

Abstract
Silk fibroin-derived polypeptides (FDPs) are polypeptides resulting from the enzymatic separation of the hydrophobic crystalline (Cp) and hydrophilic electronegative amorphous (Cs) components of silk fibroin (SF). The role of these polypeptides in promoting the nucleation of hydroxyapatite (HA) has been previously investigated, yet is still not fully understood. Here we study the potential of HA mineralization via FDPs incorporated at 1:10, 1:2 and 1:1 in a plastically compressed (PC) and dense collagen (DC) scaffold. Scaffolds were immersed in simulated body fluid (SBF) at physiological conditions (pH = 7.4, 37°C) to promote biomineralization. The effect of Cs and Cp to promote HA nucleation was investigated at different time points, and compared to pure DC scaffolds. Characterization of Cs and Cp fragments using Liquid Chromatography–Mass Spectrometry (LCMS) showed little difference in the amino acid composition of the FDPs. Results obtained in vitro using Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR), Scanning Electron Microscopy (SEM) X-Ray Diffraction (XRD) and mass analysis showed little difference between scaffolds that incorporated Cs, Cp, and DC hydrogels. These results demonstrated that silk FDPs incorporation are not yet suitable to promote HA nucleation in vivo without further refining the collagen-FDP system.
Introduction
Native bone tissue is composed of organic collagen fibrils and inorganic hydroxyapatite (HA) crystals arranged in a strict hierarchical structure. The collagen fibrils provide a template for HA, with crystals nucleating and growing within the gaps of collagen [1,2]. While the nucleation of HA crystals occurs within collagen fibrils, collagen itself does not take an active role in HA nucleation, and only a small amount of apatite will form after a long period of time [3,4]. Instead, it is the non-collagenous proteins (NCPs) that are responsible for HA formation [5]. In the literature, it is demonstrated that NCPs having calcium binding and HA nucleating properties contain glutamic acid [6–8]. Glutamic acid has the ability to bind Ca2+ ions to negatively charged carboxylate groups, which in turn attract PO43- ions and increase the local concentration past the point of supersaturation [9], with a small amount (4–8 wt% [10]) of carbonate ions remaining within the HA crystal. This supersaturation reaches a critical level, and leads to nucleation sites for HA crystals [8,11,12]. However, without the NCPs, the mineralization of collagen generally does not proceed [1]. As a consequence, collagen scaffolds used in bone implants without mineralization require more than 2–3 weeks for osteointegration [13]. Currently, an increasing number of bioactive materials are being developed to replace NCPs and promote the nucleation of HA and mineralization of collagen for bone defects repair [2,13–15].
Silk is a natural protein fiber that is used by certain insects such as spiders and silkworms (Bombyx mori), with the former using it to make webs and the latter to form cocoons. Though spider silk is known for its outstanding mechanical properties [16], Bombyx mori silk from domesticated silkworms has the advantage of being available in higher quantities [17]. The latter is composed of two main proteins, sericin and fibroin. The silk fibroin (SF) is the core structural component of silk, which exhibits high tensile strength [18], while the sericin surrounds the fibroin and acts as a glue, or “gum” [19]. Due to its ease of processing, biodegradability and high tensile strength, silk fibroin is especially attractive for biomedical applications [20–22]. In particular, due to the high concentration of glutamic acid in SF [23], it has been proposed that nucleating HA on SF is possible in vitro, and is considered as a promising material for repairing bone defects [14,24,25].
