

Abstract
The newly identified tick-borne Alongshan virus (ALSV), a segmented Jingmen virus group flavivirus, was recently associated with human disease in China. We report the detection of ALSV RNA in Ixodes ricinus ticks in south-eastern Finland. Screening of sera from patients suspected for tick-borne encephalitis for Jingmen tick virus-like virus RNA and antibodies revealed no human cases. The presence of ALSV in common European ticks warrants further investigations on its role as a human pathogen.
Keywords: Alongshan virus, ALSV, Jingmen tick virus, flavivirus, Ixodes ricinus
Recent reports have associated two members of Jingmen virus group, Alongshan virus (ALSV) and Jingmen tick virus (JMTV), to febrile disease in humans [1,2]. Here we report the presence and genetic characterisation of ALSV in Ixodes ricinus ticks in Kotka archipelago, south-eastern Finland.
In 2010, a novel segmented tick-borne RNA virus, JMTV, was detected in Rhipicephalus microplus ticks in Hubei Province, China [3]. Subsequently, similar viruses have been identified in R. microplus and cattle in Brazil, i.e. the Mogiana tick virus (MGTV) [4-6]; human Crimean-Congo haemorrhagic fever (CCHF) cases in Kosovo* [7]; Amblyomma javanense, Dermacentor silvarium and I. persulcatus ticks as well as humans in China [1]; and a red colobus monkey in Uganda [8]. Recent reports associate novel JMTV strains from China with human disease [1,2]. A retrospective study conducted by Jia et al. reported identification of JMTV from skin biopsies and blood of febrile patients [1]. Meanwhile, ALSV was detected from I. persulcatus and isolated from febrile patient sera in Heilongjian Province [2]. These viruses share the genome organisation of four segments, two of which show similarity to the NS3 and NS5 proteins of non-segmented RNA viruses in the genus Flavivirus. The other two segments appear to originate from an unknown ancestor. Together, the viruses form a separate and diverse group tentatively called the Jingmen virus group in the family Flaviviridae [9].
Detection of Jingmen-like virus in Kotka archipelago
In 2019, while performing a metatranscriptomic analysis of ticks collected in 2011 from Haapasaari island, Kotka archipelago, south-eastern Finland, we detected a full genome of JMTV-like virus together with tick-borne encephalitis virus (TBEV) genome. Thereafter, we used RT-PCR to screen 198 I. ricinus ticks collected from the Kotka archipelago in 2017 and 2018 for the presence of JMTV-like RNA. We found another positive tick from a neighbouring Kuutsalo island in the Kotka archipelago, and obtained the full genome using next-generation sequencing. The viruses (GenBank accession numbers MN107153 to MN107160) cluster together with ALSV (MH158415 to MH158418) from Heilongjian Province, China, and form a cluster distinct from the other members JMTV group, including the strains found in Kosovo (MH133313 to MH133324) [2,7] (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5). Nucleotide and amino acid identities between the Finnish strains and the other tick-borne JMTV-like viruses are shown in Table 1. The virus isolation trials in Vero, SK-N-SH and CRL-2088 cells were unsuccessful.
Figure 1.
The phylogenetic tree of NS3 segment of JMTV-like viruses
JMTV: Jingmen tick virus.
The NS3 segment amino acid sequences of all available tick-borne Jingmen tick virus-like viruses were aligned using the ClustalW algorithm. The phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19]. The GenBank accession number of the strains sequenced in this study are MN107155 and MN107159.
