

Abstract
Currently, prostate cancer (PCa) remains the most commonly diagnosed solid tumor and the second leading cause of cancer-related deaths in U.S. men. Most of these deaths are attributed to the development of castration-resistant (CR) PCa. ErbB-2 and ErbB family members have been demonstrated to contribute to the progression of this lethal disease. In this review, we focus on updating the role of ErbB-2 in advanced PCa progression and its regulation, including its regulation via ligand activation, microRNAs and protein phosphorylation. We also discuss its downstream signaling pathways, including AKT, ERK1/2 and STATs, involved in advanced PCa progression. Additionally, we evaluate the potential of ErbB-2, focusing on its protein hyper-phosphorylation status, as a biomarker for aggressive PCa as well as the effectiveness of ErbB-2 as a target for the treatment of CR PCa via a multitude of approaches, including orally available inhibitors, intratumoral expression of cPAcP, vaccination and immunotherapy.
Keywords: ErbB-2, Castration-Resistant Prostate Cancer, ErbB-2-targeting Therapies, ErbB-2 Regulation
1. Introduction
Prostate cancer (PCa), one of the most common solid tumors, remains the second leading cause of cancer-related death in United States men with an estimated 174,650 new cases in 2019 (Siegel et al. 2019). Early stage localized PCa is well managed by surgery, radiation, or a combination of both treatments; nevertheless, about 30% of the cases show regression and progression towards metastasis. Both localized and early stages of metastatic PCa cells rely on androgens for cell growth and progression; the removal of androgen via androgen deprivation therapy (ADT) results in cell cycle arrest or apoptosis of PCa cells, thereby these PCa cells are classified as androgen-sensitive (AS). ADT, therefore, is the standard-of-care treatment for the management of metastatic PCa in its early stages. However, in most cases via a multitude of mechanisms, the metastasized cancer relapses, progresses, and obtains the castration-resistant (CR) phenotype. Currently, patients with CR PCa only have limited treatment options that often fail shortly after implementation (Debes and Tindall. 2004, Sartor and Gillessen. 2014). Therefore, understanding the molecular networks underlying CR PCa progression will be beneficial for the design and development of therapeutic strategies for improving the management of CR PCa.
Protein tyrosine kinases (PTKs) are a large multigene family involved in key cellular processes regulating cellular development, differentiation, and multicellular communication (Segaliny et al. 2015). Moreover, the perturbation of PTK signaling can result in the development of numerous disease conditions, including cancer (Regad et al. 2015). One such mechanism that PCa cells utilize to obtain the CR phenotype is aberrant activation of androgen receptor (AR) and its downstream signaling pathways. Results of recent advances show that the activation of receptor tyrosine kinase (RTK) signaling, including ErbB-2, enhances CR PCa cell growth through subsequent effector pathways, such as mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K) (Muniyan et al. 2015, McKay and Morrison. 2007). Further, the autocrine growth factor loop of ErbB-1, ErbB-2 and ErbB-3 can activate AR and enhance CR PCa progression (Gao et al. 2016). This review focuses on discussing the oncogenic contribution of PTK ErbB-2 in mediating CR PCa progression and provides an update on our understanding of ErbB-2 regulation and signaling, and its potential as a therapeutic target and/or biomarker in CR PCa.
2. Regulation of ErbB-2 Protein-Tyrosine Kinase
2.1. Ligands Associated with Asymmetric Dimerization and Activation of ErbB-2
Current knowledge clearly shows that in a normal non-cancerous cell, ErbB-2 can only be activated through its heterodimerization with other ligand-bound ErbB family members. The binding of growth factors to ErbB-1/3/4 extracellular sub-domains I and III induces the conformational change, i.e., closed conformation to open conformation, by exposing the dimerization interface of sub-domain II, which is otherwise buried within sub-domain IV, and promotes dimerization (Dawson et al. 2005, Dawson et al. 2007) as shown in Figure 1. In contrast, ErbB-2 maintains a naturally open conformation in which the dimerization arm of sub-domain II is exposed (Fig. 1). This is due to the substitution of a glycine residue with proline in sub-domain II as well as a histidine residue with phenylalanine in sub-domain IV, preventing sub-domain II and IV association in ErbB-2 (Cho et al. 2002).
Figure 1. Structure of the ErbB Receptor.
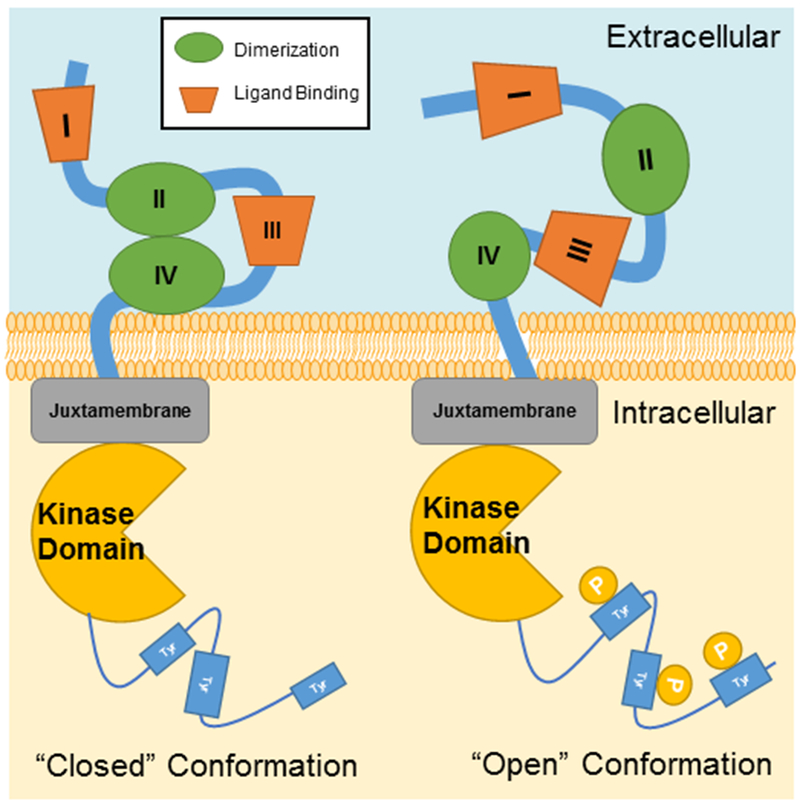
ErbB receptors transmembrane proteins with both intracellular and extracellular domains. The extracellular domain is composed of four sub-domains, including two leucine-rich sub-domains (I and III) for ligand binding and two cysteine-rich sub-domains (II and IV) for dimerization with activated ErbB members. Inactivated ErbB receptors remain in the ‘closed’ conformation, while ErbB-2 is constitutively in the ‘open’ confirmation. The dimerization with other ligand-bound ErbB family members will lead to activate ErbB-2 for signaling. Intracellularly, ErbB receptors have a juxtamembrane region, a kinase domain, and a cytoplasmic tail that is regulated by tyrosine phosphorylation.
As shown in Figure 2, many known ligands include epidermal growth factor (EGF), epigen (EPG), transforming growth factor (TGFα), amphiregulin (AREG), Betacelluin (BTC), heparin-binding epidermal growth factor (HB-EGF) and epiregulin (EPR), which all can bind to ErbB-1/EGFR. Neuregulin (NRG)-1/2/3/4 are known ligands to both ErbB-3 and ErbB-4; while ErbB-4 can also bind BTC, HB-EGF and EPR. Ligand binding to ErbB-1/3/4 induces a conformational change resulting in heterodimerization with ErbB-2 and protein kinase activation. The phosphorylation of multiple tyrosine residues creates docking sites for various adapter proteins such as Src homology 2 domain containing (Shc), Growth factor receptor-bound protein 2 (Grb2), Src, Protein tyrosine phosphatase-2c (PTP-2c), Spleen tyrosine kinase (Syk), Phospholipase Cγ (PLCγ), Src homology region 2 domain containing phosphatase-1 (SHP1), etc. The phosphorylation and subsequently docking with different adapter proteins initiates the diverse downstream signaling cascades, which can result in various cellular processes such as cell growth, differentiation, migration, adhesion, and apoptosis (El Sheikh et al. 2004, Tome-Garcia et al. 2014).
Figure 2. ErbB-2 Regulation by Ligands and Proteins.
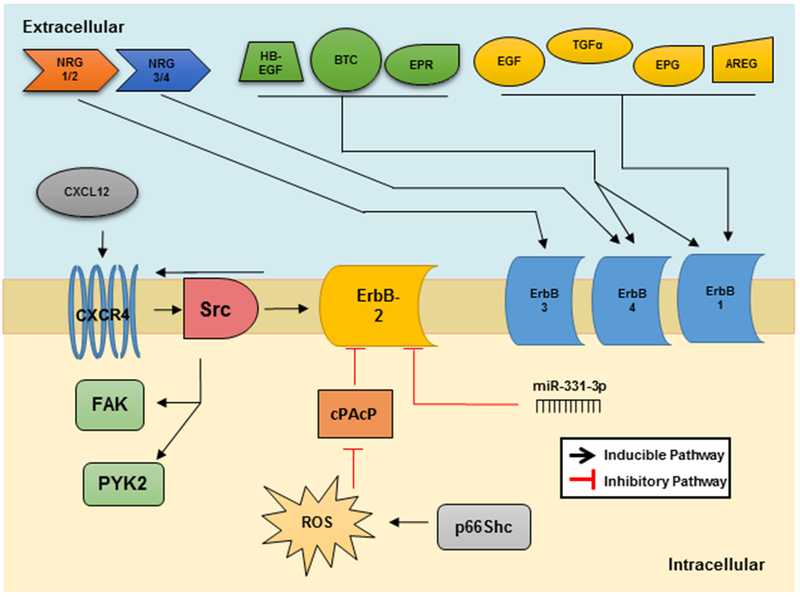
There are numerous ligands that can activate ErbB-1/3/4. ErbB-3 can only be activated by NRG1/2, while ErbB-4 is activated upon binding of NRG3/4, HB-EGF, EPR and BTC. ErbB-1 has the most numerous known ligands including HB-EGF, BTC, EPR, EGF, TGF-α, EPG, and AREG. ErbB-2 can be activated via CXCR4 when this protein is bound to its ligand, CXCL12, via Src kinase. Interestingly, ErbB-2 can also promote the activation of CXCR4 as well. Negative regulators of ErbB-2 include microRNA miR-331-3p and cPAcP. PAcP, like other protein tyrosine phosphatases, can be inactivated by ROS and is a downstream of oxidase p66Shc.
Overproduction of ligand is one alternate method through which tumor cells achieve aberrant ErbB-2 activation, and the source of ligand can be either tumor cells or surrounding stroma. Elevated expression of ErbB ligands, including TGF-α, HB-EGF and AREG, are often associated with clinical prognosis in a number of cancers, including PCa (Salomon et al. 1995), which correlates with a poor clinical outcome (Normanno et al. 2003). Recent studies further demonstrated that PCa cells are capable of overexpressing ErbB-associated ligands EGF, BTC and NRG-1 to combat treatment with ErbB family inhibitors, including cetuximab, erlotinib or gefitinib (Carrion-Salip et al. 2012). Recently, phase I/II trials of rilotumumab, a monoclonal antibody inhibiting hepatocyte growth factor (HGF), have shown increased survival rates in patients with non-small cell lung cancer (Tarhini et al. 2017). Therefore, investigation of competitive inhibitors of ErbB family ligands as well as inhibitors of ErbB ligand synthesis is suggested for potential treatment of PCa.
