cheprakov AV, The Heck Reaction as a Sharpening Stone of Palladium Catalysis. Chem. Rev 2000, 100, 3009–3066 [DOI] [PubMed] [Google Scholar]; (b) Hassan J; Sévignon M; Gozzi C; Schulz E; Lemaire M, Aryl−Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev 2002, 102, 1359–1470 [DOI] [PubMed] [Google Scholar]; (c) Miyaura N; Suzuki A, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95, 2457–2483 [Google Scholar]; (d) Ming Ge H; Yun Zhang W; Ding G; Saparpakorn P; Chun Song Y; Hannongbua S; Xiang Tan R, Chaetoglobins A and B, two unusual alkaloids from endophytic Chaetomium globosum culture. Chem. Comm 2008, 1, 5978–5980. [DOI] [PubMed] [Google Scholar]
- (4).(a) Esguerra KVN; Fall Y; Lumb J-P, Catalytic aerobic oxidation of halogenated phenols. Inorganica Chim. Acta 2018, 481, 197–200 [Google Scholar]; (b) Esguerra KVN; Lumb J-P, Synthesis of orthoAzophenols by Formal Dehydrogenative Coupling of Phenols and Hydrazines or Hydrazides. Chem. Eur. J 2017, 23, 8596–8600 [DOI] [PubMed] [Google Scholar]; (c) Esguerra KVN; Lumb J-P, A Bioinspired Catalytic Aerobic Functionalization of Phenols: Regioselective Construction of Aromatic C–N and C–O Bonds. ACS Cat 2017, 7, 3477–3482. [Google Scholar]
- (5).(a) Pal T; Pal A, Oxidative phenol coupling: A key step for the biomimetic synthesis of many important natural products. Curr. Sci 1996, 71, 106–108 [Google Scholar]; (b) Bringmann G; Gulder T; Gulder TAM; Breuning M, Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev 2011, 111, 563–639 [DOI] [PubMed] [Google Scholar]; (c) Solinski AE; Ochoa C; Lee YE; Paniak T; Kozlowski MC; Wuest WM, Honokiol-Inspired Analogs as Inhibitors of Oral Bacteria. ACS Infec. Dis 2018, 4, 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jaracz S; Kozlowski M,C; Eun Lee Y; Kim SM (April 27, 2017) Improved Synthesis of Honokiol, WO/2017/070568 ; (e) Xu W; Huang Z; Ji X; Lumb J, Catalytic Aerobic Cross-Dehydrogenative Coupling of Phenols and Catechols. ACS Catal 2019, 9, 3800–3810. [Google Scholar]; (f) Huang Z; Lumb J, Phenol-Directed C-H Functionalization. ACS Catal 2019, 9, 521–555. [Google Scholar]
- (6).(a) Elsler B; Schollmeyer D; Dyballa KM; Franke R; Waldvogel SR, Metal- and Reagent-Free Highly Selective Anodic Cross-Coupling Reaction of Phenols. Angew. Chem. Int. Ed 2014, 53, 5210–5213. [DOI] [PubMed] [Google Scholar]; (b) Kirste A; Elsler B; Schnakenburg G; Waldvogel SR, Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J. Am. Chem. Soc 2012, 134, 3571–3576 [DOI] [PubMed] [Google Scholar]; (c) Dahms B; Franke R; Waldvogel Siegfried R, Metal- and Reagent-Free Anodic Dehydrogenative Cross-Coupling of Naphthylamines with Phenols. ChemElectroChem 2018, 5, 1249–1252 [Google Scholar]; (d) Quell T; Beiser N; Dyballa Katrin M; Franke R; Waldvogel Siegfried R, Facile and Selective Cross-Coupling of Phenols Using Selenium Dioxide. Eur. J. Org. Chem 2016, 4307–4310; (e) Riehl B; Dyballa KM; Franke R; Waldvogel SR, Electro-organic Synthesis as a Sustainable Alternative for Dehydrogenative Cross-Coupling of Phenols and Naphthols. Synthesis 2017, 49, 252–259. [Google Scholar]; (f) Dahms B; Kohlpaintner PJ; Wiebe A, Breinbauer R; Schollmeyer D and Waldvogel SR, Selective Formation of 4,4′‐Biphenols by Anodic Dehydrogenative Cross‐ and Homo‐Coupling Reaction. Chem. Eur. J 2019, 25, 2713–2716. [DOI] [PubMed] [Google Scholar]