As SF is composed of peptides that are hydrophilic (Cs) and hydrophobic (Cp), it is fairly straightforward to separate SF into its constituents by initially degumming (removing the sericin/silk gum by cleaving its peptide bonds) the Bombyx mori silk and then hydrolyzing the SF [26]. Then, by dissolving the SF with α-chymotrypsin (a digestive enzyme that breaks down proteins into polypeptides), and centrifuging the resultant, the supernatant, Cs, and precipitate, Cp can be separated [27]. Once separated, it has been shown that Cs and Cp have different compositions, with Cs having more anionic amino acids than Cp [23]. Furthermore, the Cs component of SF has excellent biomineralization properties in vitro. DC hydrogels that incorporate Cs have been shown to form HA under physiological temperature and pH, when immersed in simulated body fluid (SBF), unlike Cp-containing DC gels, while Cp showed almost no mineralization [27]. It is thought that the high concentration of glutamic acid (glu) in Cs [23] allows it to play a role similar to calcium-binding non-collagenous proteins (NCPs), which act as modulators for the nucleation and growth of HA nanocrystals [28–30] by attracting more Ca2+ ions, as well as providing more nucleation sites for HA crystals [31].
Recent research has focused on using three dimensional (3D) dense collagen scaffolds that can incorporate silk fibroin-derived polypeptides (FDPs) [27]. Previous work has shown that the compression of collagen gels [32–34] and the incorporation of FDPs have more desirable properties, such as the mineralization of hydroxyapatite (HA) [27] (uniformly, in both intra- and extrafibrillar locations [35]), increased cell proliferation and adhesion [32,33], biocompatibility with the surrounding tissue [36], reduced inflammatory response, and increased bone regeneration [37–39], and similar diffusion coefficients for oxygen and glucose to that of native collagen tissue [40,41].
While the nucleation of HA crystals occur within collagen fibrils, it is the non-collagenous proteins (NCPs) which are responsible for HA formation, and account for less than 10% of proteins found in bone. Herein, we aim at promoting the increased biomineralization of HA in DC scaffolds by incorporating FDPs. By investigating both the FDPs and the nucleation of HA in SBF [42], the potential of FDPs as biomaterials for bone grafts was also determined in vitro. The amino acid composition of FDPs was also assessed and compared to previous work [23]. It was hypothesized that FDP can serve as an analogue to NCPs that serves to accumulate Ca2+ and PO43- ions that form HA. While NCPs account for less than 10% of the proteins found in bone, they play an important role in bone mineralization by acting as HA nucleation centers [43]. Additionally, plastic compression (PC) removes excess water and forms a dense collagen (DC) hydrogel, in which the collagen fibril density (CFD) increases from <0.5 wt.% to approximately 8 wt.% [44]. This results in a scaffold that closely mimics collagen in native bone tissue [32,33,36,44] with a greater potential to nucleate HA. The objective was to obtain a hydrogel that has the composite structure of flexible collagen and tough HA, which gives bone its requisite strength and high fracture toughness [45–47]. Thus, in this paper we study the potential for including FDPs in DC scaffolds for bone tissue engineering (BTE).
Methodology
Preparation of hydrogels and bioactive additives
Silk fibroin polypeptides
Soluble silk fibroin-derived peptides were produced from raw Bombyx Mori (silkworm) silk provided by Stazione Sperimentale per la Seta (Milan, Italy). The silk sericin was degummed and the fibroins were cleaved and separated into their soluble (Cs) and non-soluble phases (Cp), using a method reported in the literature [27,48,49].
Silk fibroin fibers were produced through a heating cycle that removed the silk sericin. The resulting fibroin was dissolved in a concentrated aqueous solution of LiBr (Sigma) [50,51], due to LiBr being a chaotropic salt, which disrupts the bonds in protein molecules [52,53]. The dissolved fibroin results in both α-helical and β-folded structures [51], heated, then separated using dialysis to produce a 2% silk fibroin solution [48].
The resulting silk fibroin was then diluted with a 0.1 M ammonium bicarbonate solution (NH4CO3, Fisher Scientific), then enzymatically digested with α-chymotrypsin (Sigma) at an enzyme-to-substrate ratio of 1:100 to generate Cp and Cs in solution, which was incubated at 37°C for 24 hours then centrifuged to separate the supernatant (containing Cs) from the pellet (containing Cp). Both the supernatant and the pellet were freeze-dried to produce Cs and Cp [27].
