

Abstract
Abasic (AP) sites are one of the most common DNA lesions that block replicative polymerases. HMCES recognizes and processes these lesions in the context of single-stranded DNA (ssDNA). A HMCES DNA-protein crosslink (DPC) intermediate is thought to shield the AP site from endonucleases and error-prone polymerases. The highly evolutionarily conserved SRAP domain of HMCES and its Escherichia coli ortholog YedK mediate lesion recognition. Here we discover the basis of AP site protection by SRAP domains from a crystal structure of the YedK DPC. YedK forms a stable thiazolidine linkage between a ring-opened AP site and the α-amino and sulfhydryl substituents of its N-terminal cysteine residue. The thiazolidine linkage explains the remarkable stability of the HMCES DPC, its resistance to strand cleavage, and the proteolysis requirement for resolution. Furthermore, its structure reveals that HMCES has specificity for AP sites in ssDNA at junctions found when replicative polymerases encounter the AP lesion.
Keywords: protein-DNA crosslink, abasic site, SRAP, HMCES, thiazolidine, DNA replication, DNA repair, replication stress, stalled replication fork
Introduction
Apurinic and apyrimidinic (abasic or AP) site repair via base excision repair (BER) depends on an intact DNA duplex1–3. While most AP sites form in double-stranded DNA (dsDNA), base loss is actually more rapid in single-stranded (ssDNA)4. Furthermore, the action of the DNA helicase in replicating cells will convert dsDNA AP sites that have not been repaired into ssDNA AP sites. In this case, the replicative polymerases will stall at the AP site leaving a 3′ dsDNA-ssDNA junction. Until recently, the major mechanism to overcome this replication challenge was thought to be translesion synthesis by error-prone polymerases including Polζ5. However, we recently discovered an alternative pathway dependent on the SRAP (SOS-Response Associated Peptidase) domain protein HMCES (5-Hydroxymethylcytosine Binding, ES Cell Specific) that improves cell viability and reduces mutation frequency6.
SRAP proteins are conserved in organisms from bacteria to humans, and in bacteria SRAP encoding genes are often spatially linked to DNA repair genes7. Human HMCES and E. coli YedK are similar in both sequence (29% identity and 43% similarity) and structure (Cα r.m.s.d. of 1.29 Å between PDB entries 5KO9 and 2ICU). Both HMCES and YedK preferentially bind ssDNA and efficiently form DNA-protein crosslinks (DPCs) to AP sites in ssDNA6. DPC formation requires conserved DNA binding residues and an invariant cysteine that is almost always encoded as the second amino acid in SRAP proteins. The HMCES DPC is also formed in cells, increases in abundance in response to AP site inducing agents, and is resolved over time by a mechanism that is at least partially proteasome-dependent6. Despite the importance of the HMCES AP site DPC to this mechanism, the chemical nature of the crosslink and how the SRAP domain detects the AP site are unknown.
To better understand this unusual mechanism of DNA repair, we examined the nature of the HMCES-DNA interaction. Our results indicate that SRAP proteins crosslink to AP sites via a stable thiazolidine DNA-protein linkage formed with the N-terminal cysteine and the aldehyde form of the AP deoxyribose. This linkage and its solvent inaccessibility explain why the crosslink shields the AP site from endonucleases and likely necessitates a proteolysis-dependent mechanism for resolution. Furthermore, the structure of the SRAP DPC explains the ssDNA specificity, but suggests HMCES could accommodate a dsDNA-ssDNA 3′ junction as might be expected when a replicative polymerase stalls at the AP site. As predicted, we show that HMCES has a preference for exactly this type of DNA structure.
Results
The SRAP domains of both human HMCES and E. coli YedK form covalent linkages to AP sites in ssDNA, but the nature of the DPC is unknown. The ease of detecting a HMCES DPC in cells suggests it may be a stable chemical linkage6. Indeed, incubating the human HMCES SRAP domain DPC at 4°, 25°, or 37°C for up to six days did not change the percentage of crosslinked protein (Fig. 1a). We noticed while doing these experiments that boiling the DPC hydrolyzed the crosslink but incubation at 50°C did not (Fig. 1b). Protein denaturation is not sufficient for hydrolysis since the DPC amount does not change over time when it is incubated at room temperature after denaturing the protein by boiling for a short time (Fig. 1c). Furthermore, extensive proteolysis of the DPC with proteinase K left a small peptide-DNA linkage that remains stable (Fig. 1d) and resistant to cleavage by APE1 (Fig. 1e). Thus, the HMCES-AP DPC is unlikely to be reversible in physiological conditions and resolution almost certainly requires proteolysis followed by either an unidentified enzymatic action to remove the linkage or nucleotide excision repair.
Fig. 1. Stability analysis of the human HMCES SRAP-abasic site DNA protein crosslink.