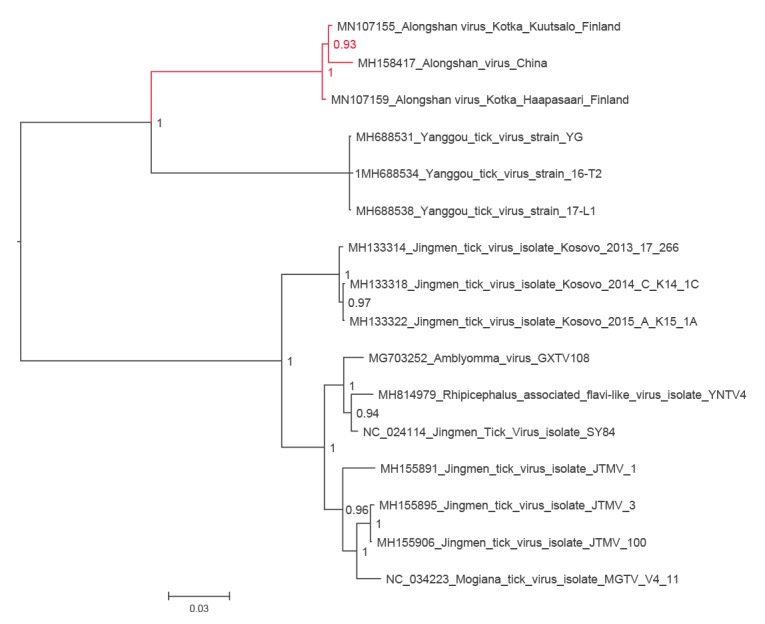
Figure 2.
The phylogenetic tree of NS5 segment of JMTV-like viruses
JMTV: Jingmen tick virus.
The NS5 segment amino acid sequences of all available tick-borne Jingmen tick virus-like viruses were aligned using the ClustalW algorithm. The phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19]. The GenBank accession number of the strains sequenced in this study are MN107156 and MN107160.
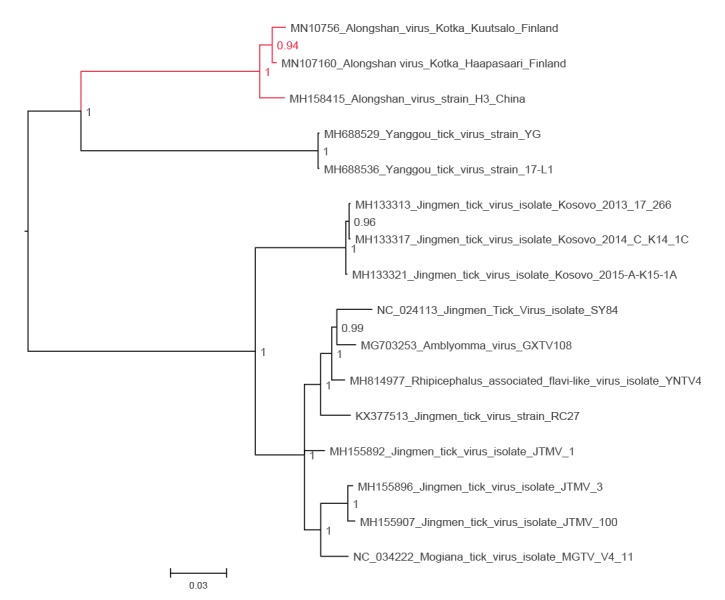
Figure 3.
The phylogenetic tree of putative capsid/membrane segment of JMTV-like viruses
JMTV: Jingmen tick virus.
The putative capsid/membrane segment amino acid sequences of all available tick-borne Jingmen tick virus-like viruses were aligned using the ClustalW algorithm. The phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19]. The GenBank accession number of the strains sequenced in this study are MN107153 and MN107157.
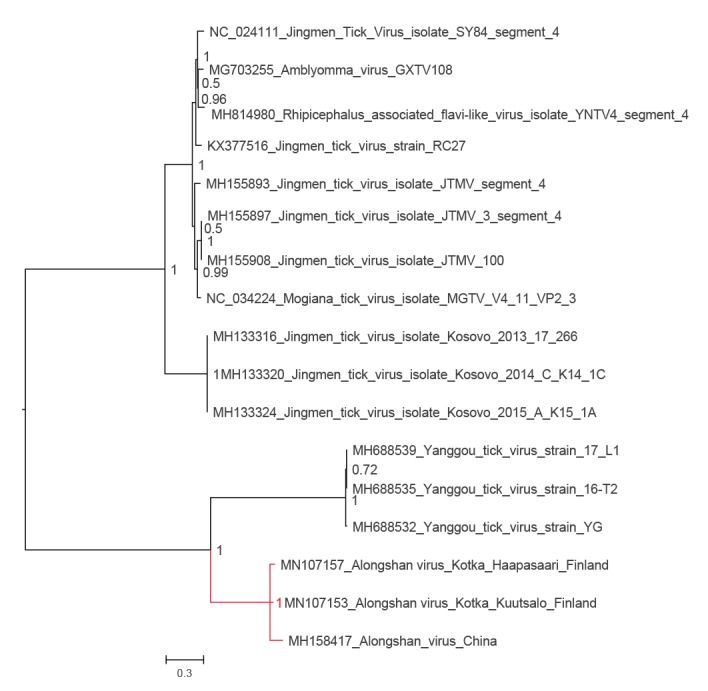
Figure 4.