Preparation of simulated body fluid
Kokubo’s SBF was created based on a protocol developed for analysing apatite formation [42]. Reagents were mixed in in 3 L of distilled water in the following order: NaCl (Fisher Scientific) (24.105 g), NaHCO3 (Fisher Scientific) (1.065 g), KCl (Sigma) (0.675 g), K2HPO4·3H2O (0.693 g), MgCl2·6H2O (Fisher Scientific) (0.933 g) 1.0M HCl (117 mL), CaCl2 (Sigma) (0.876 g), Na2SO4 (Sigma) (0.216 g), Tris(hydroxymethyl)aminomethane (Tris) (Fisher Scientific) (18.354 g), 1.0M HCl (Fisher Scientific) (0–15 mL). Tris and HCl were added concurrently to maintain a pH of ~7.4.
Plastically compressed dense collagen hydrogels
A solution of 13:1 10X Dulbeccos’s Modified Eagle Medium (DMEM) (Gibco) to 5N NaOH (Fisher Scientific, Sodium Hydroxide Solution 5N) was created. A 5.83 mg/mL bovine dermis (BD) collagen solution collagen solution (Devro Medical, Purified Soluble Collagen) was added at 4:1 by volume to the DMEM. The pH was adjusted to 7.4 by adding NaOH while kept at ~4 ºC using an icepack or a box filled with ice. [27,36].
To produce Dense Collagen (DC) gels, a 48-well plate (10.5 mm diameter per well) was filled with 1 mL of the above solution (DMEM, NaOH and collagen) and, similar to a previously established protocol [27], left in an incubator (Thermo Scientific, Forma Series II) at 37°C for 30 minutes. Collagen gels were then subjected to a plastic compression method [27,33,34,36] by applying 1 kPa (40 g per 350 mm2; 160 g for four gels) for five minutes to collagen gels placed between two nylon meshes and on top a steel mesh and blot paper (the latter to collect expelled water). The load was applied to expel water and retain collagen, and produce DC gels.
Hydrogels containing a FDP additive (Cs or Cp) were created following the previously outlined protocol [27]. The process for creating DC hydrogels containing Cs (DC-Cs), and Cp (DC-Cp) was identical to that outlined above, with an interim step of mixing the additive in the DMEM then ultrasonicating the solution prior to adding NaOH and collagen. Collagen was added to the DMEM at 4:1, and FDPs to polymer ratios of 1:10 1:2 and 1:1 were used to determine effect of FDPs in the biomineralization of collagen gels, with pure collagen gels used as control (see Table 1). After adding collagen, the solution was magnetically stirred to ensure a homogenous solution.
Table 1. Solution of 10 mL collagen with additives (added prior to plastic compression).
| Additive: polymer | Mass of collagen (mg) | Mass of Cs/Cp (mg) | Mass of polymer and additive in solution (mg) |
|---|---|---|---|
| Collagen (ctrl) | 46.8 | 0.0 | 46.8 |
| 1:10 | 46.8 | 4.7 | 51.5 |
| 1:2 | 46.8 | 23.4 | 70.2 |
| 1:1 | 46.8 | 46.8 | 93.6 |
Mineralization of hydroxyapatite within additive-incorporated hydrogels
Both Cs and Cp were immersed in SBF at a 1:3 ratio (3 mg/mL) and placed in an incubator for 24 hours before being removed and analysed. The resulting gels were immersed in SBF (pH 7.4, 37°C) for up to two weeks, using a hydrogel to SBF ratio of 1:3 (mg/mL). The solution was replaced at two or three-day intervals by fresh SBF. Samples were taken at days 0, 3, 7, 10 and 14. The results were compared to gels that had no additives incorporated. HA nucleation in bone occurs within the gaps of collagen fibrils [48], though for scaffolds constructed in vitro, a high concentration of carboxyl groups was shown to lead to HA nucleation. Charged amino acids act as nucleation sites, where calcium ions are gathered through electrostatic attraction, which then attract phosphate ions until a critical concentration is reached, leading to HA formation [49–52]. The nucleation of HA is thus expected to occur around the charged Cs particles within the collagen scaffold.