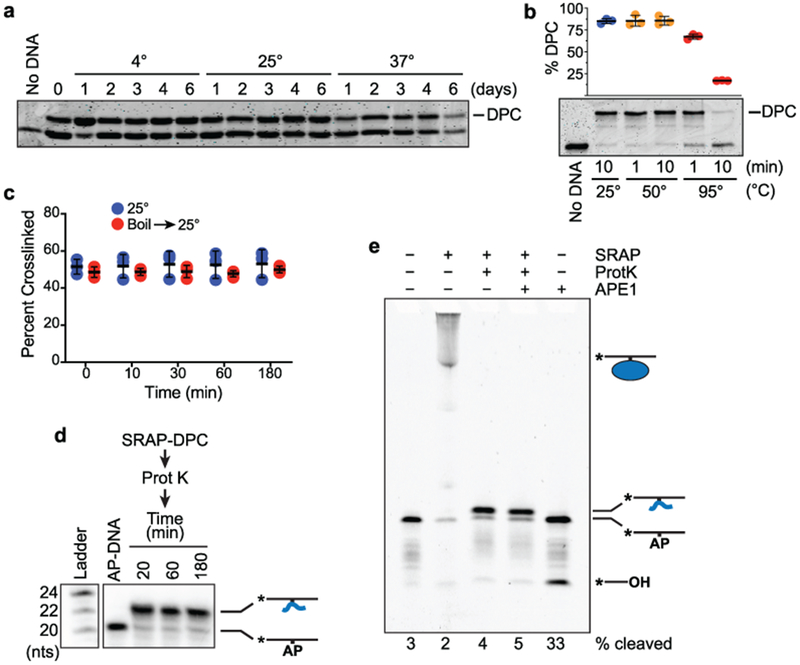
a, HMCES SRAP DPC stability measured at the indicated temperatures. Free and DNA-crosslinked HMCES was detected by coomassie blue staining. The HMCES-DPC percentage in this experiment is approximately 50% because uncrosslinked DNA was removed by dialysis after a short reaction time. b, Boiling the HMCES DPC causes hydrolysis (mean ± S.D., n=3 independent measurements) c, HMCES DPC stability measured before or after denaturation by boiling for two minutes. d, HMCES SRAP domain was incubated with a 20-mer AP-site containing oligonucleotide to form a crosslink, digested with proteinase K followed by heat inactivation of the protease, and then incubated at 37°C for the times indicated. Electrophoresis and autoradiography was used to visualize the DNA. e, HMCES SRAP was incubated with 31-mer AP-DNA and digested with proteinase K, and the peptide DPC incubated with APE1 for 2 hours. Bands were visualized by Cy5 fluorescence. Uncropped gel images are shown in Supplementary Data Set 1. Source data for b,c are available online.
To understand the molecular basis for the stability of the SRAP DPC, we determined a 1.6 Å crystal structure of E. coli YedK covalently crosslinked to 7-mer ssDNA containing an AP site (Table 1). The entire DNA ligand is visible in the electron density (Fig. 2a). The protein does not undergo any appreciable conformational change upon binding DNA, with an r.m.s.d of 1.16 Å for all atoms between unbound and DPC forms of YedK (Supplementary Fig. 1d). The core β-sheet forms an extended, positively charged channel that cradles the ssDNA phosphoribosyl backbone along one face of the protein (Fig. 2b–d). The conformation of the DNA is further constrained by nucleobase π-stacking and van der Waals interactions from random coil and α-helical motifs at each end of the binding channel that were disordered in the unbound structure (Supplementary Fig. 1d). The hydrogen-bonding edges of every nucleobase are exposed to solvent, and thus recognition of the AP site would not be sequence-dependent. Most strikingly, the DNA backbone is severely kinked and twisted by 90° at the AP site, placing the nucleobases of each flanking trinucleotide orthogonal to one another (Fig. 2b). This sharp distortion precludes pairing of a complementary DNA strand in the vicinity of the AP site, and explains why SRAP disfavors binding to dsDNA6. The residues lining the DNA binding channel are the most highly conserved among SRAP domains (Fig. 2d–e, Supplementary Fig. 1), suggesting conservation of DNA binding modality. Indeed, both YedK and HMCES have similar preferences to bind ssDNA and mutation of conserved amino acids in the channel abrogate DNA binding for both proteins6.
Table 1.
Data collection and refinement statistics
| YedK/AP-DNA Covalent DPC (6NUA) | YedK/C3spacer-DNA Non-covalent complex (6NUH) | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 61.26, 41.89, 81.42 | 47.54, 44.13, 55.09 |
| α, β, γ (°) | 90.00, 95.79, 90.00 | 90.00, 102.34, 90.00 |
| Resolution (Å) | 50.00–1.64 (1.67–1.64)a | 100.00–1.60 (1.66–1.60) |
| Rsym | 0.098 (0.500) | 0.075 (0.397) |
| Rmeas | 0.110 (0.595) | 0.086 (0.455) |
| I/σ(I) | 14.8 (1.9) | 21.3 (2.6) |
| CC1/2 | 0.989 (0.823) | 0.990 (0.869) |
| Completeness (%) | 97.4 (95.2) | 98.5 (91.1) |
| Redundancy | 4.4 (2.9) | 4.1 (4.0) |
| Refinement | ||
| Resolution (Å) | 40.50–1.64 (1.67–1.64) | 39.60–1.59 (1.65–1.59) |
| No. reflections | 49,681 (2,331) | 29,612 (2,391) |
| Rwork / Rfree | 0.770 (0.873) | 0.803 (0.829) |
| No. atoms | ||
| Protein | 3,627 | 1,802 |
| DNA | 268 | 131 |
| Bis-Tris | - | 14 |
| Water | 280 | 210 |
| B factors | ||
| Protein | 26.0 | 17.1 |
| DNA | 28.7 | 64.3 |
| Bis-Tris | - | 38.3 |
| Water | 29.3 | 24.4 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.008 |
| Bond angles (°) | 1.035 | 0.959 |
Data for each structure were generated from a single crystal.