- (7).(a) More NY; Jeganmohan M, Oxidative Cross-Coupling of Two Different Phenols: An Efficient Route to Unsymmetrical Biphenols. Org. Lett 2015, 17, 3042–3045 [DOI] [PubMed] [Google Scholar]; (b) More Nagnath Y; Jeganmohan M, Oxidative Cross-Coupling of Substituted Phenols with Unactivated Aromatics. Eur. J. Org. Chem 2017, 4305–4312; (c) More NY; Jeganmohan M, Solvent-controlled selective synthesis of biphenols and quinones via oxidative coupling of phenols. Chem. Comm 2017, 53, 9616–9619. [DOI] [PubMed] [Google Scholar]
- (8).(a) Libman A; Shalit H; Vainer Y; Narute S; Kozuch S; Pappo D, Synthetic and Predictive Approach to Unsymmetrical Biphenols by Iron-Catalyzed Chelated Radical–Anion Oxidative Coupling. J. Am. Chem. Soc 2015, 137, 11453–11460 [DOI] [PubMed] [Google Scholar]; (b) Shalit H; Libman A; Pappo D, meso-Tetraphenylporphyrin Iron Chloride Catalyzed Selective Oxidative Cross-Coupling of Phenols. J. Am. Chem. Soc 2017, 139, 13404–13413. [DOI] [PubMed] [Google Scholar]
- (9).Lee YE; Cao T; Torruellas C; Kozlowski MC, Selective Oxidative Homo- and Cross-Coupling of Phenols with Aerobic Catalysts. J. Am. Chem. Soc 2014, 136, 6782–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Narute S; Parnes R; Toste FD; Pappo D, Enantioselective Oxidative Homocoupling and Cross-Coupling of 2Naphthols Catalyzed by Chiral Iron Phosphate Complexes. J. Am. Chem. Soc 2016, 138, 16553–16560. [DOI] [PubMed] [Google Scholar]
- (11).(a) Park J; Lee J; Jung E; Park Y; Kim K; Park B; Jung K; Park E; Kim J; Park D In vitro antibacterial and anti-inflammatory effects of honokiol and magnolol against Propionibacterium sp., Eur. J. Pharmacol 2004, 496, 189–195. [DOI] [PubMed] [Google Scholar]; (b) Kim Y-S; Lee J-Y; Park J; Hwang W; Lee J; Park D, Synthesis and Microbiological Evaluation of Honokiol Derivatives as New Antimicrobial Agents. Arch. Pharm. Res 2010, 33, 61–65. [DOI] [PubMed] [Google Scholar]; (c) Jones JR; Lebar MD; Jinwal UK; Abisambra JF; Koren J III, Blair L; O’Leary JC; Davey Z; Trotter J; Johnson AG; Weeber E; Eckman CB; Baker BJ; Dickey CA, The Diarylheptanoid (+)-aR,11S-Myricanol and Two Flavones from Bayberry (Myrica cerifera) Destabilize the Microtubule-Associated Protein Tau. J. Nat. Prod 2011, 74, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Calcul L; Jinwal UK; Dickey CA; Baker BJ; The Diarylheptanoid(+)-S-myricanol from Myrica cerifera (Bayberry) and Its Derivatives Destabilize the Microtubule-Associated Protein Tau. Planta Med 2012, 78-PI472. [Google Scholar]; (e) Tasler S; Bringmann G, Biarylic Biscarbazole Alkaloids: Occurrence, Stereochemistry, Synthesis, and Bioactivity. The Chemical Record, 2002, 2, 113–126. [DOI] [PubMed] [Google Scholar]; (f) Wube AA; Bucar F; Asres K; Gibbons S; Adams M; Streit B; Bodensieck A; Bauer R, Knipholone, a selective inhibitor of leukotriene metabolism. Phytomedicine 2006, 13, 452–456. [DOI] [PubMed] [Google Scholar]; (g) Tamehiro N; Sato Y; Suzuki T; Hashimoto T; Asakawa Y; Yokoyama S; Kawanishi T; Ohno Y; Inoue K; Nagao T; Nishimaki-Mogami T, Riccardin C: A natural product that functions as a liver X receptor (LXR)α agonist and an LXRβ antagonist. FEBS Lett 2005, 579, 52995–304. [DOI] [PubMed] [Google Scholar]