Liquid chromatography–mass spectrometry
Liquid Chromatography–Mass Spectrometry (LCMS) was carried to determine the amino acid composition of the silk fibroin derived polypeptides on a QTOF (Agilent). LCMS was carried out at IRIC-Université de Montréal Proteomics facilities. The results were analysed in Microsoft Excel.
Particle size analysis
Particle Size Analysis (PSA) (Horiba LA-920) was performed on Cs and Cp to determine the size distribution. Isopropanol was used to create a solution that was then ultrasonicated before analysis. A refractive index of nD22 1.55 (nD22 1.13 in isopropanol) was used to calculate the size distribution based on values obtained from the literature [54,55].
Fourier transform infrared spectroscopy
Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR) (Perkin Elmer, Spectrum 400) was used to characterize the mineralization that occurred in the sample, as well as any changes in the chemical composition of the sample itself. The bands associated with PO43-, indicative of the presence of HA, were compared between timepoints and samples to monitor HA mineralization [27,56]. Additionally, IR is sensitive to the substitution of PO43- ions by CO32- ions, and can detect the presence of small amounts of carbonate, indicating the formation of carbonated hydroxyapatite [57].
Samples were prepared for ATR-FTIR analysis by first washing in dH2O to remove any excess SBF, then freezing in liquid nitrogen, then freeze-drying (lyophilizing) the sample until all the excess moisture had been removed. The sample was then analysed at a resolution of 2 cm-1 in the IR range of 4000 to 650 cm-1 at 32 scans per sample. The resulting spectra were normalized against the Amide I band found between 1800 to 1650 cm-1 for comparison (Spectrumsoftware, Perkin-Elmer) [36,58].
X-Ray diffraction
X-Ray Diffraction (XRD) (Bruker D8, Bruker-AXS Corp.) was performed on Cp and Cs fragments, as well as freeze-dried hydrogels that had been immersed in SBF. Samples were compressed and taped onto glass slides to produce a flat, fixed surface, and XRD analysis was performed using a Cu-Kα source. XRD patterns were recorded from 3 to 104° 2 theta at 40 kV and 40 mA. Four frames of 25° were recorded for 150 seconds and then merged during data post processing. The resulting patterns were analysed (EVA 14.0.0.0, Bruker) and compared with spectra with peaks identified in the International Centre for Diffraction Data (ICDD) database.
Scanning electron microscopy
Samples for Scanning Electron Microscopy (SEM) imaging were prepared by immersing the hydrogels in 30, 50, 70, 80, 90, 95 and 100% ethanol solutions to remove all the water [27,36]. The gels were then immersed in 1,1,1,3,3,3-hexamethyldisilazane (HMDS) and left overnight until all the HMDS had evaporated. All samples were sputter-coated with a layer of Au/Pd then analysed by SEM (FEI Inspect F-50 FE-SEM, FEI).
Statistical analysis
The data was analyzed for statistical significance using a one-way ANOVA with a statistical significance level of 0.05. Tukey and Holm-Bonferroni methods were used for comparison (Origin Pro v9.0 software, OriginLab).
Results
Materials characterization
Characterization of the starting materials (collagen, Cs and Cp) was carried out using ATR-FTIR to determine their chemical composition, which can serve as a reference (control) for future tests. While the ATR-FTIR spectra of Cp and Cs are quite similar, still there are slight differences, such as the shoulder at 1695 cm-1 in Cp and the amide I band at 1650 cm-1 for Cs. This can be attributed to Cp polypeptides being composed of the crystalline regions of silk fibroin and having a β-sheet type structure, while Cs has an α-helix type structure [16,23]. The spectra for Cp (see Fig 1A) exhibit a shoulder on the amide I band at 1695 cm-1 that is typically associated with a β-sheet structure of SF, as well as Amide I, II and III absorptions at 1622, 1515 and 1230 cm-1 that are characteristic of SF [27,59]. The spectra for Cs (see Fig 1B) differed slightly in the amide I band at 1650 cm-1 that is typical of an α-helix type structure, though it still have the Amide II and III bands at 1530–1515 cm-1 and 1239 cm-1, respectively [27].