The phylogenetic tree of putative glycoprotein segment of JMTV-like viruses
JMTV: Jingmen tick virus.
The putative glycoprotein segment amino acid sequences of all available tick-borne Jingmen tick virus-like viruses were aligned using the ClustalW algorithm. The phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19]. The GenBank accession number of the strains sequenced in this study are MN107154 and MN107158.
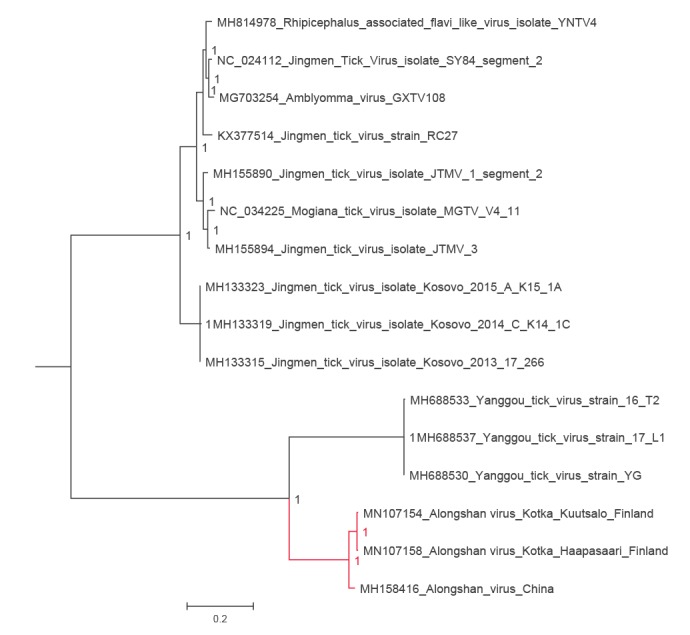
Figure 5.
The phylogenetic tree of NS5 of all species in the family Flaviviridae
NS5 sequences of the representatives of all flavivirus species were retrieved from NCBI Reference Sequence Database (RefSeq) and aligned with MAFFT programme version 7 [17] using E-INS-i algorithm, followed by removal of ambiguously aligned amino acid sites using TrimAl programme [18]. The phylogenetic tree was constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19].
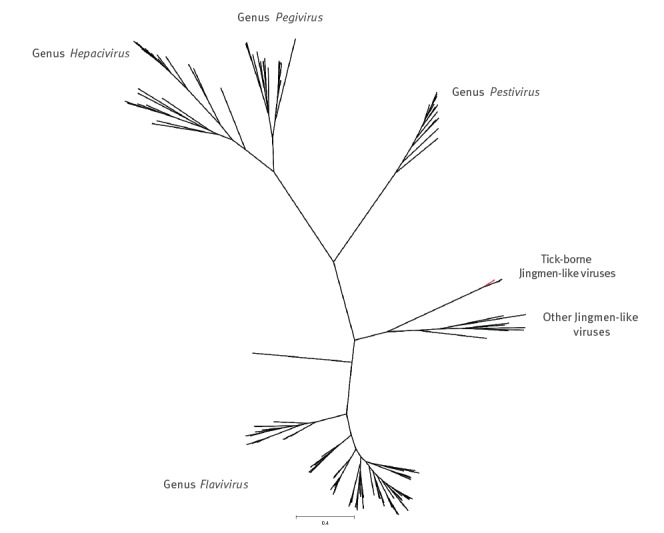
Table 1. Nt and amino acid identities between Finnish strains of Alongshan virus and other tick-borne JMTV-like viruses, Finland, 2019.