- (12).(a) Woo LK, Intermetal oxygen, sulfur, selenium, and nitrogen atom transfer reactions. Chem. Rev 1993, 93, 1125–1136 [Google Scholar]; (b) Schuster C; Möllmann E; Tompos A; Hölderich WF, Highly stereoselective epoxidation of (−)-α-pinene over chiral transition metal (salen) complexes occluded in zeolitic hosts. Cat. Lett 2001, 74, 69–75 [Google Scholar]; (c) Cho J; Woo J; Eun Han J; Kubo M; Ogura T; Nam W, Chromium(V)-oxo and chromium(III)-superoxo complexes bearing a macrocyclic TMC ligand in hydrogen atom abstraction reactions. Chemical Science 2011, 2, 2057–2062. [Google Scholar]
- (13).(a) Premsingh S; Venkataramanan NS; Rajagopal S; Mirza SP; Vairamani M; Rao PS; Velavan K, Electron Transfer Reaction of Oxo(salen)chromium(V) Ion with Anilines. Inorg. Chem 2004, 43, 5744–5753 [DOI] [PubMed] [Google Scholar]; (b) Siddall TL; Miyaura N; Huffman JC; Kochi JK, Isolation and molecular structure of unusual oxochromium(V) cations for the catalytic epoxidation of alkenes. J. Chem. Soc., Chem. Comm 1983, 1, 1185–1186. [Google Scholar]
- (14).(a) Choi CH; Kwon HC; Yook S; Shin H; Kim H; Choi M, Hydrogen Peroxide Synthesis via Enhanced Two-Electron Oxygen Reduction Pathway on Carbon-Coated Pt Surface. J. Phys. Chem. C 2014, 118, 30063–30070 [Google Scholar]; (b) Liu ZX; Li ZP; Qin HY; Liu BH, Oxygen reduction reaction via the 4-electron transfer pathway on transition metal hydroxides. J. Power Sources 2011, 196, 4972–4979 [Google Scholar]; (c) Fukuzumi S; Lee Y-M; Nam W, Mechanisms of Two-Electron versus Four-Electron Reduction of Dioxygen Catalyzed by Earth-Abundant Metal Complexes. ChemCatChem 2018, 10, 9–28 [Google Scholar]; (d) Song C; Zhang J, Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers, Springer: London, 2008. [Google Scholar]
- (15).(a) Venkataramanan NS; Premsingh S; Rajagopal S; Pitchumani K, Electronic and Steric Effects on the Oxygenation of Organic Sulfides and Sulfoxides with Oxo(salen)chromium(V) Complexes. J. Org. Chem 2003, 68, 7460–7470 [DOI] [PubMed] [Google Scholar]; (b) O’Reilly ME; Del Castillo TJ; Falkowski JM; Ramachandran V; Pati M; Correia MC; Abboud KA; Dalal NS; Richardson DE; Veige AS, Autocatalytic O2 Cleavage by an OCO3– Trianionic Pincer CrIII Complex: Isolation and Characterization of the Autocatalytic Intermediate [Cr(IV)]2(μ-O) Dimer. J. Am. Chem. Soc 2011, 133, 13661–13673 [DOI] [PubMed] [Google Scholar]; (c) Bryliakov KP; Talsi EP, Cr(III)(salen)Cl Catalyzed Asymmetric Epoxidations: Insight into the Catalytic Cycle. Inorg. Chem 2003, 42, 7258–7265 [DOI] [PubMed] [Google Scholar]; (d) Daly AM; Renehan MF; Gilheany DG, High Enantioselectivities in an (E)-Alkene Epoxidation by Catalytically Active Chromium Salen Complexes. Insight into the Catalytic Cycle. Org. Lett 2001, 3, 663–666 [DOI] [PubMed] [Google Scholar]; (e) McGarrigle EM; Gilheany DG, Chromium− and Manganese−salen Promoted Epoxidation of Alkenes. Chem. Rev 2005, 105, 1563–1602 [DOI] [PubMed] [Google Scholar]; (f) Acevedo O; Jorgensen WL, Cope Elimination: Elucidation of Solvent Effects from QM/MM Simulations. J. Am. Chem. Soc 2006, 128, 6141–6146 [DOI] [PubMed] [Google Scholar]; (g) Joergensen KA, Transition-metal-catalyzed epoxidations. Chem. Rev 1989, 89, 431–458. [Google Scholar]