Interestingly, it has been shown that in advanced cancer cells, the ErbB-2 protein can homodimerize via its open subdomains II and IV, leading to self-activation upon its overexpression or lack of expression of its regulatory phosphatase (Muniyan et al. 2015, Meng et al. 1998). Consistently, in the absence of ligand activation, overexpression of either EGFR or ErbB-2 in non-cancerous cells leads to the malignant transformation (Di Fiore et al. 1987). Several studies show that overexpression, mutation or loss of ErbB-2 expression results in dysregulation of dimerization and the downstream signaling of other ErbB proteins, indicating the importance of ErbB-2 in ErbB members signaling (Dawson et al. 2005, Worthington et al. 2017, Holbro et al. 2003). Nevertheless, the specific activation of respective signaling pathways requires further elucidation.
2.2. Cellular Prostatic Acid Phosphatase (cPAcP) Dephosphorylates ErbB-2
Upon heterodimerization, the cytoplasmic domain of ErbB-2 is hyper-phosphorylated at multiple tyrosine residues, leading to protein kinase activation. Therefore, the phosphatases that can reverse ErbB-2 tyrosine phosphorylation are capable of inhibiting its signaling cascade. Cellular prostatic acid phosphatase (cPAcP) is a dual specificity prostate-specific phosphatase that can dephosphorylate tyrosine, serine and threonine residues with a preference for phosphotyrosine (p-Tyr) residues (Lin and Clinton 1986). Importantly, results of several analyses on prostate adenocarcinoma archival specimens clearly show decreased cPAcP expression at both mRNA and protein levels, compared to adjacent non-cancerous cells (Veeramani et al. 2005). Among several PCa cell lines and within sublines of the same origin, in low PAcP-expressing PCa cells, ErbB-2 is hyper-activated by tyrosine phosphorylation, supporting a correlative of low cPAcP activity and high ErbB-2 activation by tyrosine phosphorylation (Meng et al. 1998, Muniyan et al 2015). Consistently, several lines of evidence further show that in PCa cells, ErbB-2 is a direct target of cPAcP including co-immunoprecipitation experiments, demonstrating the potential interaction between these two proteins for dephosphorylation (Meng et al. 1998, Chuang et al. 2010, Veeramani et al. 2012).
The loss of cPAcP expression is an early event in PCa (Veeramani et al. 2005), therefore, it is proposed to be a prominent source of ErbB-2 hyper-activation commonly observed in PCa cells. Interestingly, when LNCaP-AS and MDA PCa2b-AS cells progress to the androgen-independent (AI) stage, i.e., proliferation in androgen-reduced culture conditions and obtaining the CR phenotype seen in clinical PCa by expression of functional AR and PSA, ErbB-2 is hyper-phosphorylated and cPAcP protein levels decrease (Meng and Lin 1998, Muniyan et al. 2015). Additionally, cPAcP knockdown in LNCaP-AS cells results in enhanced ErbB-2 tyrosine phosphorylation and AI cell proliferation in cultures (Muniyan et al. 2015, Chuang et al. 2010). In those cells, there is no observed EGFR/ErbB-2 heterodimerization by co-immunoprecipitation (Veeramani et al. 2005). Conversely, PAcP cDNA transfection of cPAcP-deficient LNCaP-AI or cPAcP-null PC-3 PCa cells results in decreased ErbB-2 tyrosine phosphorylation as well as the reacquisition of the AS phenotype, indicating the role of cPAcP and ErbB-2 interaction is involved in regulating androgen-sensitivity (Meng et al. 1998, Chuang et al. 2010, Muniyan et al. 2015). Importantly, PAcP-knockdown of AS PCa cells via shRNA, but not the parental control cells, develop xenograft PCa tumors in female mice, androgen-deprived environments (Muniyan et al. 2015, Chuang et al. 2010), and the PAcP gene-knockout mice spontaneously develop prostate adenocarcinoma (Quintero et al. 2013). Thus, in PAcP-deficient prostate epithelial cells, ErbB-2 is hyperactivated by tyrosine phosphorylation, contributing to enhanced PCa tumorigenicity. Increased cPAcP expression is thus a pending novel therapeutic strategy for the treatment of CR PCa.
2.3. Androgens, p66Shc, and ROS Enhance ErbB-2 Specific Activity
In AS PCa cells, androgen treatments induce tyrosine phosphorylation and activation of ErbB-2 which is associated with increased cell proliferation. Recent advances reveal that this is achieved in part via androgen-induced elevation of p66Shc protein, a 66 kDa proto-oncogene Src and collagen homologue (Shc), and one of three members of the Shc family including isoforms p52Shc and p46Shc (Rajendran et al. 2010, Alam et al. 2009). p66Shc differs from the other Shc isoforms in that it possesses oxidase activity, its protein half-life is increased by androgens, and importantly, its protein level is elevated in PCa archival specimens and correlates with the CR phenotype of PCa cells (Veeramani et al. 2012, Lee et al. 2004, Kumar et al. 2011, Veeramani et al. 2008). In PCa cells, p66Shc is localized in both cytosol and mitochondria. In androgen-treated PCa cells, p66Shc protein levels in mitochondria are elevated, where p66Shc binds to and oxidizes cytochrome C, uncoupling the electron transport chain to promote the production of cellular reactive oxygen species (ROS) (Kumar et al. 2011, Veeramani et al. 2008). In the cytosol of mouse embryonic fibroblasts (MEFs), p66Shc displaces Son of Sevenless 1 (SOS1), promotes Rac1 activity, and stimulates NADPH oxidase (NOX) complex formation, leading to the generation of superoxide (Khanday et al. 2006). In AI PCa cells, elevated p66Shc protein results in enhanced Rac1 activation and cell proliferation as well as migration (Ingersoll et al. 2018).
ErbB-2 can be activated by cellular ROS. Veeramani et. al. (Veeramani et al. 2012, Veeramani et al. 2008) demonstrated that ErbB-2 is one of several downstream targets of p66Shc, and elevated expression of p66Shc, but not its oxidase-deficient mutant, in PCa cells results in increased ROS production, ErbB-2 tyrosine phosphorylation, and cell growth rate. Similarly, treatment of AS LNCaP cells with hydrogen peroxide at physiological levels was shown to decrease cPAcP activity and increase p-Tyr1221/1222 levels of ErbB-2 and cell proliferation, which is counteracted by antioxidant N-acetyl cysteine (NAC). It was further determined that p66Shc/ROS-mediated inactivation of cPAcP phosphatase activity is at least one mechanism through which ErbB-2 activation is achieved (Veeramani et al. 2012, Veeramani et al. 2008). In summary, androgen up-regulation of p66Shc protein levels (Kumar et al. 2011) results in increased cellular levels of ROS which oxidize and inactivate cPAcP, preventing cPAcP from dephosphorylating ErbB-2, which leads to ErbB-2 activation and PCa cells obtain the CR PCa phenotype.
2.4. microRNAs Downregulate ErbB-2 Protein Levels
MicroRNAs are short, non-coding RNAs which post-transcriptionally regulate gene expression and are often aberrantly expressed in tumors. These molecules can regulate protein expression by binding to the 3’-untranslated regions of target mRNAs to suppress translation or induce degradation. While miR-125a and miR-125b can directly regulate ErbB-2 in breast cancer cells; they do not directly regulate ErbB-2 in PCa cells (Shi et al. 2008, Scott et al. 2007). miR-331-3p binds to the 3’-untranslated region of ErbB-2 in two target sites to regulate ErbB-2 protein expression in multiple PCa cell lines (Epis et al. 2009). Moreover, miR-331-3p is expressed at a lower level in cancer cells compared to benign cell lines, and induction of miR-331-3p in cancer cells suppresses tumor phenotype through inhibition of PI3K/AKT signaling (Epis et al. 2009). Interestingly, human antigen R (HuR) is an RNA binding protein with elevated levels in PCa, which competes with miR-331-3p for 3’-UTR binding sites on ErbB-2 mRNA. It is thus proposed that loss of miR-331-3p and elevation of HuR can lead to increased ErbB-2 protein in the absence of gene amplification observed in a subset of advanced PCa tumors (Epis et al. 2011).
2.5. Cholesterol and Lipid-Rafts Enhance ErbB-2 Signaling
Hypercholesterolemia is associated with an increased risk of aggressive PCa via a multitude of mechanisms, including increased steroidogenesis, inflammation, proliferation, and alterations in lipid rafts (Pelton et al. 2013). Lipid rafts are specialized domains located within the plasma membrane enriched with cholesterol, sphingolipids, and various signaling proteins. G-protein coupled receptors (GPCRs), glycosylphosphatidylinositol (GPI)-anchored proteins, Src family kinases, and G-proteins such as Ras are associated with lipid rafts where they initiate signal transduction and amplification. ErbB family members are also shown to be associated with lipid rafts (Zhuang et al. 2002). In PCa cells, a small sub-population of ErbB-2 was found to be associated with lipid rafts, despite that the majority of ErbB-2 molecules are localized within the cytoplasm (Chinni et al. 2008). It should also be noted that a small subset of cPAcP was obtained by detergent extraction from the lipid fraction of non-cancerous prostate cells and that fraction is diminished in cancerous cells (Veeramani et al. 2005). Interestingly, the subpopulation of ErbB-2 located within lipid rafts of PCa cells has higher phosphorylation levels than ErbB-2 in non-raft membranes, and ErbB-2 signaling to downstream effectors is abrogated when cholesterol levels are reduced (Zhuang et al. 2002). For ErbB-2 targeting therapy, further investigation on lipid raft-associated ErbB-2 is warranted.
2.6. CXCR4 Transactivates ErbB-2 in Lipid Rafts
C-X-C chemokine receptor type 4 (CXCR4) is a seven-transmembrane trimeric GPCR expressed in epithelial cancer cells. Currently, the only known ligand of CXCR4 is the C-X-C motif chemokine ligand 12 (CXCL12), an 11 kDa peptide expressed locally in the microenvironment of common metastatic sites, such as lung, bone, and liver. Furthermore, binding of CXCL12 to CXCR4 has been shown to play a crucial role in site-specific metastasis to lymph nodes, lung, and bone (Taichmann et al. 2002, Chinni et al. 2006). GPCRs can transactivate ErbB family members by ectodomain shedding of membrane-bound ErbB family receptor ligands by proteases (Fischer et al. 2003) or by intracellular phosphorylation of ErbB family members via Src kinase (Fig. 2) (Luttrell and Luttrell 2004). CXCR4 and ErbB-2 are often co-localized at cell surface in lipid raft domains. CXCR4 overexpression can enhance ErbB-2 phosphorylation and is proposed to promote metastasis and invasion of PCa cells to the bone (Chinni et al. 2006, Conley-LaComb et al. 2016). Interestingly, in breast cancer, ErbB-2 has been shown to transactivate CXCR4 as well, leading to activation of Rac1 and subsequent cell migration (Li et al. 2004). Further elucidation on the interaction of CXCR4 and ErbB-2 may lead to alternate targeting therapies.