Values in parentheses are for highest-resolution shell.
Fig. 2. YedK DPC crystal structure.
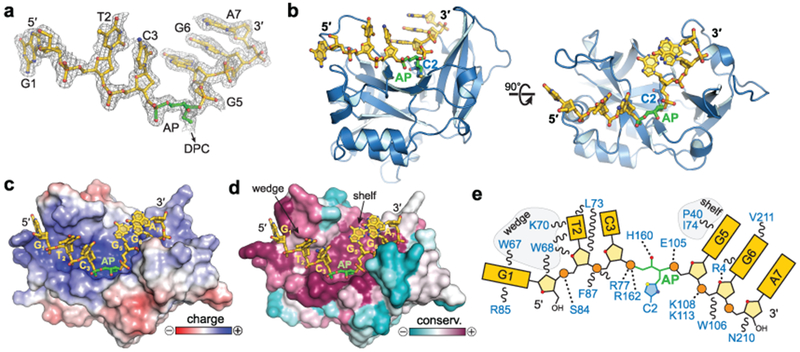
a, DNA fit to 2Fo-Fc composite annealed omit electron density contoured at 1σ. b, Orthogonal views of E. coli YedK (blue) crosslinked to AP-DNA (gold). c,d, YedK solvent-accessible surface colored by electrostatic potential from −5 to +5 kBT/eC (c) and sequence conservation from 158 unique SRAP orthologs (d). e, Schematic of protein-DNA interactions.
The AP site is positioned directly above Cys2, previously implicated in SRAP DPC formation6. This cysteine is at the N-terminus of the protein since the methionine is likely removed by aminopeptidases. The electron density clearly shows the AP site in the ring-opened form, with continuous density between C3′ and the Cys2 side chain (Fig. 3a). The anomeric C1′ carbon of the AP site is covalently bonded to both the α-amino nitrogen and the side chain sulfur of Cys2 to form a thiazolidine ring (Fig. 3a). Such a linkage would be generated by nucleophilic attack of the AP aldehyde C1′ carbon by Cys2 α-NH2 to form a Schiff base intermediate, followed by subsequent attack of C1′ by the Cys2 sulfhydryl group (Fig. 3b)8. Consistent with crosslinking by Cys2, YedK DPC formation is abrogated by removal of the thiol in a C2A mutant6, and by a C2S mutant, which potentially forms an oxazolidine ring that would not be as stable as a thiazolidine (Fig. 3c,d)9,10.
Fig. 3. The SRAP DPC forms a thiazolidine linkage stabilized by conserved residues.
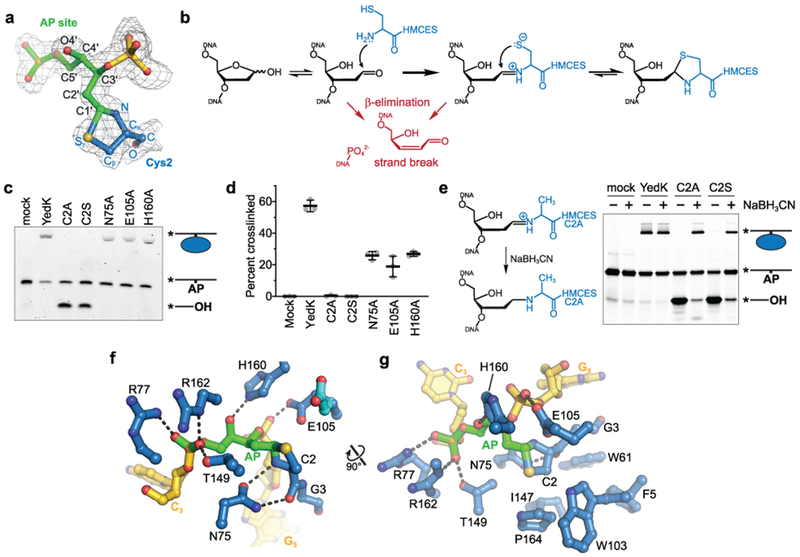
a-h, a, The DPC between the AP site (green) and Cys2 (blue) superimposed against 2Fo-Fc composite annealed omit electron density contoured at 1σ. b, Proposed chemical mechanism of the crosslinking reaction with competing lyase reactions in red. c, Representative denaturing PAGE gel showing crosslinking and lyase activity of YedK mutants. Bands were visualized by FAM fluorescence. d, Crosslinking efficiencies of YedK mutants (mean ± SD, n=3 independent measurements). e, NaBH3CN was added to crosslinking reactions to trap the Schiff base intermediates of YedK C2A and C2S mutants. The NaBH3CN-reduced Schiff base is refractory to β-elimination. Bands were visualized by FAM fluorescence. f, Residues contacting the DPC (DNA, gold; AP site green; protein, blue). The alternate Glu105 conformer is cyan. Dashed lines denote hydrogen bonds. g, Orthogonal view showing hydrophobic residues cradling Cys2. The second Glu105 conformer is not shown for clarity. Uncropped gel images are shown in Supplementary Data Set 1. Source data for d are available online.