- (16).Venkataramanan NS; Rajagopal S, Effect of added donor ligands on the selective oxygenation of organic sulfides by oxo(salen)chromium(V) complexes. Tetrahedron 2006, 62, 5645–5651. [Google Scholar]
- (17).(a) Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S, Oxidation Mechanism of Phenols by Dicopper−Dioxygen (Cu2/O2) Complexes. J. Am. Chem. Soc 2003, 125, 11027–11033 [DOI] [PubMed] [Google Scholar]; (b) Kundu S; Miceli E; Farquhar ER; Ray K, Mechanism of phenol oxidation by heterodinuclear Ni Cu bis(µ-oxo) complexes involving nucleophilic oxo groups. Dalton Trans 2014, 43, 4264–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lansky DE; Goldberg DP, Hydrogen Atom Abstraction by a High-Valent Manganese(V)−Oxo Corrolazine. Inorg. Chem 2006, 45, 5119–5125. [DOI] [PubMed] [Google Scholar]
- (18).Hansch C; Leo A; Hoekman D, Exploring QSAR: Fundamentals and Applications in Chemistry and Biology ACS: Washington, DC, 1995. [Google Scholar]
- (19).Hansch C; Leo A; Taft RW, A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev 1991, 91, 165–195. [Google Scholar]
- (20).Samsel EG; Srinivasan K; Kochi JK Mechanism of the Chromium-Catalyzed Epoxidation of Olefins. Role of Oxochromium(V) Cataions. J. Am. Chem. Soc 1985, 107, 7606–7617. [Google Scholar]
- (21).Brandt P; Norrby P-O; Daly AM; Gilheany DG, Chromium–Salen-Mediated Alkene Epoxidation: A Theoretical and Experimental Study Indicates the Importance of Spin-Surface Crossing and the Presence of a Discrete Intermediate. Chem. Eur. J 2002, 8, 4299–4307. [DOI] [PubMed] [Google Scholar]
- (22).Hehre WJ, A Guide to Molecular Mechanics and Quantum Chemical Calculations Wavefunction, Inc.: 2003. [Google Scholar]
- (23).Singleton DA; Thomas AA, High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc 1995, 117, 9357–9358. [Google Scholar]
- (24).Meyer MP; DelMonte AJ; Singleton DA Reinvestigation of the Isotope Effects for the Claisen and Aromatic Claisen Rearrangements: The Nature of the Claisen Transition States. J. Am. Chem. Soc 1999, 121, 10865–10874. [Google Scholar]
- (25).(a) Measures of local nucleophilicities have been reported, but are thermodynamic in nature or require more complex calculations. Waymouth RM; Santarsiero BD; Grubbs RH, Trigonal-bipyramidal methyl group bridging two zirconocene-ketene centers. J. Am. Chem. Soc 1984, 106, 4050–4051 [Google Scholar]; (b) Mendez F; Gazquez JL, Chemical Reactivity of Enolate Ions: The Local Hard and Soft Acids and Bases Principle Viewpoint. J. Am. Chem. Soc 1994, 116, 9298–9301 [Google Scholar]; (c) Senet P, Kohn-Sham orbital formulation of the chemical electronic responses, including the hardness. J. Chem. Phys 1997, 107, 2516–2524 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.