2.7. Src Activates ErbB-2, FAK, and PYK2 to Enhance PCa Tumorigenicity
Src kinase is a non-receptor tyrosine kinase localized in both lipid rafts and nonlipid rafts of PCa cells. Within the lipid raft, Src kinase activation is required for serving as the intermediate for CXCL12/CXCR4–induced ErbB-2 phosphorylation (Fig. 2). Src kinase associates with the carboxyl-terminal region of ErbB-2 through its SH2 domain and also promotes the heterodimerization of ErbB-2/ErbB-3 complex formation (Ishizawar et al. 2007). In addition, Src can be an upstream kinase of ErbB-2 in the nonlipid raft domain in PCa cells. The interaction of Src with ErbB-2 is shown to be required for ErbB-2–mediated invasive and migratory properties of epithelial cells (Conley-LaComb et al. 2016). Src can also be a downstream target of ErbB-2 and is upregulated in PAcP-knockdown cells in which ErbB-2 is activated by tyrosine hyperphosphorylation (Veeramani et al. 2005). ErbB-2 can promote Src stabilization and protein synthesis (Tan et al. 2005) as well as increase Src-specific activity (Muthuswamy et al. 1994) to promote cell adhesion, migration and invasion via Focal adhesion kinase (FAK) or Protein tyrosine kinase 2 (PYK2) (Yuan et al. 2007). FAK and PYK2 are structurally related non-receptor protein tyrosine kinases that can compensate for one another if either protein is lost. Both proteins interact with integrins and GPCRs to promote cell motility or adhesion via c-Src kinase, Extracellular-signal-regulating kinase (ERK)/MAPK, p38/MAPK and paxillin. Thus, both FAK and PYK2 contribute to cancer cell adhesion, proliferation, and invasion. Nevertheless, in PCa cells it was clearly shown that in AR-positive PCa cells, ErbB-2 expression and activation correlates with PYK2 activation, leading to cell adhesion (Yuan et al. 2007), whereas FAK activity correlates with ErbB-2 expression in AR-negative PCa cells (Yuan et al. 2007, Johnson et al. 2008). The differential roles of PYK2 vs. FAK in mediating ErbB-2 signaling in advanced PCa progression requires further investigation.
3. Downstream Targets of ErbB-2 in PCa Progression.
3.1. ErbB-2 Promotes PCa Cell Proliferation and Migration via PI3K/AKT
Deregulation of protein kinase B (PKB), commonly referred to as AKT, is pervasive in PCa progression due to its regulation of mechanisms critical to metastasis. AKT plays a role in diverse cellular signaling, including cell survival, proliferation, migration, invasion and other biological events associated with carcinogenesis and cancer progression (Saxena et al. 2007, Roy et al. 2002). Further, AKT activation via T308/S473 phosphorylation is primarily regulated by the PI3K/phosphatase and tensin homolog (PTEN) axis as shown in Figure 3 (Bellacosa et al. 1998). Once activated by RTKs such as ErbB-2, PI3K converts phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3) which activates a number of downstream signaling components, the most notable of which is AKT (Hodgkin et al. 2000). Conversely, PTEN catalyzes the opposite reaction, hydrolyzing PIP3 to PIP2 and preventing the activation of AKT (Maehama et al. 1998). In PCa, loss of PTEN function is occurs in advanced PCa progression (Ozen et al. 2008, Wang et al. 2003). Interestingly, cPAcP (Section 3.2) can also hydrolyze PIP3 (Muniyan et al. 2014). In PTEN-active, PAcP-deficient DU145 PCa cells, AKT is hyper-activated. Furthermore, in PTEN-inactive, cPAcP-expressing LNCaP-AS cells, AKT with low or no phosphorylation or activation; while in PAcP-deficient LNCaP-AI cells, AKT is hyper-activated by T308/S473 phosphorylation, similar to PTEN- and PAcP-null PC-3 cells. It is thus proposed that cPAcP is a PTEN-functional homologue in prostate epithelia (Muniyan et al. 2014). Because knockout of PAcP expression in mice results in the development of prostate adenocarcinoma (Quintero et al. 2014) and cPAcP expression is significantly reduced in primary PCa (Veeramani et al. 2005, Muniyan et al. 2014), it is hypothesized that decreased cPAcP expression in part by epigenetic modifications is involved in PCa development and the early stages of PCa progression, while PTEN plays a role in advanced PCa progression (Muniyan et al. 2014, Chou et al. 2015).
Figure 3. ErbB-2 Downstream Targets to Promote PCa Tumorigenicity.
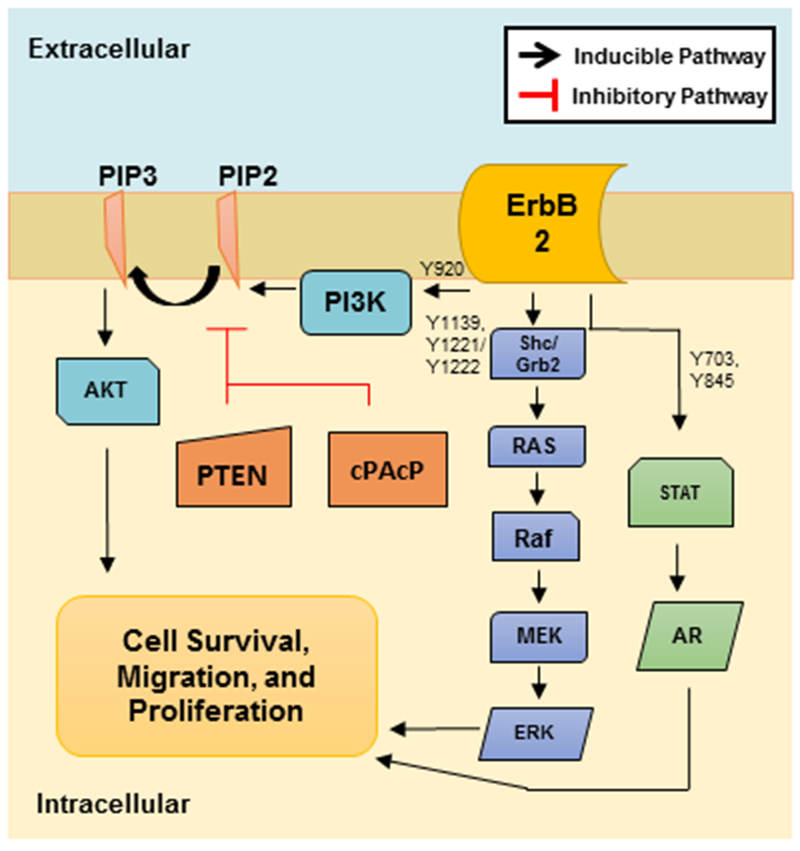
ErbB-2 can regulate numerous signaling pathways in the cell to positively influence cell survival, migration and proliferation. ErbB-2 tyrosine phosphorylation of Y703 or Y845 can activate STATs, which promote AR nuclear translocation and transcription of survival and proliferative genes. ErbB-2 can also activate the Ras-Raf-MEK-ERK pathway upon Y1139 or Y1221/Y1222 phosphorylation to promote proliferation and migration via Grb2 or Shc, respectively. Additionally, ErbB-2 Y920 phosphorylation can induce phosphorylation of PI3K to promote AKT activation via phosphorylation of PIP2 to PIP3 to enhance PCa proliferation and survival. The PI3K/AKT pathway can be inhibited by both PTEN and cPAcP.
Upon activation, AKT regulates a number of cellular processes critical to PCa survival and progression. For example, AKT promotes cell survival by inactivating pro-apoptotic proteins, such as p53, BAX, BAD, YAP and Caspase-9, while inducing anti-apoptotic proteins such as Bcl-2, BcL-XL and Survivin (Dhanasekaran et al. 2008, Pelicano et al. 2006). AKT also promotes cell proliferation by enabling the cell to overcome cell cycle arrest at the G1 and G2 phases through activation of Cyclin D1, Cyclin-dependent kinase A (CDKA), and CDK6 as well as activating other pro-growth signaling molecules such as mammalian target of rapamycin (mTOR) (Liang et al. 2003). Additionally, AKT promotes angiogenesis through Vascular endothelial growth factor (VEGF) activation as well as cell migration and invasion through regulation of cadherin proteins, MYC, matrix metalloproteinases, Snail and Forkhead box protein M1 (FOXM1) (Ingersoll et al. 2018, Julien et al. 2007, Jin et al. 2015, Gera et al. 2004). AKT also promotes cholesterol synthesis, which is utilized by over 50% of advanced prostate tumors for de novo androgen synthesis (Zhuang et al. 2005, Dillard et al. 2008). Thus, AKT is capable of promoting the aggressive metastatic phenotype of PCa.
In PCa, AKT and AR share an interconverting complex signaling network in which each protein is capable of regulating the other. Through p66Shc elevation, AR indirectly increases cellular ROS levels that leads to the inactivation of phosphatases, such as cPAcP (Veeramani et al. 2012). By preventing cPAcP-mediated dephosphorylation and inhibition of ErbB-2, AR also promotes the activation of a number of ErbB-2-regulated pathways such the PI3K/AKT pathway (Veeramani et al. 2012). Conversely, AKT can modulate the transcriptional activity of AR through direct phosphorylation. For example, phosphorylation of AR at S210 and S790 by AKT suppresses AR-mediated apoptosis and contributes to PCa survival (Lin et al. 2003). Further, AKT can bind and phosphorylate AR at S213, which increases AR ligand-binding and promotes AR activation and translocation to the nucleus (Wen et al. 2000). Moreover, in the event of AR inhibition or androgen-deprivation, the PI3K/AKT pathway is reported to be activated in PCa cells as a mechanism of androgen-independence. Clearly, AKT is a useful therapeutic target for CR PCa treatment (Wen et al. 2000).
3.2. ErbB-2 Activates the MAPK/ERK Pathway to Promote PCa Tumorigenicity
In addition to AKT, activated ErbB-2 also activates the MAPK pathway, which includes ERK1/2 (Chuang et al. 2010, Agus et al. 2002, Zhang et al. 2001). These proteins are frequently reported to be activated in aggressive PCa and are key regulatory kinases for processes vital to PCa development and progression (Veeramani et al. 2012, Zhang et al. 2001, Oshikawa et al. 2012). Figure 3 demonstrates that at the cell membrane, activated RTKs initially recruit Shc and/or Grb2 with SOS1 to facilitate GDP-GTP nucleotide exchange on Ras (McKay and Morrison 2007). Ras then activates Raf kinase that subsequently activates MAPK/ERK kinase (MEK), which finally activates ERK1/2 via T202 and Y204 phosphorylation (McKay and Morrison 2007, Schlessinger 2000). ERK is a serine-threonine kinase and its isoforms include ERK1 and ERK2 according to their coding sequences. ERK1/2 are highly activated in advanced AI PCa (Gioeli et al. 1999). Activated ERK regulates several processes critical to PCa progression including survival, proliferation and migration via activation of transcription factors such as Elk1 (Kinkade et al. 2008, Gao et al. 2006, Kue et al. 2002). Similar to AKT, ERK and AR signaling pathways undergo dynamic crosstalk and feedback loops (Recchia et al. 2009, Ballare et al. 2003). Through ErbB-2 regulation, AR is able to promote downstream activation of ERK (Recchia et al. 2009, Chia et al. 2011). In turn, ERK can regulate AR through induction of cAMP-responsive element binding protein 1 (CREB1) transcription factor that binds to the AR promoter and enhances its transcription (Chia et al. 2011). ERK is also capable of direct phosphorylation of AR and its coregulators, resulting in AR activation and promoting its translocation to the nucleus (Recchia et al. 2009, Chia et al. 2011, Gioeli et al. 2006). Thus, due to its promotion of the metastatic phenotype as well as induction of AR signaling, ERK is a potential therapeutic target for CR PCa treatment (Zelivianski et al. 2003).