Fig 1.

A) ATR-FTIR spectra showing secondary structures and B) XRD patterns showing amorphous/crystalline structure for as-made i) Cs and ii) Cp. arrows in the XRD patterns indicate crystalline regions and amorphous regions.
XRD patterns for solubilized Cs and Cp (see Fig 1B) fragments revealed a crystalline structure in Cp that is similar to XRD patterns of SF reported elsewhere [60], while Cs fragments resulted in spectra confirming the amorphous nature. SEM images of Cs and Cp also indicate that Cp has a crystalline structure, while Cs has an amorphous structure (see Fig 2). These results confirmed that the structure of SF is composed of both the crystalline phase of Cp and the amorphous phase of Cs.
Fig 2.

SEM images of A) Cs and B) Cp particles.
The size distribution of Cp and Cs particles indicates that Cp is significantly larger than Cs (see Fig 3). The mean particle size of Cp is 91.02 μm, while Cs is only 22.46 μm. The difference in size can be attributed to Cp having a molecular weight 4–20 times higher than Cs [27], but is more likely due to the structure of Cp being semi-crystalline, whereas Cs is amorphous [61–64]. The use of α-chymotrypsin to separate the two phases attacks the amorphous Cs phase, while not affecting the crystalline Cp, resulting in larger crystalline Cp and smaller chunks of Cs [22,64,65]
Fig 3. Size distribution of i) Cp and ii) Cs particles.

LCMS results contrast with the previously assumed composition of the silk fibroin-derived polypeptides (see Table 2). LCMS showed that Cs and Cp have a similar composition to silk fibroin. The composition of Cp is similar to values found previously [23]. In contrast, Cs has a slightly more varied composition, though it has a lower concentration of sequences rich in aspartic acid and glutamic acid residues compared to similar results reported in the literature [23,55]. The values obtained from LCMS match the structure seen previously, with the Glycine amino acids alternating with other amino acids (except for one Ala-Ala link) [66]. The NCBI database for the amino acid composition of silk fibroin polypeptides that are composed of a mixture of Heavy Chains (HC) (NCBI Reference Sequence: NP_001106733.1) and Light Chains (LC) (NCBI Reference Sequence: NP_001037488.1) shows that Cs and Cp have a similar composition to silk fibroin as well, though with a lower concentration of glutamic acid (HC– 0.57%, LC– 1.91%) and aspartic acid (HC– 0.48%, LC– 6.49%).
Table 2. Comparison of amino acid composition for fibroin-derived polypeptides, Cp and Cs, obtained from LCMS (Right–from literature, left–as-made).
| Amino Acid | Cp (%) [55] | Cs (%) [55] | Cp (%) | Cs (%) |
|---|---|---|---|---|
| Alanine | 32.03 | 24.12 | 29.5±1.7 | 27.6±2.4 |
| Arginine | 0.10 | 1.31 | 0.5±0.1 | 0.4±0.4 |
| Asparagine | 0.00 | 0.00 | 1.3±0.7 | 1.8±1.2 |
| Aspartic acid | 0.42 | 4.53 | 0.9±0.4 | 1.3±0.8 |
| Cysteine | - | - | 0.0±0.1 | 0.0±0.0 |
| Glutamic acid | 0.34 | 3.12 | 0.4±0.3 | 0.4±0.1 |
| Glutamine | 0 | 0 | 1.1±0.6 | 2.2±1.1 |
| Glycine | 49.13 | 36.50 | 43.7±2.5 | 40.7±3.8 |
| Histidine | 0.04 | 0.54 | 0.4±0.3 | 0.7±0.2 |
| Isoleucine | 0.19 | 1.61 | 1.4±0.6 | 1.9±0.9 |
| Leucine | 0.10 | 1.71 | 1.3±1.0 | 2.5±1.4 |
| Lysine | 0.12 | 0.95 | 0.2±0.1 | 0.4±0.2 |
| Methionine | 0.03 | 0.31 | 0.1±0.1 | 0.4±0.2 |
| Phenylalanine | 0.34 | 1.47 | 0.5±0.0 | 0.6±0.1 |
| Proline | 0.13 | 0.96 | 0.1±0.0 | 0.2±0.1 |
| Serine | 11.79 | 9.20 | 10.8±1.6 | 10.2±0.4 |
| Threonine | 0.47 | 1.91 | 1.6±0.5 | 1.7±0.4 |
| Tryptophan | 0.00 | 0.00 | 0.0±0.1 | 0.0±0.1 |
| Tyrosine | 3.66 | 7.55 | 4.5±0.8 | 4.7±0.7 |