| Segment | Nt identity (%) | Amino acid identity (%) | ||||
|---|---|---|---|---|---|---|
| ALSV (Finland) | ALSV (China) | Other JMTV-like viruses | ALSV (Finland) | ALSV (China) | Other JMTV-like viruses | |
| Putative capsid/membrane | 5.3 | 9.5–9.7 | 33.1–37.7 | 1.4 | 2.4–2.8 | 29.0–36.3 |
| Putative glycoprotein | 1.5 | 8.0–8.1 | 35.9–43.1 | 0.5 | 4.4a | 38.3–51.2 |
| NS3 | 4.8 | 8.8–9.0 | 28.1–31.1 | 0.5 | 1.4–1.6 | 16.1–21.7 |
| NS5 | 4.0 | 10.5–10.6 | 28.3–32.1 | 0.9 | 2.1–2.5 | 19.8–21.6 |
ALSV: Alongshan virus; JMTV: Jingmen tick virus; Nt: nucleotide.
a Both ALSV strains from Finland have the same number of amino acid differences compared to the Chinese strain.
Human and tick samples
The emerging reports on the association of JMTV-like viruses with human disease in China [1,2] led us to investigate sera of TBE-suspected cases for JMTV-like virus RNA or antibodies against recombinant proteins of ASLV in 2019. The sera panel included 974 serum samples from 879 individuals. These samples were originally sent for TBEV antibody testing to Helsinki University Hospital laboratory (Helsinki, Finland) from May to November 2018. All samples were tested for JMTV RNA by RT-PCR, with 304 from 283 individuals (median age: 48 years, range: 1–88 years) for antibodies to JMTV VP1a, VP1b, membrane and capsid proteins. For the RNA detection, we could verify that the RT-PCR detects local Finnish strains of ALSV, but we had no human ALSV positive control samples available for the antibody tests. We also studied three serial samples from two patients positive in an earlier sample for JMTV RNA from Kosovo at dilutions 1:20 and 1:80 for reference. These two patients shown to be infected with Kosovo strains of JMTV (capsid/membrane 63.5–64.0% amino acid identity, glycoprotein 49.9–50.1% amino acid identity) did not exhibit clear reactivity to the ALSV recombinant protein [7]. The 90 and 108 ticks collected in 2017 and 2018, respectively, from Kuutsalo island, Kotka archipelago in south-eastern Finland were tested for JMTV RNA (Table 2).
Table 2. Summary of JMTV-like virus RT-PCR, NGS and antibody tests of I. ricinus ticks and sera from suspected human TBE cases., Finland, 2019.
| Sample | Year | Sample origin | Number Studied (N) |
Number of positives (n) | Method | Sequencing result of positive sample |
|---|---|---|---|---|---|---|
| I. ricinus panel | 2011 | Haapasaari island, Kotka archipelago | 3 | 1 | NGS | Whole genome ALSV |
| I. ricinus panel | 2017 | Kuutsalo island, Kotka archipelago | 90 | 1 | Conventional RT-PCR | Whole genome ALSV |
| I. ricinus panel | 2018 | Kuutsalo island, Kotka archipelago | 108 | 0 | Conventional and real-time RT-PCR | NA |
| Human sera from suspected TBE case | 2018 | Throughout Finland | 974 | 0 | Real-time RT-PCR | NA |
| Human sera from suspected TBE case | 2018 | Throughout Finland | 304 | 0 | Recombinant JMTV protein IFA | NA |
ALSV: Alongshan virus; IFA: immunofluorescence assay; JMTV: Jingmen tick virus; NA: not applicable; NGS: next generation sequencing; TBE: tick-borne encephalitis.