Studies on the reaction of cysteine and aldehydes show that the equilibrium between Schiff base and thiazolidine greatly favors the latter11,12, explaining why we do not see any evidence for DNA lyase activity that can result from β-elimination of the Schiff base intermediate, such as found in bifunctional DNA glycosylases that initiate BER (Fig. 3b)13–15. In contrast to the wild-type protein, both the C2A and C2S mutant exhibited DNA lyase activity when incubated with ssDNA containing an AP site (Fig. 3c). This lyase activity was significantly reduced by performing the crosslinking reaction in the presence of sodium cyanoborohydride (NaBH3CN), which acts as a reducing agent to stabilize the Schiff base intermediate (Fig. 3e)16. These results further support a reaction mechanism that includes capture of the Schiff base intermediate by nucleophilic attack of the cysteine thiol and explains why this residue is invariant in all SRAP proteins.
Cys2 belongs to a cluster of three conserved residues that includes Glu105 and His160 implicated in SRAP function7,17. These and several other evolutionarily conserved residues stabilize the DNA and protein sides of the thiazolidine linkage (Figs. 3f,g and Supplementary Fig. 1). The AP site is stabilized by His160, which forms a hydrogen bond with the O4′ hydroxyl group (Fig. 3f). Similarly, Arg77 and Arg162, previously shown to be essential for DNA binding6, and Thr149, interact with the AP site 5′-phosphate (Fig. 3f,g). The Glu105 side chain fluctuates between two conformations at the crosslink (Fig. 3f, Supplementary Fig. 2). One conformer places one carboxylate oxygen 3.5 Å from the thiazolidine C1′ and the second within hydrogen bonding distance to the phosphate 3′ to the AP site, strongly implying that the carboxylate is protonated to avoid electrostatic repulsion with the DNA. The second conformer points back toward the core of the protein and sits further away from the thiazolidine ring. On the protein side of the crosslink, the carboxamide side chain from a highly conserved asparagine (Asn75) helps position the crosslinking nucleophile by forming two hydrogen bonds with the backbone amide nitrogen and carbonyl oxygen of Cys2 (Fig. 3f). Consistent with their roles in stabilizing the crosslink, individual substitutions of Glu105, His160, or Asn75 with alanine reduced crosslinking efficiency (Fig. 3c,d). In addition to the direct contacts to the DNA and the thiazolidine linkage, there are several highly conserved residues that create a hydrophobic pocket to cradle Cys2 from underneath (Figs. 3g and Supplementary Fig. 1). Thus, the SRAP structure guides the AP site into a specific, solvent inaccessible environment suited for thiazolidine formation and protected from AP endonuclease cleavage.
We also determined a crystal structure of YedK bound non-covalently to a ssDNA oligomer containing a C3-spacer in place of the AP site (Supplementary Fig. 3). The protein in the non-covalent complex is virtually identical to that of the DPC, except for modest repositioning of a β-hairpin (β7-β8) that was disordered in the unbound YedK structure (PDB ID 2ICU) and that stabilizes the backbone of the DNA 3′ to the AP site in the DPC (Supplementary Fig. 3a). In the non-covalent complex, the DNA at the 5′-end is positioned as in the DPC structure. However, the 3′ end of the DNA in the non-covalent complex is more mobile, as evidenced by weaker electron density and higher B-factors for the 3′ nucleotides and including the C3-spacer (Supplementary Fig. 3b–d). The destabilized 3′-DNA end resulted in a crystal packing difference between the two complexes.
Both the DPC and non-covalent complex structures suggest that the SRAP domain can accommodate dsDNA on the 3′-side of the AP site, but would disfavor duplex formation on the 5′ side. The DNA backbone on the 5′ side of the AP site is kinked 90° by a wedge motif (residues 65–73 and 84–87), which stacks against the second and third nucleotides (G1 and T2) from the AP site (Fig. 4a,b). Trp68 wedges the nucleobases of G1 and T2 apart, and G1 is stacked between Trp67 and Arg85 (Fig. 4b). Such a distortion would prevent duplex formation with DNA 5′ to the AP site. The importance of the wedge motif to HMCES function is underscored by the strong conservation of these residues among SRAP domains (Supplementary Fig. 1).
Fig. 4. SRAP can accommodate dsDNA 3′ to the AP site.
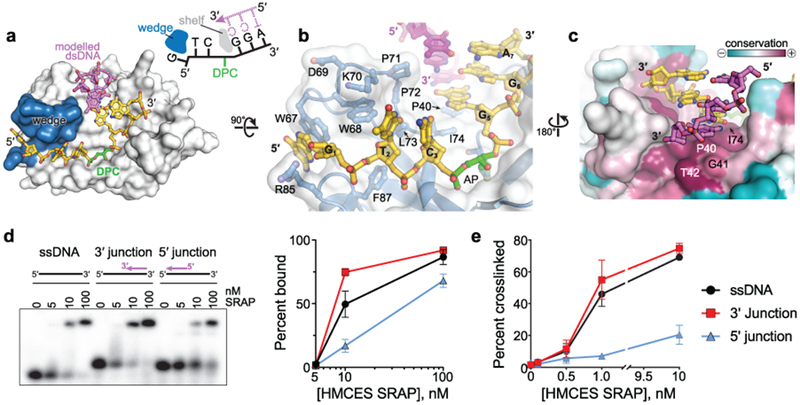
a, Model of YedK DPC with a 3′ junction at AP site. The modeled complementary DNA strand is pink. The wedge domain blocking dsDNA access 5′ to the AP site is blue. b, Wedge-DNA interactions 5′ to the AP site. c, Sequence conservation of the DNA shelf that presumably stabilizes dsDNA 3′ to the AP site. d, EMSA showing binding of human HMCES SRAP domain to the indicated DNA ligands. The plot shows mean ± S.E.M from n = 3 independent measurements. Uncropped gel image is shown in Supplementary Data Set 1. e, Percent of the indicated DNA substrates crosslinked to human HMCES SRAP domain (mean ± S.D., n=3 independent measurements). Source data for d,e are available online.