| Valine | 1.11 | 4.21 | 1.6±0.5 | 2.4±0.5 |
Regarding the charge of the FDPs (see Table 3), tabulating the LCMS results above (see Table 3) shows that overall the FDP is composed of neutral amino acids (97.6% and 96.8% for Cp and Cs, respectively). Compared to the charge calculated from the composition of the FDPs obtained from the literature [55], where 7.6% of the amino acids are negatively charged, there is a significant difference in the charge of Cs, as only 1.5% of the amino acids within the samples examined are negatively charged. It is unlikely that the Cs fragments incorporated into the hydrogel are attracted to the collagen, which is cationic in nature [27], as was previously assumed.
Table 3. Comparison of fibroin-derived polypeptides charge (right–from literature, left–as-made).
The SBF that was used to imitate a physiological environment for in vitro testing was analysed using Ion Chromatography. The results were compared to the theoretical composition of the ionic species present in solution as well as a commercial SBF solution (Hank’s Balanced Salt Solution, HBSS [67]) and Extracellular Fluid (ECF), specifically plasma [68]. The carbonate present in the SBF could not be measured due to on-column neutralisation of CO32- to HCO3- by dissolved CO2.
Polypeptides (Cs and Cp) immersed in SBF at 37 ºC showed aggregation within a day. The HA aggregate was confirmed from XRD analysis (see Fig 4). While HA can exist in different forms, depending on the condition of its formation [69], the method of nucleation remains the same. It is known that HA forms an intermediary amorphous calcium phosphate, that HA crystals exhibit “polycrystalline character of the elemental particles,” and nucleation is favoured [70], which would indicate that HA forms are determined by nucleation conditions.
Fig 4. XRD patterns of i) HA (ICDD file 00-009-0432) and polypeptides ii) Cs and iii) Cp immersed in SBF.

The resulting XRD pattern showed peaks that matched the corresponding pattern for HA (ICDD file 00-009-0432), indicating that both polypeptides lead to the nucleation of HA at physiological conditions. Given the similar composition of Cs and Cp (see Table 2), it is to be expected that both would nucleate HA. Previous work has shown that the presence of acid-rich proteins (specifically aspartic and glutamic acid) led to biomineralization in vitro [71,72] via epitactic nucleation, whereas heterogeneous nucleation occurs via the formation of critical nuclei on a surface [73]. The XRD measurements of Cs and Cp immersed in SBF results in a diffraction pattern identical to that arising from HA.
Hydroxyapatite nucleation investigation
Hydrogels incorporating Cs and Cp at a 1:10 additive to collagen ratio (by mass) were created as stated. SEM imaging was performed as described above. The results for DC, 1:10 DC-Cp and 1:10 DC-Cs gels, after seven days immersion in Kokubo’s SBF were analysed to visually determine whether HA nucleation occurred.
SEM reveals no major difference in the morphology of the as-made hydrogels, either DC, 1:10 DC-Cs or 1:10 DC-Cp (see Fig 5.). Particles that differed were seen in the DC and 1:10 DC-Cp gels at day 7, though there were no signs of particle nucleation in the 1:10 DC-Cp gels. In the DC/1:10 DC-Cs gels, the particles were located in small clumps of 1–20 particles randomly spaced across the surface of the hydrogel. Most of the hydrogel surface remained bare. No evidence of Cs is observed in the hydrogels via SEM at day 7, indicating that electrostatic interactions between collagen and the hydrophilic FDP [27] do not last.