3.3. STATs Induce Transcription upon Activation by ErbB-2
The signal transducer and activator of transcription (STAT) proteins are indispensable in the development and progression of a wide variety of cancers. In PCa cells, loss of cPAcP expression and thus constitutive activation of ErbB-2 results in the activation of STAT-3 and STAT-5 via protein phosphorylation (Chuang et al. 2010). STAT-3 can be involved in promoting cell proliferation, anti-apoptosis, angiogenesis, inflammation and also epithelial-to-mesenchymal transition (EMT) in PCa progression (Bishop et al. 2014). In PCa, STAT-3 can promote the transcription of AR downstream targeted gene, prostate-specific antigen (PSA), via STAT-3 interaction with the N-terminal domain of AR. Interleukin-6 (IL-6)-mediated activation of STAT-3 and AR is also associated with trans-differentiation of LNCaP cells to obtain the neuroendocrine (NE) phenotype (Yuan et al. 2007, Spiotto et al. 2000). Further, the IL-6 receptor can also cooperate with ErbB-2 to activate the MAPK pathway upon short-term IL-6 treatment to promote AR activity (Lin et al. 2001). IL-6 was proposed to serve as a useful biomarker for the detection of PCa as well as the extent of the disease (Nakashama et al. 2000). Unfortunately, as a cancer diagnostic marker, IL-6 fails because it can be secreted and elevated under many pathological conditions including cancers (Azevedo et al. 2011), making it difficult to deduce what type of cancer or disease a patient has developed.
4. ErbB-2 as a Prognostic Marker
4.1. ErbB-2 Hyperactivation/Tyrosine Phosphorylation in Human PCa Progression
Several lines of evidence have clearly shown the increased ErbB-2 activation, but not protein levels, upon progression from the AS to the AI stage in LNCaP and MDAPCa2b cells (Muniyan et al. 2015, Meng et al. 1998, Chuang et al. 2010). Activation of ErbB-2 via tyrosine phosphorylation is required for cell growth in both AS and AI PCa cells. In AS PCa cells, DHT-stimulation of cell proliferation is mediated by ErbB-2 tyrosine phosphorylation. In AR-positive PCa cells, ErbB-2 activation, in part due to the loss of cPAcP expression, can enhance AR activity, cell survival and AI progression, thus those cell obtain the CR phenotype (Muniyan et al. 2015, Tome-Garcia et al. 2014). While ErbB-2 phosphorylation status has not yet been analyzed in clinical PCa samples, ErbB-2 hyper-phosphorylation has the potential to be a promising biomarker for the disease state of PCa.
4.2. ErbB-2 Activation Involving the Metastatic Tumor Phenotype and Castration Resistance of PCa
Results of several studies have shown that ErbB-2 protein level is elevated in a small subset of CR PCa tissue samples (Minner et al. 2010, Montironi et al. 2006, Gregory et al. 2005) and correlates with poor disease prognosis (Edwards et al. 2006, Carles et al. 2004). A consensus on the role of ErbB-2 activation in AI PCa has emerged. Activation of ErbB kinases, particularly ErbB-2, can result in the transcription and activation of AR via MAPK (Tome-Garcia et al. 2014). Constitutively active ErbB-2 also promotes AKT activation, which can thereby activate AR to enhance the transcription of androgen-regulated genes to promote AI growth of PCa cells (Wen et al. 2010, Craft et al. 1999, Mellinghoff et al. 2004, Lee et al. 2003). Importantly, ErbB-2 activation has been demonstrated in abiraterone-resistant CR PCa as well, resulting in activation of AR via AKT (Gao et al. 2016). Additionally, ErbB-2, but not other members of the ErbB family, in AI proliferation is demonstrated in various PCa cells (Muniyan et al. 2015, Hsu et al. 2011). Interestingly, ErbB-2 activation was shown to be associated with promoting a NE-like phenotype in PCa via an AKT-independent mechanism (Cortez et al. 2012). Recent advances link EGFR/ErbB-2 signaling to promotion of PCa stem cell renewal and stem cell-mediated PCa metastasis to the bone (Day et al 2017, Rybak et al. 2013). Clearly, ErbB-2 hyper-activation, but not ErbB-2 protein elevation, plays a critical role in advanced PCa progression.
5. ErbB-2 as a Therapeutic Target for PCa/CR PCa
5.1. Inhibitors of ErbB Protein Kinase Domain Have Limited Efficacy against PCa
Due to the lack of a known ligand for ErbB-2 and the general requirement of heterodimerization of ErbB-2 with other ErbB family members for its activation, targeting EGFR is proposed to be a potential approach for treating cancers that require ErbB-2 signaling as shown in Table 1. EGFR inhibitors erlotinib and gefitinib target the kinase domain of EGFR and thus prevent tyrosine phosphorylation and block downstream signaling. Unfortunately, PCa cells are capable of overexpressing ErbB-associated ligands to combat treatment with ErbB family inhibitors, including erlotinib and gefitinib, leading to limited efficacy of these agents. Furthermore, erlotinib-resistant PCa cells were shown to have increased ErbB-2 and ErbB-3 mRNA and protein levels to promote the activation of the PI3K/AKT pathway. Erlotinib resistance in PCa cells could be overcome upon monoclonal antibody blockage of ErbB-3 (Carrion-Salip et al. 2012). While other EGFR inhibitors, such as ZD1839 (Iressa), have shown some promising results in clinical trials for treatment of a variety of cancers further investigation is required in PCa (de Bono et al. 2002).
Table 1. ErbB-2 Therapies Utilized for PCa.
Several attempts have been made to target ErbB-2 in PCa through a variety of methods. Results of in vivo mouse studies as well as patient clinical trials have shown some therapies can effectively reduce PCa growth while others have limited anti-tumor activity. Furthermore, many of these studies have demonstrated that the utilization of ErbB-2-targeting therapies require a specific patient sub-population in which the tumors rely on ErbB-2 signaling for proliferation and/or survival.
| Treatment | Method of Action | Efficacy Against PCa (in vivo and clinical trials) | Source |
|---|---|---|---|
| Erlotinib | EGFR inhibitor | Limited antitumor activity in vivo | Carrion-Salip et al. 2012 |
| Gefitinib | EGFR inhibitor | Limited antitumor activity in vivo | Carrion-Salip et al. 2012 |
| ZD1839 (Iressa) | EGFR inhibitor | Effectively reduced PCa growth in patients | Bono et al. 2002 |
| CI-1033 | Pan-ErbB inhibitor | Limited antitumor activity in patients | Campos et al. 2005 |
| Lapatinib | Dual EGFR/ErbB2 inhibitor | N/A | Blackwell et al. 2010 |
| HKI-272 | Dual EGFR/ErbB2 inhibitor | Undergoing phase I trials; effectively reduced xenograft tumors | Wissner et al. 2003, Rabindran et al. 2004 |
| BIBW-2992 | Dual EGFR/ErbB2 inhibitor | Limited anticancer activity in patients | Reid et al. 2004, Molife et al. 2014 |
| ErbB-2 vaccine | ErbB-2-targeted T cell response | N/A | Bhattacharya et al. 2002, Piechocki et al. 2003 |
| cPAcP cDNA intratumoral injection | Overexpression of negative regulator | Effectively reduced PCa xenograft tumors | Igawa et al. 2003 |
| Trastuzumab (Herceptin) | Anti-ErbB-2 antibody; reduces ErbB-2 protein | Effective in PCa if ErbB-2 overexpressed in tumors | Ziada et al. 2004 |
| Pertuzumab | Anti-ErbB-2 antibody; blocks dimerization | Delays CR PCa disease progression; clinical trial terminated | Agus et al. 2007 |
Because small molecule inhibitors cannot specifically target ErbB-2 alone, dual EGFR/ErbB-2 inhibitors are currently being developed. CI-1033 is a pan-ErbB irreversible inhibitor which was well tolerated by patients in phase I clinical trials; however, this molecule was not as effective as desired and is no longer involved in clinical trials (Campos et al. 2005). Lapatinib is a tyrosine kinase inhibitor that obstructs both EGFR and ErbB-2 signaling and has had great efficacy with reducing tumor growth in breast cancer patients (Blackwell et al. 2010); however, further studies are required in treating CR PCa patients. At this time lapatinib is not being pursued in a clinical setting, however previous studies suggest analyzing patients for predictive biomarkers and combination with other anti-cancer agents (Whang et al. 2013). Recently, a potent orally available dual EGFR/ErbB-2 inhibitor HKI-272 (Neratinib) was developed using a homology model for the catalytic domain of ErbB-2 and has since entered phase I clinical trials (Wissner et al. 2003). This molecule effectively inhibits both ErbB-2 and EGFR irreversibly in vitro, resulting in abrogated signaling through MAPK and AKT. HKI-272 also has great efficacy against xenograft breast tumors, and oral administration of this compound was well tolerated in mice (Rabindran et al. 2004). Results of clinical trials on the efficacy of HKI-272 in PCa have not been reported at this time. BIBW-2992 (Afatinib) is another potent oral dual EGFR/ErbB-2 inhibitor underwent clinical trials in PCa in combination with an anti-angiogenic agent as well as in combination with docetaxel (Reid et al. 2007). Unfortunately, the phase II study in CR PCa patients revealed that BIBW-2992 only exhibits limited anti-tumor activity (Molife et al. 2011). As such, ErbB-2 TKIs alone may not be a suitable method for treating CR PCa (Table 1).
5.2. ErbB-2 Targeting Vaccine can Treat ErbB-2-Overexpressing PCa
Peptide-based vaccines targeting the extracellular domain of ErbB-2 were one of the initial therapies for ErbB-2-expressing cancers (Disis et al. 1999). Using a peptide vaccine approach with granulocyte macrophage-colony-stimulating factor (GMCS) as an adjuvant, ErbB-2 protein-specific T-cell responses are generated in patients with breast and ovarian tumors that overexpress ErbB-2 (Disis et al. 1999). A study by Bhattacharya et al. showed a vector encoding the fusion protein of a truncated sequence of the ErbB-2 extracellular domain and the N-terminal sequence of EGFR effectively induces enhanced antitumor immunity against PCa cells expressing the cognate tumor-associated antigen (Bhattacharya et al. 2002). While many studies have since been focused on ErbB-2 vaccine developments; the availability of a suitable preclinical model is a major challenge. Rat ErbB-2 (neu) transgenic models are inadequate due to the 10% difference in sequences between neu and ErbB-2, while subcutaneous transplant xenograft models do not readily metastasize (Yamamoto et al. 1986, Piechocki et al. 2003). Moreover, because ErbB-2 is infrequently overexpressed in PCa, a vaccine would likely only be effective against a small subpopulation of PCa tumors that have elevated ErbB-2 protein levels. At this time, ErbB-2 vaccines have yet to enter clinical trials in PCa patients.