In contrast to the distorted 5′ side of the DPC, all three nucleobases on the 3′ side of the AP site are stacked in a B-DNA conformation (Fig. 4b). The residue adjacent to the AP site (guanine G5) stacks against Pro40 and Ile74 on the surface of the protein (Fig. 4b,c). The exposure of the hydrogen bonding faces of the G5, G6, and A7 nucleobases 3′ to the AP site would allow for base pairing of a second strand up to the 3′-side of the AP site. Modelling shows that a complementary strand fits against the protein surface with no steric clashes (Fig. 4a–c). The 3′-end of the modeled strand stacks against Gly41 and Thr42, which together with Pro40 and Ile74 form a highly conserved “shelf” that would stabilize a base pair 3′ to the AP site (Fig. 4c, Supplementary Fig. 1). Conservation of this shelf region implies that binding to AP sites in the context of a 3′-truncated ssDNA-dsDNA junction is an important feature. This is the exact context in which SRAP proteins should operate at a stalled replication fork since DNA polymerase stalling at an AP site leaves a 3′-truncated nascent strand with a 5′-overhaning template. Consistent with this prediction we found that HMCES is just as efficient at binding and crosslinking to an AP site immediately adjacent to the 3′ ssDNA-dsDNA junction as to ssDNA (Figs. 4d,e, Supplementary Fig. 4). In contrast, binding and crosslinking is less efficient when the dsDNA is present on the 5′-side of the AP site, consistent with the effect of the wedge motif.
Discussion
The YedK-AP-DNA crosslink structure reveals how the unique DNA binding surface and N-terminal cysteine facilitates recognition and covalent crosslinking of HMCES and SRAP-containing proteins to AP sites in the context of ssDNA. Furthermore, the results explain the stability of this crosslink and the substrate preferences that correspond to DNA structures formed when polymerases stall at abasic sites.
The thiazolidine linkage acts as a sink for abasic sites and prevents strand breaks resulting from (1) non-enzymatic β-elimination at C2′, (2) lyase activity from enzyme-catalyzed β-elimination of the Schiff base, or (3) APE1 incision. This contrasts with unstable, transient protein-DNA Schiff base crosslinks that rapidly proceed to β-elimination as part of enzymatic strand cleavage reactions catalysed by bifunctional glycosylases and DNA polβ as part of the BER pathway13–15. Other proteins, including PARP-1, Histone H4, and Ribosomal protein uS3 can crosslink to AP sites, but in each case the DPC leads to strand scission18–21.
HMCES is named 5-Hydroxymethylcytosine (5hmC) Binding, ES Cell Specific because it was identified in a proteomics experiment using duplex DNA containing multiple 5hmC residues as a bait to purify proteins from embryonic cell lysates22. Furthermore, the HMCES SRAP domain was shown to autoproteolyze itself and incise duplex DNA containing 5hmC17. The DNA-bound SRAP structure suggests SRAP is unlikely to recognize 5hmC in the context of duplex DNA and we have not observed either the proteolysis or duplex DNA incision activity reported.
A single SRAP domain protein exists in organisms in all three domains of life, indicating a critical function even though knockouts in human, yeast, and bacterial cells are viable. The stability of the SRAP-AP-DNA crosslink and unique thiazolidine DPC linkage supports the conclusion that these proteins act to maintain genome stability during DNA replication and thereby improve organism fitness.
Methods
Protein purification.
Escherichia coli YedK was expressed in a modified pBG101 vector containing a Rhinovirus 3C (PreScission) protease cleavable hexahistidine tag. E. coli BL21 (DE3) cells were grown in Luria broth (LB) containing 15 ng/mL kanamycin at 37 °C to 0.8 OD600, and YedK overexpression was induced at 16 °C for 16 hr after addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were collected by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl pH 8.0 at 4 °C, 500 mM NaCl, 10% glycerol, 10 mM imidazole) with 1 mM each of leupeptin, pepstatin, and aprotinin. The lysate was homogenized using dounce and pressure homogenizers (Avestin Emulsiflex), centrifuged at 20,500 RPM for 30 min and passed through a 22-gauge needle prior to loading onto a 5 mL Ni-NTA column. The column was washed with 6-column volumes lysis buffer with 20 mM imidazole, and bound proteins were eluted with lysis buffer with 300 mM imidazole. The N-terminal His-tag was removed by overnight incubation with PreScission protease (1:30 w/w) at 4 °C during dialysis (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM TCEP). The solution was passed over 2 mL Ni-NTA resin, and the flow-through further purified using gel filtration on a 16/300 Superdex 200 column (GE Healthcare) in S200 buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% glycerol, 2 mM TCEP). YedK-containing fractions were concentrated to 4 mg/mL with Amicon MWCO 10 kDa centrifugal filters. Protein aliquots were flash-frozen in liquid nitrogen and stored at −80 °C.