Fig 5.

SEM of Plastically compressed A) DC, B) 1:10 DC-Cs and C) 1:10 DC-Cp hydrogels immersed in SBF for 7 days (scale bar is 10 μm, arrows indicate particles).
Spectroscopic and XRD analysis was conducted on DC hydrogels, both with and without additives incorporated, as described above. The resulting ATR-FTIR spectra were plotted and showed that bands indicative of the presence of HA; υ3 PO43- (1030 and 1080 cm-1) [58,74,75], as well as the bands for υ3 and υ2 CO32- (1450 and 1400 cm-1 and 850 cm-1 respectively) [58,74] were present in all samples, post-immersion in SBF. The presence of type I collagen is confirmed from the bands at 1630, 1550 and 1240 cm-1, corresponding to the amide I, II and II groups respectively [58,76] (see Fig 6) present in all samples.
Fig 6.

ATR-FTIR Spectra for A) DC, B) 1:10 DC-Cs and C) 1:10 DC-Cp, gels immersed in SBF at day i) 0, ii) 3, c) 7, iii) 10 and iv) 14.
The spectra obtained show that there was an increase in the υ3 PO43- over the first 10 days compared to the initial band, indicating the occurrence of HA nucleation/growth. This was particularly apparent in DC and 1:10 DC-Cs gels (see Fig 6A and 6B), as well as in the 1:10 DC-Cp gels (see Fig 6C).
The spectra indicated that all hydrogels showed a presence of phosphate and carbonate groups, suggesting some HA nucleation occurs within the hydrogel. Comparing the value of the peaks associated with phosphate and carbonate showed the bands associated with phosphate increased with time (see Fig 7A), those associated with carbonate did not (see Fig 7B), indicating that phosphate absorbance within the collagen increases over time. This change can be attributed to the mineralization of non-carbonated HA, and is consistent with previous findings [58]. The hydrogels that had Cs incorporated did not show significantly higher values associated with the phosphate peak, contrary to earlier work [27], indicating that it does not serve to nucleate HA at a higher rate. The ratio of phosphate and carbonate peaks to the Amide I band remained nearly constant over time, indicating that little change takes place within the 1:10 DC-Cp or Cs gels. It is likely that the FDPs distributed within the hydrogel lack the acidic amino acids, particularly glutamic acid, necessary for the nucleation of HA [71,72,77]. Additionally, the lack of electrostatic interaction between collagen and the mainly neutral FDP is also a barrier to HA formation, as it was shown to be necessary for mineralization to occur [73,78].
Fig 7.
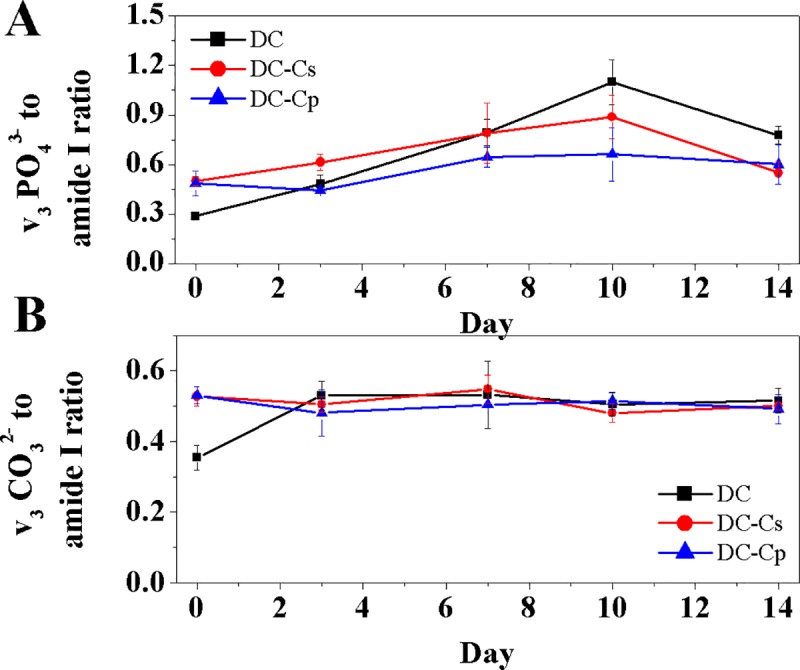
Ratio of A) ν3 PO43- and B) ν3 CO32- bands to amide I bands from ATR-FTIR spectra of DC gels containing no additives, Cs and Cp (at a 1:10 ratio to collagen) immersed in SBF (SD, n = 4, p<0.05).