RNA and antibody detection
Ticks were homogenised and RNA was extracted as described previously [10]. Total nucleic acids from human serum samples were extracted using MagNa Pure LC 2.0 instrument and Total Nucleic Acid Isolation Kit (Roche, Basel, Switzerland). Viral RNA was detected with real-time or conventional reverse transcription (RT)-PCR targeting the NS5 gene. Primers and the probe were designed based on sequences available to us in August 2018, and we used JM F1312 as the forward primer (5’-TTCGGRGCMTGGCAMCTSACCT-3’), JM1548 as the reverse primer (5’-CCKGTTDTCCATYTGGTADCCCAT-3’), and JM2 as the probe (FAM-CTCCTAAAGATGTTAAACACTGC-BHQ). Conventional RT-PCR without the probe was initially used for tick samples with SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, California, United States (US)). Patient and tick samples were screened with real-time RT-PCR using the TaqMan Fast Virus 1-Step Master Mix (Thermo Scientific, Waltham, Massachusetts, US). An in vitro transcribed RNA served as the positive control.
Synthetic gene constructs encoding JTMV glycoproteins VP1a, VP1b, membrane and capsid proteins were cloned into pCAGGS/MCS [11]. The recombinant and empty plasmids were transfected into Vero E6 cells using Fugene HD according to the manufacturer’s instructions. The transfected cells were fixed onto microscopic slides with acetone, serum samples were diluted 1:20 in phosphate-buffered saline and immunofluorescence assay was performed as described previously [12].
Next generation sequencing and phylogenetic analysis
Tick homogenates were treated with a mixture of micrococcal nuclease (New England BioLabs Ipswich, Massachusetts, US) and benzonase (Millipore, Burlington, Massachusetts, US) for 1 hour at 37 °C, followed by RNA extraction using TriPure Isolation reagent (Roche, Basel, Switzerland). RRNA was removed using a NEBNext rRNA Depletion Kit (New England BioLabs) according to the manufacturer’s protocol. The sequencing library was prepared using a NEBNext Ultra II RNA Library Prep Kit (New England BioLabs). The library fragment sizes were measured using agarose gel electrophoresis and the concentrations using Qubit dsDNA BR Assay Kit (Life Technologies, Carlsbad, California, US) and NEBNext Library Quant Kit for Illumina (New England BioLabs). Sequencing was conducted using MiSeq Reagent Kit V2 with 150 bp reads.
Raw sequence reads were trimmed and low-quality, quality score < 15, and short, < 36 nt, sequences were removed using Trimmomatic [13]. Thereafter, de novo assembly was conducted using MegaHit [14]. Open reading frames were sought using MetaGeneAnnotator [15], followed by taxonomic annotation using SANSparallel [16].
Complete genome sequences of all available tick-borne JMTV-like viruses were downloaded from GenBank (accessed June 2019). The amino acid sequences were aligned using the ClustalW algorithm followed by manual refinement. In addition, NS5 sequences of the representatives of all flavivirus species were retrieved from NCBI Reference Sequence Database (RefSeq) and aligned with MAFFT programme version 7 [17] using E-INS-i algorithm, followed by removal of ambiguously aligned amino acid sites using TrimAl programme [18].
The phylogenetic trees were constructed using the Bayesian Markov chain Monte Carlo (MCMC) method, implemented in MrBayes version 3.2 [19] with two independent runs and four chains per run. The analysis was run for 5 million states and sampled every 5,000 steps.
Conclusion
Our findings show that ALSV, a newly described tick-borne human pathogen, is also present in south-eastern Finland. Notably, ALSV was detected in I. ricinus ticks, a tick species that is common across the European continent. Despite apparent ALSV circulation in the south-eastern archipelago of Finland, no ALSV RNA or antibodies to selected recombinant ALSV proteins were found in ca 900 Finnish patients suspected for TBEV infection in recent years. While our results suggest low human infection pressure, further research using other methods, including properly evaluated ALSV antibody tests, and focusing on other geographic areas and patient cohorts beyond meningitis or encephalitis cases is needed.
Acknowledgements
We are very grateful to Johanna Martikainen and Mira Utriainen for excellent laboratory assistance.
Funding statement: This work was supported by the Jane and Aatos Erkko Foundation (Jane ja Aatos Erkon Säätiö), the Academy of Finland (Suomen Akatemia), Sigrid Jusélius Foundation (Sigrid Juséliuksen Säätiö) and Helsinki University Central Hospital (Helsingin ja Uudenmaan Sairaanhoitopiiri).