5.3. cPAcP Intratumoral Expression Reduces ErbB-2 Activity and Tumorigenicity in PCa Tumors
As described above in Section 2.2, several lines of evidence have clearly demonstrated that cPAcP is a negative regulator of ErbB-2 phosphorylation and activation. Accordingly, suppression of ErbB-2 activity through intratumoral expression of cPAcP has the potential as a PCa therapy (Igawa et al. 2003, Table 1). Studies in PCa xenograft tumor models show that abrogation of ErbB-2 activity in established subcutaneous AI LNCaP C-81 tumors with an intratumoral injection of expression vector containing the wild-type cPAcP cDNA results in concomitant suppression of tumor growth. Nevertheless, a PTPase enzymatically-dead mutant of cPAcP, which lacks its ability of dephosphorylating ErbB-2, has reduced tumor-suppressive activity (Igawa et al. 2003). Together, the data indicates that inhibition of ErbB-2 activity is critical for the suppression of prostate adenocarcinoma, and restoration of PAcP expression is an alternate approach for PCa/CR PCa therapy.
5.4. Immunotherapy Impedes ErbB-2 Signaling in ErbB-2-Overexpressing PCa
Various studies have investigated the potential of anti-ErbB-2 monoclonal antibodies to reduce ErbB-2 expression in ErbB-2-transformed NIH3T3 cells and reverse their transformed phenotype including tumorigenicity and metastasis in xenograft mouse models (Drebin et al. 1986, Drebin et al. 1988, Yu et al. 2000). Importantly, anti-ErbB-2 murine monoclonal antibody (muMAb 4D5) has been used to develop the humanized antibody trastuzumab (Herceptin) that binds to the extracellular domain of ErbB-2 to reduce cell-surface ErbB-2 proteins (Shepard et al. 1991). In ErbB-2 amplified breast cancer, trastuzumab has demonstrated to effectively inhibit tumor growth and sensitize to several chemotherapeutic agents, such as docetaxel, in preclinical studies as well as in phase II and phase III clinical trials, and is now FDA-approved for treating ErbB-2 overexpressing breast cancers (Pegram et al. 1999, Pietras et al. 1998. Agus et al. 1999, Table 1).
Studies of trastuzumab in PCa xenograft models have demonstrated antitumor activity, while clinical trials in men with advanced PCa showed little efficacy because PCa cells rarely overexpress ErbB-2 and trastuzumab is most effective when cells express high levels of ErbB-2 (Baselga et al. 1999, Ziada et al. 2004). Another ErbB-2 targeting monoclonal antibody, pertuzumab, targets a different extracellular domain than trastuzumab and strictly blocks the association of ErbB-2 with other ErbB members thus preventing signaling in both high and low ErbB-2 expressing tumor cells (Agus et al. 2002). Importantly, pertuzumab has shown to delay disease progression in CR PCa patients (Agus et al. 2007). Currently, pertuzumab is approved by FDA only for breast cancer treatment; nevertheless, one clinical trial analyzing the efficacy of pertuzumab in PCa patients was terminated due to insufficient therapeutic response (de Bono et al. 2007). Although this study ultimately failed, screening of PCa tumor samples for the reliance of the cancer on ErbB-2 signaling and directing the study towards the CR PCa patient population may increase the effectiveness of pertuzumab treatment for a subpopulation of PCa patients. Currently, clinical trials on ErbB-2-targeted immunotherapies continue to recruit PCa and CR PCa patients, thus suggesting the potential of this avenue of treatment can be beneficial for a particular patient population and/or in combination with other therapeutic agents.
5.5. Promising ErbB-2 Combination Treatments for CR PCa
Although many clinical trials analyzing the effectiveness of ErbB-2 as a single therapeutic target have ended with unfavorable results, combination therapies including these molecules have shown promise and should be subjected to further analysis in clinical trials. Upon termination of the phase II clinical trial of peruzumab in CR PCa, de Bono et al. (de Bono et al. 2007) suggested that the lack of efficacy of pertuzumab could likely be due to intraprostatic androgen signaling. As such, several lines of study have analyzed the effects of inhibition of ErbB-2 in the presence of anti-androgens. Interestingly, the risk of recurrence of PCa xenograft tumors was significantly reduced when castration or anti-androgens were combined with trastuzumab or mTOR inhibitor everolimus (Guyader et al. 2012). The combination of lapatinib and enzalutamide also has shown synergistic anti-tumor effects in CR PCa (Shiota et al. 2015), and castration in combination with herbal extract of Wedelia chinensis greatly reduced CR PCa growth via disruption of ErbB-2, AKT and AR signaling (Tsai et al. 2017, Table 1). Together, these data suggest that clinical trials would be more successful with combination of ErbB-2 inhibitors and anti-androgens and thus warrants further investigation.
6. Conclusion and Future Perspectives
In summary, ErbB receptors are vital to many signaling events and are indispensable for cancer cell survival in a variety of cancer subsets, including those in PCa. ErbB-2 is a unique member within this family as it currently does not have a known ligand; ErbB-2 activation in general requires heterodimerization with another activated ErbB family member. Therefore, ErbB-2 can be regulated by a variety of ligands binding to other ErbB family members as well as a variety of intracellular proteins. Conversely, protein phosphatase cPAcP has been demonstrated to negatively regulate ErbB-2 enzymatic activity via protein dephosphorylation, and miR-331-3p reduces ErbB-2 mRNA levels. ErbB-2 activity can also be positively regulated by androgens, oxidase p66Shc, CXCR4, and Src. ErbB-2 downstream targets include AKT, ERK and STAT proteins, and upon activation of these proteins, there is promotion of tumor progression, including cell growth, survival and migration, as demonstrated in Figure 3. As such, results of several studies have indicated that ErbB-2 protein or its hyper-phosphorylation levels could be analyzed for potential utilization as a biomarker for the progression of PCa and other steroid hormone-regulated cancers. In parallel, several approaches to target ErbB-2 activity have been investigated, including dual EGFR/ErbB-2 inhibitors, ErbB-2 vaccine, and immunotherapy as shown in Table 1. Unfortunately, most of these ErbB-2-targeted single therapies have limited antitumor activity in clinical trials for PCa. Thus, development of anticancer agents that target ErbB-2 regulatory molecules or downstream proteins may be a more promising method to reduce deaths due to CR PCa, in addition to combination therapies with anti-androgens. For example, dual targeting of ErbB-2 and PI3K has been shown to be an effective means of growth suppression in breast cancer (Junttila et al. 2009, Rexer et al. 2014, Choi et al. 2018) that could be translated to CR PCa.
Moving forward, ErbB-2 remains an important molecule in the context of PCa, especially in advanced disease progression. Significantly, the analysis on ErbB-2 phosphorylation status in patient PCa samples should be performed to determine if altered phosphorylation of ErbB-2 is seen upon disease progression to the CR stage, as observed in the PCa cell progressive model (Muniyan et al. 2015, Meng et al. 1998, Chuang et al. 2010, Wu et al. 2010). As such, restoration of cPAcP expression may serve as an alternative for CR PCa treatment. Furthermore, the role of ErbB-2 in PCa stem cell and NE-like cell differentiation is still poorly understood and warrants further investigation. Understanding the unique signaling pathway of ErbB-2 in CR PCa cells will help identify novel targets for CR PCa therapy, such as the novel approach to restore cPAcP expression or knockdown of p66Shc protein.
Acknowledgements
This study was supported in part by the National Institutes of Health [CA88184], the US Department of Defense PCRP Grant [PC121645, and PC141559], the University of Nebraska Food for Health Grant, the University of Nebraska Medical Center Bridge Fund, the Buffet Cancer Center Pilot Project Grant, the University of Nebraska Medical Center Cancer Biology Training Grant [T32CA009476], the University of Nebraska Medical Center Graduate Fellowship, and the Purdue Pharma Scholars Award.
Abbreviations
- ADT
Androgen deprivation therapy
- AI
Androgen-independent
- AR
Androgen receptor
- AREG
Amphiregulin
- AS
Androgen-sensitive
- BTC
Betacellulin
- CR
Castration-resistant
- CREB1
cAMP responsive element binding protein 1
- CDKA
Cyclin-dependent kinase A
- CXCR4
C-X-C chemokine receptor type 4
- CXCL12
C-X-C motif chemokine ligand 12
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial-to-mesenchymal transition
- EPG
Epigen
- EPR
Epiregulin
- ERK
Extracellular-signal-regulated kinase
- FAK
Focal adhesion kinase
- FOXM1
Forkhead box protein M1
- GPCR
G-protein coupled receptor
- GMCS
Granulocyte macrophage-colony-stimulating
- GPI
glycosylphosphatidylinositol
- Grb2
Growth factor receptor bound protein 2
- Hsp
Heat shock protein
- HB-EGF
Heparin-binding EGF-like growth factor
- HGF
hepatocyte growth factor
- HuR
Human antigen R
- IL-6
Interleukin-6
- MAPK
Mitogen-activated protein kinase
- MEFs; MEK; miRNA
microRNA, MAPK/ERK kinase, mouse embryonic fibroblasts
- mTOR
mammalian target of rapamycin
- NE
Neuroendocrine
- NOX
NADPH oxidase
- NRG
Neuregulin
- cPAcP
cellular prostatic acid phosphatase
- PCa
Prostate cancer
- PI3K
Phosphoinositide 3-kinase
- PIP3
Phosphatidylinositol (3,4,5)-triphosphate
- PIP2
Phosphatidylinositol (,4,5)-bisphosphate
- PLCγ
Phospholipase C γ
- PKB
Protein kinase B
- PSA
Prostate specific antigen
- PTEN
Phosphatase and tensin homolog
- PTK
Protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- PYK2
Protein tyrosine kinase 2
- ROS
reactive oxygen species
- RTK
Receptor tyrosine kinase
- STAT
Signal transducer and activator of transcription
- Shc
Src homology 2 domain containing
- SHP1
Src homology region 2 domian containing phosphatase-1
- SOS1
Son-of-Sevenless 1
- Syk
Spleen tyrosine kinase
- TGF-α
Transforming growth factor alpha
- VEGF
Vascular endothelial growth factor
Footnotes
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- Agus DB, Scher HI, Higgins B, Fox WB, Heller G, Fazzari M, Cordon-Cardo C, Golde DW 1999. Response of prostate cancer to anti-HER-2/neu antibody in androgen-dependent and –independent human xenograft model. Cancer Research 59(19) 4761–4764. (PMID: ) [PubMed] [Google Scholar]
- Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI 2002. Targeting ligand-activated ErbB-2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2(2) 127–137. ( 10.1016/S1535-6108(02)00097-1) [DOI] [PubMed] [Google Scholar]