YedK point mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent), in which forward and reverse PCR reactions were performed separately to improve mutagenic primer annealing, and the corresponding single stranded copies of the plasmid combined. Mutant plasmids were sequence verified. Mutant proteins were overexpressed and purified the same as wild type without the size exclusion step. Mutant YedK was buffer exchanged in S200 buffer, flash-frozen in liquid nitrogen, and stored at −80 °C.
Human HMCES SRAP domain (amino acids 1–270) was purified similar to YedK with the following modifications. After repass over the Ni-NTA column, HMCES SRAP was purified via anion exchange via a HiTrap Q column prior to S200 size exclusion chromatography in 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, and 10 mM DTT.
DNA binding.
Sequences of oligonucleotides used in the biochemical assays are listed in Supplementary Table 1. Relative binding affinity was measured by EMSA using 32P-labeled DNAs containing a deoxyuracil. 1 nM DNA was incubated with the indicated concentration of HMCES SRAP protein in reaction buffer (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 10 mM MgCl2, 5 mM DTT, 100 µg/ml BSA) at 37 °C for 1 hr. Ficoll was added to a final concentration of 1.25% and the samples were resolved on a 10% polyacrylamide gel in 1X TBE buffer (100 mM Tris-HCl pH 8, 90 mM boric acid, 2 mM EDTA) at 40 V for 180 min at 4 °C. Fluorescence anisotropy was used to measure binding of HMCES SRAP to ssDNA-dsDNA junctions containing a tetrahydrofuran (THF) abasic site analog. The THF strand contained 6-carboxfluorescein (FAM) at the 5′-end. Protein was titrated against 25 nM DNA in binding buffer (20 mM Tris-HCl pH 8.0, 100 nM NaCl, 10 mM MgCl2, 5 mM DTT) in a 384-well plate for 20 min at 4°C. Fluorescence was measured using a BioTek Synergy H1 Hybrid Reader with a filter cube containing 485/20 nm excitation and 528/20 nm emission filters.
DNA-protein crosslinking assays.
For the experiments shown in Figs. 1a–c, AP-DNA was prepared by incubating 50 µM uracil-containing oligonucleotides with 25 units of uracil DNA glycosylase23 (UDG, New England Biolabs) in Buffer X1 (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 10 mM MgCl2, 5 mM DTT) at 37 °C for 30 min. Human HMCES SRAP was incubated with AP-DNA in Buffer X1 at the following concentrations: 20.8 µM protein + 25 µM DNA (Fig. 1a) and 0.75 µM protein + 1.5 µM DNA (Fig. 1b,c). For the experiment shown Fig. 1c, DPCs were formed at 37 °C for 12 hr and treated with either no heat or 95 °C for 2 min prior to incubation at 25 °C. Free and DNA-crosslinked HMCES were separated on 10% polyacrylamide Tris-glycine gels.
For the experiments shown in Figures 1d–e, 3c-e, and 4d, reaction products were separated on 15% polyacrylamide urea gels in 1X TBE buffer. In Fig. 1d, AP-DNA was prepared by incubating 100 nM uracil-containing ssDNA with 1 unit of UDG in Buffer X1, crosslinks formed with 10 nM AP-DNA and 100 nM SRAP in 20 mM Tris-acetate pH 8.0, 50 mM potassium acetate, 10 mM magnesium acetate, and 5 mM DTT at 37 °C for 1 hr, followed by proteinase K (Sigma Aldrich) digestion for 5 min. In Fig. 1e, DPC was formed using 1 µM human HMCES SRAP and 10 nM 3′-Cy5-labeled oligonucleotide in 20 mM Tris-HCl pH 6.0, 50 mM NaCl, 10 mM MgCl2 and 5 mM DTT at 37 °C for 1 hr. DPC was then digested with proteinase K at 37 °C for 5 min. APE1 (NEB) was added where indicated and incubated at 37 °C for 120 min.
E. coli YedK DPCs (Fig. 3c,d) were formed from incubation of 1 µM protein and 10 nM 5′-FAM-labeled oligonucleotide in 20 mM Tris-HCl pH 6.0, 1 mM EDTA, and 5 mM DTT at 37 °C for 1 hr. Schiff base intermediates (Fig. 3e) were trapped by incubating 2 µM YedK with 6 µM 5′-FAM-labeled oligonucleotide in 20 mM HEPES-NaOH pH 7.0, 100 mM NaCl, 1 mM DTT at 25 °C for 5 min, after which NaCNBH3 was added to a final concentration of 50 mM and reactions incubated at 25 °C for 18 hr.
DNA binding reactions with ssDNA-dsDNA junctions (Fig. 4d) were carried out with 10 nM DNA and increasing concentrations of HMCES SRAP at 37 °C for 1 hr in Buffer X1. Crosslinking reactions with ssDNA-dsDNA junctions (Fig. 4e) were carried out with 1 nM AP-DNA and increasing concentration of HMCES SRAP at 37 °C for 1 hr in Buffer X1.