Note
*This designation is without prejudice to positions on status, and is in line with United Nations Security Council Resolution 1244/99 and the International Court of Justice Opinion on the Kosovo Declaration of Independence.
Conflict of interest: None declared.
Authors’ contributions: SK drafted the manuscript, and performed RT-PCR and virus isolation. LL produced the antigens and performed the serological screen. LK performed RT-PCR, TS designed the RT-PCRs, AJJ collected the diagnostic data, FZ performed serological screening and JH designed the protein constructs. PE and JSC provided samples, performed serological screen and revised the manuscript. IP and TS performed sequencing and data analysis. TS revised the manuscript, and OV designed the study and revised the manuscript. All authors read and approved the final manuscript.
References
- 1.Jia N, Liu HB, Ni XB, Bell-Sakyi L, Zheng YC, Song JL, et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine. 2019;43:317-24. 10.1016/j.ebiom.2019.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang ZD, Wang B, Wei F, Han SZ, Zhang L, Yang ZT, et al. A New Segmented Virus Associated with Human Febrile Illness in China. N Engl J Med. 2019;380(22):2116-25. 10.1056/NEJMoa1805068 [DOI] [PubMed] [Google Scholar]
- 3.Qin XC, Shi M, Tian JH, Lin XD, Gao DY, He JR, et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc Natl Acad Sci USA. 2014;111(18):6744-9. 10.1073/pnas.1324194111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maruyama SR, Castro-Jorge LA, Ribeiro JM, Gardinassi LG, Garcia GR, Brandão LG, et al. Characterisation of divergent flavivirus NS3 and NS5 protein sequences detected in Rhipicephalus microplus ticks from Brazil. Mem Inst Oswaldo Cruz. 2014;109(1):38-50. 10.1590/0074-0276130166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Souza WM, Fumagalli MJ, Torres Carrasco AO, Romeiro MF, Modha S, Seki MC, et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci Rep. 2018;8(1):16315. 10.1038/s41598-018-34630-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pascoal JO, Siqueira SM, Maia RDC, Juan Szabó MP, Yokosawa J. Detection and molecular characterization of Mogiana tick virus (MGTV) in Rhipicephalus microplus collected from cattle in a savannah area, Uberlândia, Brazil. Ticks Tick Borne Dis. 2019;10(1):162-5. 10.1016/j.ttbdis.2018.10.002 [DOI] [PubMed] [Google Scholar]
- 7.Emmerich P, Jakupi X, von Possel R, Berisha L, Halili B, Günther S, et al. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect Genet Evol. 2018;65:6-11. 10.1016/j.meegid.2018.07.010 [DOI] [PubMed] [Google Scholar]
- 8.Ladner JT, Wiley MR, Beitzel B, Auguste AJ, Dupuis AP, Lindquist ME, et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe. 2016;20(3):357-67. 10.1016/j.chom.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, Chen LJ, et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J Virol. 2016;90(2):659-69. 10.1128/JVI.02036-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smura T, Tonteri E, Jääskeläinen A, von Troil G, Kuivanen S, Huitu O, et al. Recent establishment of tick-borne encephalitis foci with distinct viral lineages in the Helsinki area, Finland. Emerg Microbes Infect. 2019;8(1):675-83. 10.1080/22221751.2019.1612279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193-9. 10.1016/0378-1119(91)90434-D [DOI] [PubMed] [Google Scholar]
- 12.Levanov L, Kuivanen S, Matveev A, Swaminathan S, Jääskeläinen-Hakala A, Vapalahti O. Diagnostic potential and antigenic properties of recombinant tick-borne encephalitis virus subviral particles expressed in mammalian cells from Semliki Forest virus replicons. J Clin Microbiol. 2014;52(3):814-22. 10.1128/JCM.02488-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114-20. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674-6. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 15.Noguchi H, Taniguchi T, Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008;15(6):387-96. 10.1093/dnares/dsn027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somervuo P, Holm L. SANSparallel: interactive homology search against Uniprot. Nucleic Acids Res. 2015;43(W1):W24-9. 10.1093/nar/gkv317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772-80. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972-3. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539-42. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]