- Agus DB, Sweeney CJ, Morris MJ, Mendelson DS, McNeel DG, Ahmann FR, Wang J, Derynck MK, Ng K, Lyons B, et al. 2007. Efficacy and safety of single-agent pertuzumab (rhuMAb 2C4), a human epidermal growth factor receptor dimerization inhibitor, in castration-resistant prostate cancer after progression from taxane-based therapy.Journal of Clinical Oncology 25(6) 675–681. (doi: 10.1200/JCO.2006.07.0649) [DOI] [PubMed] [Google Scholar]
- Alam SM, Rajendran M, Ouyang S, Veeramani S, Zhang L, Lin MF 2009. A novel role of Shc adaptor proteins in steroid hormone-regulated cancers. Endocrine Related Cancer 16(1) 1. (doi: 10.1677/ERC-08-0179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo A, Cunha V, Teixeira AL, Medeiros R 2011. IL-6/IL-6R as a potential key signaling pathway in prostate cancer development. World Journal of Clinical Oncology 2(12) 384–396. (doi: 10.5306/wjco.v2.i12.384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballaré C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M 2003. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Molecular and Cell Biology 23(6) 1994–2008. (doi: 10.1128/MCB.23.6.1994-2008.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, et al. 1999. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Seminars in Oncology 26 (4 Suppl 12) 78–83. (PMID:) [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P 1998. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17(3) 313–325. (doi: 10.1038/sj.onc.1201947) [DOI] [PubMed] [Google Scholar]
- Bhattacharya R, Bukkapatnam R, Praoko I, Soto J, Morgan M, Salup RR 2002. Efficacy of vaccination with plasmid DNA encoding for HER2/neu or HER2/neu-EGFP fusion protein against prostate cancer in rats. International Immunopharmacology 2(6) 783–796. (doi: 10.1016/S1567-5769(02)00017-6) [DOI] [PubMed] [Google Scholar]
- Bishop JL, Thaper D, Zoubeidi A 2014. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers (Basel) 6(2) 829–859. (doi: 10.3390/cancers6020829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, Ellis C, Casey M, Vukelja S, Bischoff, et al. 2010. Randomized Study of Lapatinib Alone or in Combination With Trastuzumab in Women With ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. Journal of Clinical Oncology 28(7) 1124–1130. (doi: 10.1200/JCO.2008.21.4437) [DOI] [PubMed] [Google Scholar]
- Campos S, Hamid O, Sedein MV, Oza A, Plante M, Potkul RK, Lenehan PF, Kaldjian EP, Varterasian ML, Jordan C, et al. 2005. Multicancer, randomized phase II trial of oral CI-1033 for previously treated advanced ovarian cancer. Journal of Clinical Oncology 23(24) 5597–5604. (doi: 10.1200/JCO.2005.08.091) [DOI] [PubMed] [Google Scholar]
- Carles J, Lloreta J, Salido M, Font A, Suarez M, Baena V, Nogue M, Domenech M, Fabregat X 2004. Her-2/neu expression in prostate cancer: a dynamic process? Clinical Cancer Research 10(14) 4742–4745. (doi: 10.1158/1078-0432.CCR-04-0115) [DOI] [PubMed] [Google Scholar]
- Carrion-Salip D, Panosa C, Menendez JA, Puiq T, Oliveras G, Pandiella A, De Llorens R, Massaquer A 2012. Androgen-independent prostate cancer cells circumvent EGFR inhibition by overexpression of alternative HER receptors and ligands. International Journal of Oncology 41(3) 1128–1138. (doi: 10.3892/ijo.2012.1509) [DOI] [PubMed] [Google Scholar]
- Chia KM, Liu J, Francis GD, Naderi A 2011. A feedback loop between androgen receptor and ERK signaling in estrogen receptor-negative breast cancer. Neoplasia 13(2) 154–166. (doi: 10.1593/neo.101324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD, Cher ML 2006. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12.Prostate 66(1) 32–48. (doi: 10.1002/pros.20318) [DOI] [PubMed] [Google Scholar]
- Cho HS, Leahy DJ 2002. Structure of the extracellular region of HER2 reveals interdomain tether. Science 297(5585) 1330–1333. (DOI: 10.1126/science.1074611) [DOI] [PubMed] [Google Scholar]
- Choi JH, Kim KH, Roh KH, Jung H, Lee A, Lee JY, Song JY, Park SJ, Kim I, Lee WS, Seo SK, Choi IW, Yea SS, Park SG 2018. A PI3K p110α-isoform-selective inhibitor enhances the efficacy of anti-HER2/neu antibody therapy against breast cancer in mice. Oncoimmunology 7(5) e1421890 (doi: 10.1080/2162402X.2017.1421890) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YW, Lin FF, Muniyan S, Lin FC, Chen CS, Wang J, Huang CC, Lin MF 2015. Cellular prostatic acid phosphatase (cPAcP) serves as a useful biomarker of histone deacetylase (HDAC) inhibitors in prostate cancer cell growth. Cell Bioscience 5 38. (doi: 10.1186/s13578-015-0033-y.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TD, Chen SJ, Lin FF, Veeramani S, Kumar S, Batra SK, Tu Y, Lin MF 2010. Human prostatic acid phosphatase, an authentic tyrosine phosphatase, dephosphorylates ErbB-2 and regulates prostate cancer cell growth. Journal of Biological Chemistry 285(31) 23598–23606. (doi: 10.1074/jbc.M109.098301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley-LaComb MK, Semaan L, Singareddy R, Li Y, Kim S, Cher ML, Chinni SR 2016. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate bone metastasis. Molecular Cancer 15(1) 68. (doi: 10.1186/s12943-016-0552-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Cariaga-Martinez AE, Lobo MVT, Martin Orozco RM, Motino O, Rodriguez-Ubreva FJ, Angulo J, Lopez-Ruiz P, Colas B 2012. EGF promotes neuroendocrine-like differentiation of prostate cancer cells in the presence of LY294002 through increased ErbB-2 expression independent of phosphatidylinositol 3-kinase-AKT pathway. Carcinogenesis 33(6) 1169–1177. (doi: 10.1093/carcin/bgs139) [DOI] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL 1999. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. National Medicine 5 280–285. (doi: 10.1038/6495) [DOI] [PubMed] [Google Scholar]
- Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM 2005. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Molecular and Cellular Biology 25(17) 7734–7742. (doi: 10.1128/MCB.25.17.7734-7742.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson JP, Bu Z, Lemmon MA 2007 Ligand-induced structural transitions in ErbB receptor extracellular domains. Structure 15(8) 942–954. ( 10.1016/j.str.2007.06.013) [DOI] [PubMed] [Google Scholar]
- Day KC, Lorenzatti Hiles G, Kozminsky M, Dawsey SJ, Paul A, Broses LJ, Shah R, Kunja LP, Hall C, Palanisamy N, et al. (2017) HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Research 77(1) 74–85. (doi: 10.1158/0008-5472.CAN-16-1656) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono JS, Rowinsky EK 2002. The ErbB receptor family: a therapeutic target for cancer. Trends in Molecular Medicine 8(4) S19–S26. ( 10.1016/S1471-4914(02)02306-7) [DOI] [PubMed] [Google Scholar]
- de Bono JS, Bellmunt J, Attard G, Droz JP, Miller K, Flechon A, Sternberg C, Parker C, Zugmaier G, Hersberger-Gimenez V, et al. 2007. Open-label phase II study evaluating the efficacy and safety of two doses of pertuzumab in castrate chemotherapy-naïve patients with hormone refractory prostate cancer. Journal of Clinical Oncology 25(3) 257–262 (doi: 10.1200/JCO.2006.07.0888) [DOI] [PubMed] [Google Scholar]
- Debes JD, Tindall DJ 2004. Mechanisms of androgen-refractory prostate cancer. New England Journal of Medicine 315 1488–1490. (doi: 10.1056/NEJMp048178) [DOI] [PubMed] [Google Scholar]
- Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, Gross GJ, Falck JR, Patel PK, Jacobs ER, Medhora M 2008. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. American Journal of Physiology-Heart and Circulatory Physiology 294(2) H724–H735. (doi: 10.1152/ajpheart.00979.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, Fleming TP, Hazan R, Ullrich A, King CR, Schlessinger J, Aaronson SA 1987. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell 51(6) 1063–1070. ( 10.1016/0092-8674(87)90592-7) [DOI] [PubMed] [Google Scholar]
- Dillard PR, Lin MF, Khan SA 2008. Androgen-independent prostate cancer cells aquire the complete steridogenic potential of synthesizing testosterone from cholesterol. Molecular and Cellular Endocrinology 295 115–20. (doi: 10.1016/j.mce.2008.08.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disis ML, Grabstein KH, Sleath PR, Cheever MA 1999. Generation of immunity to the HER-2/neu oncogenic protein in patients with breast and ovarian cancer using a peptide-based vaccine. Clinical Cancer Research 5(6) 1289–1297. (PMID: ) [PubMed] [Google Scholar]
- Drebin JA, Link VC, Weinberg RA, Greene MI 1986. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proceedings of the National Academy of Science U.S.A 83(23) 9129–9133. (PMID: ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drebin JA, Link VC, Greene MI 1988. Monoclonal antibodies specific for the neu oncogene product directly mediate anti-tumor effects in vivo. Oncogene 2(4) 387–394. (PMID:) [PubMed] [Google Scholar]
- Edwards J, Traynor P, Munro AF, Pirret CF, Dunne B, Bartlett JM 2006. The role of HER1–4 and EGFRvIII in hormone-refractory prostate cancer. Clinical Cancer Research 12(1) 123–130. (doi: 10.1158/1078-0432.CCR-05-1445) [DOI] [PubMed] [Google Scholar]
- El Sheikh SS, Domin J, Abel P, Stamp G, Lalani EN 2004. Phosphorylation of both EGFR and ErbB-2 is a reliable predictor of prostate cancer cell proliferation in response to EGF. Neoplasia 6(6) 846–853. ( 10.1593/neo.04379) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epis MR, Giles KM, Barker A, Kendrick TS, Leedman PJ 2009. miR-331–3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. Journal of Biological Chemistry 284(37) 24696–24704. (doi: 10.1074/jbc.M109.030098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ 2011. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331–3p in prostate cancer cells. Journal of Biological Chemistry 286(48) 41442–41454. (doi: 10.1074/jbc.M111.301481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer OM, Hart S, Gschwind A, Ullrich A 2003. EGFR signal transactivation in cancer cells. Biochemical Society Transactions 31(6) 1203–1208. (doi:10.1042/) [DOI] [PubMed] [Google Scholar]
- Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C 2006. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proccedings of the National Academy of Science 103(39) 14477–14482. (Doi: 10.1073/pnas.0606836103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Ye H, Gerrin S, Wang H, Sharma A, Chen S, Patnaik A, Sowalsky AG, Voznesensky O, Han W, et al. 2016. ErbB2 signaling increases androgen receptor expression in abiraterone-resistant prostate cancer. Clinical Cancer Research 22(14) 3672–3682. (doi: 10.1158/1078-0432.CCR-15-2309.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein AK 2004. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. Journal of Biological Chemistry 279(4) 2737–2746. (doi:1-/1-74/jbc.M3099999200) [DOI] [PubMed] [Google Scholar]