X-ray crystallography.
AP-DNA was prepared by incubating 50 µM 7-mer d(GTCUGG) ssDNA with 2.5 units of uracil DNA glycosylase (UDG, New England Biolabs) in Buffer X1 at 37 °C for 30 min. YedK DPC was generated by incubation of 20 µM YedK with 25 µM AP-DNA for 1 hr at 37 °C in MES pH 5.5, 50 mM NaCl, 10 mM MgCl2, and 5 mM DTT. YedK DPC was purified via cation exchange on a MonoS 5/50 GL column, concentrated, and buffer exchanged into 20 mM Tris pH 8.0, 80 mM NaCl, 2 mM TCEP, and 0.5 mM EDTA. YedK DPC was crystallized by hanging drop vapor diffusion at 21 ºC by mixing equal volumes of 3 mg/mL YedK DPC and reservoir solution containing 16% (w/v) PEG 3350 and 0.2 M KH2PO4. Diffraction quality crystals were grown from drops that were seeded with microcrystals produced in the same condition and that had been stabilized in 30% PEG 3350 and 0.2 M KH2PO4. Crystals were harvested 7 days after setting the drops and cryoprotected in 10% (v/v) glycerol, 30% PEG 3350, and 0.2 M KH2PO4 and flash-frozen in liquid nitrogen.
The non-covalent YedK-DNA complex was crystallized using the same 7-mer DNA sequence as in the DPC, but with a C3-spacer (Integrated DNA Technologies) in place of the AP site. The YedK-DNA complex was formed by incubating 80 μM YedK with 96 μM 7-mer C3-spacer ssDNA at 4 °C for 30 min. Crystals were grown by hanging drop vapor diffusion at 21 ºC from drops containing 2 µL protein-DNA solution, 2 µL reservoir containing 0.1 M Bis-Tris pH 5.4 and 23% (w/v) PEG 3350, and 0.5 µL DPC microcrystal seed stock stored in 30% PEG 3350 and 0.2 M KH2PO4. Crystals were harvested after 16 days into 0.1 M Bis-Tris pH 5.4, 30% PEG 3350, and 10% (v/v) glycerol, and flash-frozen in liquid nitrogen.
X-ray diffraction data were collected at the Advanced Photon Source beamlines 21-ID-D (DPC) and 21-ID-F (C3-spacer) at Argonne National Laboratory and processed with HKL2000 24. Data collection statistics are provided in Table 1. Phasing and refinement was carried out using the PHENIX suite of programs 25. Phasing of the DPC structure was carried out by molecular replacement of a previously determined structure of YedK alone (PDB accession 2ICU). The protein was subjected to simulated annealing, atomic coordinate, temperature factor, and TLS refinement prior to building the DNA model. The entirety of the 7-mer ssDNA and the Cys2-DNA crosslink was readily apparent in the density maps. All seven nucleotides and the Cys2-AP crosslink were manually built in Coot 26, guided by 2mFo-DFc and mFo-DFc electron density maps. Geometry restraints for the thiazolidine linkage were generated from idealized coordinates of (2R,4R)-1,3-thiazolidine-2,4-dicarboxylic acid (ligand 5XB) from the 1.47-Å structure of PDB ID 5FF2, and the stereochemistry of AP-site and Cys2 ring substituents verified my manual inspection of the electron density prior to model building. The protein-DNA model was iteratively refined by energy minimization and visual inspection of the electron density maps. The C3-spacer structure was phased by molecular replacement using the protein from the DPC structure, followed by simulated annealing to eliminate model bias prior to further refinement. The three nucleotides at the 5′-end of the DNA were readily apparent in the residual electron density. After several rounds of coordinate, B-factor, TLS refinement, the C3-spacer and the 3′-end of the DNA was visible, albeit with much weaker electron density. To minimize model bias in either structure, 2mFo-DFc composite omit and mFo-DFc annealed omit electron density maps with AP or C3-spacer and Cys2 removed from the structure factor calculation were used to guide placement and refinement of the crosslink or the C3-spacer. The final YedK-DNA models were validated using the wwPDB Validation Service and contained no residues in the disallowed regions of the Ramachandran plots. Structures were deposited in the Protein DataBank under accession codes 6NUA (DPC) and 6NUH (C3-spacer).
All structural biology software was curated by SBGrid 27. Structure images were created in PyMOL (https://pymol.org). Sequence conservation was mapped onto the structure using the Consurf Server 28. YedK DPC containing a ssDNA-dsDNA junction was modeled by superposition of ideal B-DNA with the sequence d(GGA/TCC) onto the three d(GGA) nucleotides at the 3′ end of the ssDNA in the YedK DPC crystal structure.
Statistics and Reproducibility
All experiments were completed at least three times unless otherwise indicated.
Data Availability statement
Structures were deposited in the Protein DataBank under accession codes 6NUA (DPC) and 6NUH (C3-spacer). Source data for Figs. 1b, 1c, 3d, 4d, 4e, and Supplementary Fig. 4 are available with the paper online as Source Data for Figures 1, 3, and 4. All other data is available upon request.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01ES030575 (D.C.), R01GM117299 (B.F.E.), and P01CA092584 (D.C. and B.F.E). P.S.T. was supported by F30CA228242, K.M.A. was supported by the Vanderbilt Molecular Biophysics Training Program (T32GM08320), and K.N.M was supported by T32CA009582. Core facilities were supported by the Vanderbilt-Ingram Cancer Center P30CA068485. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of LS-CAT Sector 21 beamline was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Footnotes
Competing Interests Statement
The authors declare no conflicts of interest.
Reporting Summary statement
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
References
- 1.Krokan HE & Bjoras M Base excision repair. Cold Spring Harb Perspect Biol 5, a012583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hitomi K, Iwai S & Tainer JA The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 6, 410–28 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Tsutakawa SE, Lafrance-Vanasse J & Tainer JA The cutting edges in DNA repair, licensing, and fidelity: DNA and RNA repair nucleases sculpt DNA to measure twice, cut once. DNA Repair (Amst) 19, 95–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kavli B, Slupphaug G, Mol CD, Arvai AS, Peterson SB, Tainer JA & Krokan HE Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J 15, 3442–7 (1996). [PMC free article] [PubMed] [Google Scholar]
- 5.Schaaper RM, Kunkel TA & Loeb LA Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc Natl Acad Sci U S A 80, 487–91 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM & Cortez D HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153 e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aravind L, Anand S & Iyer LM Novel autoproteolytic and DNA-damage sensing components in the bacterial SOS response and oxidized methylcytosine-induced eukaryotic DNA demethylation systems. Biol Direct 8, 20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kallen RG Mechanism of reactions involving Schiff base intermediates. Thiazolidine formation from L-cysteine and formaldehyde. Journal of the American Chemical Society 93, 6236–6248 (1971). [DOI] [PubMed] [Google Scholar]
- 9.Canle M, Lawley A, McManus EC & O’Ferrall RAM Rate and equilibrium constants for oxazolidine and thiazolidine ring-opening reactions. in Pure and Applied Chemistry Vol. 68 813 (1996). [Google Scholar]
- 10.Just G, Chung BY, Kim S, Rosebery G & Rossy P Reactions of oxygen and sulphur anions with oxazolidine and thiazolidine derivatives of 2-mesyloxymethylglyceraldehyde acetonide. Canadian Journal of Chemistry 54, 2089–2093 (1976). [Google Scholar]
- 11.Ratner S & Clarke HT The Action of Formaldehyde upon Cysteine. Journal of the American Chemical Society 59, 200–206 (1937). [Google Scholar]
- 12.Fife TH, Natarajan R, Shen CC & Bembi R Mechanism of thiazolidine hydrolysis. Ring opening and hydrolysis of 1,3-thiazolidine derivatives of p-(dimethylamino)cinnamaldehyde. Journal of the American Chemical Society 113, 3071–3079 (1991). [Google Scholar]
- 13.Brooks SC, Adhikary S, Rubinson EH & Eichman BF Recent advances in the structural mechanisms of DNA glycosylases. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1834, 247–271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fromme JC & Verdine GL Base Excision Repair. in Advances in Protein Chemistry, Vol. 69 1–41 (Academic Press, 2004). [DOI] [PubMed] [Google Scholar]
- 15.Beard WA & Wilson SH Structure and Mechanism of DNA Polymerase β. Chemical Reviews 106, 361–382 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Billman JH & Diesing AC Reduction of Schiff Bases with Sodium Borohydride. The Journal of Organic Chemistry 22, 1068–1070 (1957). [Google Scholar]
- 17.Kweon SM, Zhu B, Chen Y, Aravind L, Xu SY & Feldman DE Erasure of Tet-Oxidized 5-Methylcytosine by a SRAP Nuclease. Cell Rep 21, 482–494 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Prasad R, Horton JK, Dai DP & Wilson SH Repair pathway for PARP-1 DNA-protein crosslinks. DNA Repair (Amst) 73, 71–77 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prasad R, Horton JK, Chastain PD 2nd, Gassman NR, Freudenthal BD, Hou EW & Wilson SH Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Res 42, 6337–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sczepanski JT, Wong RS, McKnight JN, Bowman GD & Greenberg MM Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc Natl Acad Sci U S A 107, 22475–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grosheva AS, Zharkov DO, Stahl J, Gopanenko AV, Tupikin AE, Kabilov MR, Graifer DM & Karpova GG Recognition but no repair of abasic site in single-stranded DNA by human ribosomal uS3 protein residing within intact 40S subunit. Nucleic Acids Res 45, 3833–3843 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, Munzel M, Wagner M, Muller M, Khan F, Eberl HC, Mensinga A, Brinkman AB, Lephikov K, Muller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T & Vermeulen M Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152, 1146–59 (2013). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 23.Krokan HE, Saetrom P, Aas PA, Pettersen HS, Kavli B & Slupphaug G Error-free versus mutagenic processing of genomic uracil--relevance to cancer. DNA Repair (Amst) 19, 38–47 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ & Grosse-Kunstleve RW PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M & Sliz P Collaboration gets the most out of software. Elife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T & Ben-Tal N ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structures were deposited in the Protein DataBank under accession codes 6NUA (DPC) and 6NUH (C3-spacer). Source data for Figs. 1b, 1c, 3d, 4d, 4e, and Supplementary Fig. 4 are available with the paper online as Source Data for Figures 1, 3, and 4. All other data is available upon request.