- Gioeli D, Mandell JW, Petroni GR, Frierson HF, Weber MJ 1999. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Research 59(2) 279–284. (PMID:) [PubMed] [Google Scholar]
- Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, Paschal BM, Weber MJ 2006. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Molecular Endocrinology 20(3) 503–515. (doi: 10.1210/me.2005-0351) [DOI] [PubMed] [Google Scholar]
- Gregory CW, Whang YE, McCall W, Fei X, Liu Y, Ponguta LA, French FS, Wilson EM, Earp III HS 2005. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clinical Cancer Research 11(5) 1704–1712. (doi: 10.1158/107-0432.CCR-04-1158) [DOI] [PubMed] [Google Scholar]
- Guyader C, Ceraline J, Gravier E, Morin A, Michel S, Erdmann E, de Pinieux G, Cabon F, Bergerat JP, Poupon MF, Oudard S 2012. Risk of hormone escape in a human prostate cancer model depends on therapy modalities and can be reduced by tyrosine kinase inhibitors. PLoS One 7(8) e43352. (doi: 10.1371/journal.pone.0042252) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin MN, Masson MR, Powner D, Saqib KM, Ponting CP, Wakelam MJ 2000. Phospholipase D regulation and localisation is dependent upon a phosphatidylinositol 4, 5-bisphosphate-specific PH domain. Current Biology 10(1) 43–66. ( 10.1016/S0960-9822(99)00264-X) [DOI] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF III, Hynes NE 2003. The ErbB-2/ErbB-3 heterodimer functions as an oncogenic unit: ErbB-2 requires ErbB-3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Science. U.S.A 100(15) 8933–8938. (doi: 10.1073/pnas.1537685100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu FN, Yang MS, Lin E, Tseng CF, Lin H 2011. The significance of Her2 on androgen receptor protein stability in the transition of androgen requirement in prostate cancer cells. American Journal of Physiological Endocrinology Metabolism 3000 E902–E908. (doi: 10.1152/ajpendo.00610.2010) [DOI] [PubMed] [Google Scholar]
- Igawa T, Lin FF, Rao P, Lin MF 2003. Suppression of LNCaP prostate cancer xenograft tumors by a prostate-specific protein tyrosine phosphatase, prostatic acid phosphatase. The Prostate 55 247–258. (doi: 10.1002/pros.10240) [DOI] [PubMed] [Google Scholar]
- Ingersoll MA, Chou YW, Lin JS, Yuan TC, Miller DR, Xie Y, Tu Y, Oberley-Deegan RE, Batra SK, Lin MF 2018. p66Shc regulates migration of castration-resistant prostate cancer cells. Cellular Signaling 46, 1–14. (doi: 10.1016/j.cellsig.2018.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawar RC, Miyake T, Parsons SK 2007. c-Src modulates ErbB-2 and ErbB-3 heterocomplex formation and function. Oncogene 26 3503–3510. (doi: 10.1038/sj.onc.1210138) [DOI] [PubMed] [Google Scholar]
- Jin H, Park MH, Kim SM 2015. 3, 3’-Diindolylmethane potentiates paclitaxel-induced antitumor effects on gastric cancer cells through the Akt/FOXM1 signaling cascade. Oncology Reports 33(4) 2031–2036. (doi: 10.3892/or.2015.3758) [DOI] [PubMed] [Google Scholar]
- Johnson TR, Khandrika L, Kumar B, Venezia S, Koul S, Chandhoke R, Maroni P, Donohue R, Meacham RB, Koul HK 2008. Focal adhesion kinase controls aggressive phenotype of androgen-independent prostate cancer cells. Molecular Cancer Research. 6(10), 1639–1648. (doi: 10.1158/1541-7786.MCR-08-0052) [DOI] [PubMed] [Google Scholar]
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, Van Roy F, Dargemont C, De Herreros AG, Bellacosa A, Larue L 2007. Activation of NF-[kappa] B by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 26(53) 7445. (doi: 10.1038/sj.onc.1210546) [DOI] [PubMed] [Google Scholar]
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX 2009. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15(5) 429–440. (doi: 10.1016/j.ccr.2009.03.020) [DOI] [PubMed] [Google Scholar]
- Khanday FA, Santhanam L, Kasuno K, Yamamori T, Dericco J, Bugayenko A, Mattagajasingh I, Disanza A, Scita G, Irani K 2006. Sos-mediated activation of Rac1 by p66Shc. Journal of Cell Biology 172(6) 817–822. (doi: 10.1083/jcb.200506001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C 2008. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. Journal of Clinical Investigations 118(9) 3051. (doi: 10.1172/JCI34764) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kue PF, Taub JS, Harrington LB, Polakiewicz RD, Ullrich A, Daaka Y 2002. Lysophosphatidic acid‐regulated mitogenic ERK signaling in androgen‐insensitive prostate cancer PC‐3 cells. International Journal of Cancer 102(6) 572–579. (doi: 10.1002/ijc.10734) [DOI] [PubMed] [Google Scholar]
- Kumar S, Kumar S, Rajendran M, Alam SM, Lin FF, Cheng PW, Lin MF 2011. Steroids up-regulate p66Shc longevity protein in growth regulation by inhibiting its ubiquitination. PLoS One 6(1) e15942 ( 10.1371/journal.pone.0015942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Igawa T, Yuan TC, Zhang XQ, Lin FF, Lin MF 2003. ErbB-2 signaling is involved in regulating PSA secretion in androgen-independent human prostate cancer LNCaP C-81 cells. Oncogene 22 781–796. (doi: 10.1038/sj.onc.126066) [DOI] [PubMed] [Google Scholar]
- Lee MS, Igawa T, Cehn SJ, Van Bemmel D, Lin JS, Lin FF, Johansson SL, Christman JK, Lin MF 2004. p66Shc protein is upregulated by steroid hormones in hormone-sensitive cancer cells and in primary prostate carcinomas. International Journal of Cancer 108 672–678. (doi: 10.1002/ijc.11621) [DOI] [PubMed] [Google Scholar]
- Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC 2004. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6(5) 459–469. (doi: 10.1186/1756-9966-29-16) [DOI] [PubMed] [Google Scholar]
- Liang J, Slingerland JM 2003. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2(4) 336–342. (PMID:) [PubMed] [Google Scholar]
- Lin DL, Whitney MC, Yao Z, Keller ET 2001. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clinical Cancer Research 7(6) 1773–1781. (PMID: ) [PubMed] [Google Scholar]
- Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, Chang C 2003. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. Journal of Biological Chemistry 278(51) 50902–50907. (doi: 10.1074/jbc.M300676200) [DOI] [PubMed] [Google Scholar]
- Lin MF, Clinton GM 1986. Human prostatic acid phosphatase has phosphotyrosyl protein phosphatase activity. Biochemical Journal 235 351–357. (PMID: ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell DK, Luttrell LM 2004. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 23(48) 7969–7978. (doi: 10.1038/sj.onc.1208162) [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE 1998. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. Journal of Biological Chemistry 273(22) 13375–13378. (doi: 10.1074/jbc.273.22.13375) [DOI] [PubMed] [Google Scholar]
- McKay MM, Morrison DK 2007. Integrating signals from RTKs to ERK/MAPK. Oncogene 26(22) 3113. (doi: 10.1038/sj.onc.1210394) [DOI] [PubMed] [Google Scholar]
- Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL 2004. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 6(5) 517–527. (doi: 10.1016/j.ccr.2004.09.031) [DOI] [PubMed] [Google Scholar]
- Meng TC, Lin MF 1998. Tyrosine phosphorylation of c-ErbB-2 is regulated by the cellular form of prostatic acid phosphatase in human prostate cancer cells. Journal of Biological Chemistry 273(34) 22096–22104. (doi: 10.1074/jbc.273.34.22096) [DOI] [PubMed] [Google Scholar]
- Minner S, Jessen B, Stiedenroth L, Burandt E, Kollermann J, Mirlacher M, Erbersdobler A, Eichelberg C, Fisch M, Brummendorf TH, et al. 2010. Low level HER2 overexpression is associated with rapid tumor cell proliferation and poor prognosis in prostate cancer. Clinical Cancer Research 16 1553–1560. (doi: 10.1158/1078-0432.CCR-09-2546) [DOI] [PubMed] [Google Scholar]
- Molife LR, Omlin A, Jones RJ, Karavasilis V, Bloomfield D, Lumsden G, Fong PC, Olmos D, O’Sullivan JM, Pedley I, et al. 2014. Randomized phase II trial of nintedanib, afatinib and sequential combination in castration-resistant prostate cancer. Future Oncology 10(2) 219–231. (doi: 10.2217/fon.13.250) [DOI] [PubMed] [Google Scholar]
- Montironi R, Mazzucchelli R, Barbisan F, Stramazzotti D, Santinelli A, Scarpelli M, Lopez BA 2006. HER2 expression and gene amplification in pT2a Gleason score 6 prostate cancer incidentally detected in cystoprostatectomies: comparison with clinically detected androgen-dependent and androgen-independent cancer. Human Pathology 37 1137–1144. (doi: 10.1016/j.humpath.2006.04.004) [DOI] [PubMed] [Google Scholar]
- Muniyan S, Ingersoll MA, Batra SK, Lin MF 2014. Cellular prostatic acid phosphatase, a PTEN-functional homologue in prostate epithelia, functions as a prostate-specific tumor suppressor. Biochimica et Biophysica Acta 1846 88–98. (doi: 10.1016/j.bbcan.2014.04.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniyan S, Chen SJ, Lin FF, Whang Z, Mehta PP, Batra SK, Lin MF 2015. ErbB-2 signaling plays a critical role in regulating androgen-sensitive and castration-resistant androgen-receptor-positive prostate cancer cells. Cell Signaling 27(11) 2261–2271. (doi: 10.1016/j.cellsig.2015.08.002.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Siegel PM, Dankort DL, Webster MA, Muller WJ 1994. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Molecular Cell Biology 14(1) 735–743. (PMID:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, Murai M 2000. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clinical Cancer Research 6(7) 2702–2706. (PMID:) [PubMed] [Google Scholar]
- Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS 2003. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocrine Related Cancer 10(1) 1–21. (doi: 10.1677/ERC-08-0208) [DOI] [PubMed] [Google Scholar]
- Oshikawa J, Kim SJ, Furuta E, Caliceti C, Chen GF, McKinney RD, Kuhr F, Levitan I, Fukai T, Ushio-Fukai M 2012. Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. American Journal of Physiology-Heart and Circulatory Physiology 302(3) 724–732. (doi: 10.1152/ajpheart.00739.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen M, Creighton CJ, Ozdemir M, Ittmann M 2008. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 27(12) 1788. (doi: 10.1038/sj.onc.1210809) [DOI] [PubMed] [Google Scholar]
- Pegram MD, Lipton A, Hayes DF, Weber BL, Baselga JM, Tripathy D, Baly D, Baughman SA, Twaddell T, Glaspy JA, Slamon DJ 1998. Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment. Journal of Clinical Oncology 16(8) 2659–2671. (doi: 10.1200/JCO.1998.16.8.2659) [DOI] [PubMed] [Google Scholar]
- Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, Zhang W 2006. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. Journal of Cellular Biology 175(6) 913–923. (doi: 10.1083/jcb.200512100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton K, Freeman MR, Solomon KR 2013. Cholesterol and prostate cancer. Current Opinions in Pharmacology 12(6) 751–759. (doi: 10.1016/j.coph.2012.07.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechocki MP, Ho YS, Pilon S, Wei WZ 2003. Human ErbB-2 (Her-2) transgenic mice: a model system for testing Her-2 based vaccines. Journal of Immunology 171(11) 5787–5794. ( 10.4049/jimmunol.171.11.5787) [DOI] [PubMed] [Google Scholar]
- Pietras RJ, Pegram MD, Finn RS, Maneval DA, Slamon DJ 1998. Remission of human breast cancer xenograft on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 17(17) 2235–2249. (doi: 10.1038/sj.onc.1202132) [DOI] [PubMed] [Google Scholar]
- Quintero IB, Herrala AM, Araujo CL, Pulkka AE, Hautaniemi S, Ovaska K, Pryazhnikov E, Kulesskiy E, Ruuth MK, Soini Y, et al. 2013. Transmembrane prostatic acid phosphatase (TMPAP) interacts with snapin and deficient mice develop prostate adenocarcinoma. PLoS One 8(9) e73072. (doi: 10.1371/journal.pone.0073072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, et al. 2004. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Research 64(11) 3958–3965. (doi: 10.1158/0008-5472.CAN-03-2868) [DOI] [PubMed] [Google Scholar]
- Rajendran M, Thomes P, Zhang L, Veeramani S, Lin MF 2010. p66Shc- a longevity redox protein in human prostate cancer progression and metastasis. Cancer Metastasis Reviews 29(1) 207–222. (doi: 10.1007/s10555-010-9213-8.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia AG, Musti AM, Lanzino M, Panno ML, Turano E, Zumpano R, Belfiore A, Andò S, Maggiolini M 2009. A cross-talk between the androgen receptor and the epidermal growth factor receptor leads to p38MAPK-dependent activation of mTOR and cyclinD1 expression in prostate and lung cancer cells. International Journal of Biochemistry and Cell Biology 41(3) 603–614. (doi: 10.1016/j.biocel.2008.07.004) [DOI] [PubMed] [Google Scholar]
- Regad T 2015. Targeting RTK signaling pathways in cancer. Cancers (Basel) 7(3) 1758–1784. (doi: 10.3390/cancers7030860.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid A, Vidal L, Shaw H, de Bono J 2007. Dual inhibition of ErbB-1 (EGFR/HER1) and ErbB-2 (HER2/neu). European Journal of Cancer 43(3) 481–489. (doi: 10.1016/j.ejca.2006.11.2007) [DOI] [PubMed] [Google Scholar]
- Rexer BN, Chanthaphaychith S, Dahlman K, Arteaga CL 2014. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 16(1) R9. (doi: 10.1186/bcr3601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, Smyrk TC 2002. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 23(1) 201–205. (PMID:) [DOI] [PubMed] [Google Scholar]
- Rybak AP, Ingram AJ, Tang D 2013. Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PLoS One 8(4) e61716 ( 10.1371/journal.pone.0061716) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N 1995. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncology and Hematology 19(3) 183–232. ( 10.1016/1040-8428(94)00144-I) [DOI] [PubMed] [Google Scholar]
- Sartor O, Gillessen S 2014. Treatment sequencing in metastatic castrate-resistant prostate cancer. Asian J Androl. 16(3) 426–431. (doi: 10.4103/1008-682X.126378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA 2007. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Research 67(6) 2497–2507. (doi: 10.1158/0008-5472.CAN-06-3075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J 2000. Cell signaling by receptor tyrosine kinases. Cell. 103(2) 211–225. ( 10.1016/S0092-8674(00)00114-8) [DOI] [PubMed] [Google Scholar]
- Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC 2007. Coordinate suppression of ERBB-2 and ERBB-3 by enforced expression of micro-RNA miR-125a or miR-125b. Journal of Biological Chemistry 282(2) 1479–1486. (doi: 10.1074/jbc.M609383200) [DOI] [PubMed] [Google Scholar]
- Segaliny AI, Telez-Gabriel M, Heymann MF, Heymann D 2015. Receptor tyrosine kinases: characterization, mechanism of action and therapeutic interests for bone cancers. Journal of Bone Oncology 4(1) 1–12. (doi: 10.1016/j.jbo.2015.01.001.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard HM, Lewis GD, Sarup JC, Fendly BM, Maneval D, Mordenti J, Figari I, Kotts CE, Palladino MA Jr, Ullrich A, Slamon D 1991. Monoclonal antibody therapy of human cancer: taking the HER2 protooncogene to the clinic. Journal of Clinical Immunology 11(3) 117–127. (PMID: ) [DOI] [PubMed] [Google Scholar]
- Shi XB, Tepper CG, deVere White RW 2008. microRNAs and prostate cancer. Journal of Cellular and Molecular Medicine 12(5a) 1456–1465. (doi: 10.1111/j.1582-4934.2008.00420.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota M, Bishop JL, Takeuchi A, Nip KM, Cordonnier T, Beraldi E, Kuruma H, Gleave ME, Zoubeidi A 2015. Inhibition of HER2-YB1-AR axis with lapatinib synergistically enhances enzalutamide anti-tumor efficacy in castration resistant prostate cancer. Oncotarget 6(11) 9086–9098. (doi: 10.18632/oncotarget.3602) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A 2019. Cancer Statistics, 2019. CA: A Cancer Journal for Clinicians 69 7–34. (doi: 10.3322/caac.21551) [DOI] [PubMed] [Google Scholar]
- Spiotto MT, Chung TD 2000. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate 42(3) 186–195. (doi: 10.1002/(SICI)1097–0045(20000215)42:3<186::AID-PROS4>3.0.CO;2-E) [DOI] [PubMed] [Google Scholar]
- Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK 2002. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Research 62(6) 1832–1837. (PMID:) [PubMed] [Google Scholar]
- Tan M, Li P, Klos KS, Lu J, Lan KH, Nagata Y, Fand D, Jing T, Yu D 2005. ErbB-2 promotes Src synthesis and stability: novel mechanism of Src activation that confer breast cancer metastasis. Cancer Research 65(5) 1858–1867. (doi: 10.1158/0008-5472.CAN-04-2353) [DOI] [PubMed] [Google Scholar]
- Tarhini AA, Rafique I, Floros T, Tran P, Gooding WE, Villaruz LC, Burns TF, Friedland DM, Petro DP, Farooqui M, Gomez-Garcia J, Gaither-Davis A, Dacic S, Argiris A, Socinski MA, Stabile LP, Siegfried JM 2017. Phase 1/2 study of rilotumumab (AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer. Cancer 123(15) 2936–2944. (doi: 10.1002/cncr.30717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome-Garcia J, Li D, Ghazaryan S, Shu L, Wu L 2014. ERBB-2 increases metastatic potentials specifically in androgen-insensitive prostate cancer cells. PLOS One 9(6) e99525 ( 10.1371/journal.pone.0099525) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CH, Tzeng SF, Hsieh SC, Tsai CJ, Yang YC, Tsai MH, Hsiao PW 2017. A standard wedelia chinensis extract overcomes the feedback activation of HER2/3 signaling upon androgen-ablation in prostate cancer. Frontiers in Pharmacology 8 721. (doi: 10.3389/fphar.2017.00721) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramani S, Yuan TC, Chen SJ, Lin FF, Petersen JE, Shaheduzzaman S, Srivastava S, MacDonald RG, Lin MF 2005. Cellular prostatic acid phosphatase: a protein tyrosine phosphatase involved in androgen-independent proliferation of prostate cancer. Endocrine-Related Cancers 12 805–822. (doi: 10.1677/ERC/1.00950) [DOI] [PubMed] [Google Scholar]
- Veeramani S, Yuan TC, Lin FF, Lin MF 2008. Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells. Oncogene 27 5057–5068. (doi: 10.1038/onc.2008.143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramani S, Chou YW, Lin FC, Muniyan S, Lin FF, Kumar S, Xie Y, Lele SM, Tu Y, Lin MF 2012. Reactive oxygen species induced by p66Shc longevity protein mediate nongenomic androgen action via tyrosine phosphorylation signaling to enhance tumorigenicity of prostate cancer cells. Free Radical Biology and Medicine 53 95–108. (doi: 10.1016/j.freeradbiomed.2012.03.024.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X 2003. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer cell 4(3) 209–221. ( 10.1016/S1535-6108(03)00215-0) [DOI] [PubMed] [Google Scholar]
- Wen Y, Hu MC, Makino K, Spohn B, Bartholomeusz G, Yan DH, Hung MC 2000. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Research 60(24) 6841–6845. (PMID:) [PubMed] [Google Scholar]
- Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, Wallen EM, Crane JM, Moore DT, Grigson G, et al. 2013. A phase II study of lapatinib, a dual EGRF and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urological Oncology: Seminars and Original Investigations 31(1) 82–86. (doi: 10.1016/j.urolonc.2010.09.018) [DOI] [PubMed] [Google Scholar]
- Wissner A, Overbeek E, Reich MF, Floyd MB, Johnson BD, Mamuya N, Rosfjord EC, Discafani C, Davis R, Shi X, et al. (2003). Synthesis and structure-activity relationaship of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). Journal of Medicinal Chemistry 46(1) 49–63. (doi: 10.1021/jm020241c) [DOI] [PubMed] [Google Scholar]
- Worthington J, Spain G, Timms JF 2017. Effects of ErbB-2 overexpression on the proteome and ErbB ligand-specific phosphosignaling in mammary luminal epithelial cells. Molecular and Cellular Proteomics 16(4) 608–621. (doi: 10.1074/mcp.M116.061267.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Ikawa S, Akiyama T, Semba K, Nomura N, Miyajima N, Saito T, Toyoshima K 1986. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 319(6050) 230–234. (doi: 10.1-38/319230a0) [DOI] [PubMed] [Google Scholar]
- Yu D, Hung MC 2000. Overexpression of ErbB-2 in cancer and ErbB-2-targeting strategies. Oncogene 19(53) 6115–6121. (doi: 10.1038/sj.onc.1203972) [DOI] [PubMed] [Google Scholar]
- Yuan TC, Lin FF, Veeramani S, Chen SJ, Earp III HS, Lin MF 2007. ErbB-2 via PYK2 upregulates the adhesive ability of androgen-receptor positive human prostate cancer cells. Oncogene 26 7552–7559. (doi: 10.1038/sj.onc.1210570) [DOI] [PubMed] [Google Scholar]
- Zelivianski S, Spellman M, Kellerman M, Kakitelashvilli V, Zhou XW, Lugo E, Lee MS, Taylor R, Davis TL, Hauke R, Lin MF 2003. Erk inhibitor PD98059 enhances docetaxel-induced apoptosis of androgen-independent human prostate cancer cells. International Journal of Cancer 107 478–485. (doi: 10.1002/ijc.11413) [DOI] [PubMed] [Google Scholar]
- Zhang XQ, Lee MS, Zelivianski S, Lin MF 2001. Characterization of a prostate-specific tyrosine phosphatase by mutagenesis and expression in human prostate cancer cells. Journal of Biological Chemistry 276(4) 2544–2550. (doi: 10.1074/jbc.M006661200) [DOI] [PubMed] [Google Scholar]
- Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR 2002. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Research 62(8) 2227–22231. (PMID:) [PubMed] [Google Scholar]
- Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR 2005. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. Journal of Clinical Investigations 115(4) 959. (doi: 10.1172/JCI200519935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziada A, Barqawi A, Glode LM, Varella-Garca M, Crighton F, Majeski S, Bosenblum M, Kane M, Chen L, Crawford ED 2004. The use of trastuzumab in the treatment of hormone refractory prostate cancer; phase II trial. The Prostate 60(4) 332–337. (PMID: 10.1002/pros.20065) [DOI] [PubMed] [Google Scholar]