The XRD patterns for DC hydrogels immersed in SBF showed that the formation of HA occurs in all samples and increases over time, though the extent of mineralization differs based on whether it was a DC, DC-Cp or DC-Cs hydrogel (see Fig 8). XRD patterns for pure collagen hydrogels showed peaks at 2θ = 33° and 45° in all samples, which are associated with HA [79]. The latter also exhibit a peak at 2θ = 23° in an otherwise amorphous section of the pattern. As collagen is an amorphous material with no defined peaks, and the 2θ = 23° peak does not match any peaks associated with HA, the presence of this peak has been attributed to the polyethylene (PE) tape used to hold the samples in place. In addition, the peak corresponds to ICDD file 00-060-1505 for PE.
Fig 8.

XRD pattern for A) DC, B) 1:10 DC-Cs and C) 1:10 DC-Cp hydrogels immersed in SBF at days i) 3, ii) 7, iii) 14 (●–HA corresponding to ICDD file 00-009-0432, ■–PE corresponding to ICDD file 00-060-1505).
XRD patterns confirmed the nucleation of HA in all samples. The 1:10 DC-Cs hydrogels (see Fig 8B) initially showed a similar extent of mineralization as DC and 1:10 DC-Cp gels on days 3 and 7. However, the XRD patterns for DC and DC-Cs hydrogels (see Fig 8A and 8B) showed much higher HA peaks at day 14, indicating a greater extent of mineralization than DC-Cp (see Fig 8C) hydrogels in the long-term. In particular, the peak at 2θ = 45° was not as apparent as in other DC and DC-Cs hydrogels. In general, however, both 1:10 DC-Cs and DC-Cp hydrogels showed that their incorporation led to HA nucleation, which can be explained by the fact that both Cs and Cp have similar amino acid compositions, as seen by the LCMS results, and the fact that both Cs and Cp lead to the nucleation of HA, as confirmed by the XRD pattern from the particles were immersed in SBF (see Fig 4). The major difference between Cs and Cp is related to the high, narrow peak at 2θ = 23°. The 2θ = 23° peak is present in all samples, but the intensity is much greater in the DC and DC-Cs samples. The broad peaks in the DC-Cp samples are attributed to a relatively small crystal size [80–82] or to the poorly crystalline nature [83,84] of HA within the sample. The XRD results match ATR-FTIR results, which shows that FDPs do not have a significant impact on HA nucleation due to the lack of the necessary acidic amino acids [71,72,77] or of the electrostatic interaction between collagen and FDP [73,78].
ATR-FTIR was also conducted on hydrogels with a higher amount of FDPs additive added (1:2 and 1:1) that were immersed in SBF for 3 and 7 days (see Fig 9). The resulting spectra for the different types of hydrogels, only DC-Cp showed that bands attributed to υ3 PO43- (1030 and 1080 cm-1) [58,74,75], increased with time, while the bands for υ3 and υ2 CO32- (1450 and 1400 cm-1 and 850 cm-1 respectively) [58,74] remained relatively constant, similar to the results obtained for additives added at a 1:10 ratio. The results obtained are contrary to the expectation that the inclusion of a greater amount of FPDs would lead to previously reported increased HA nucleation [27].
Fig 9.

ATR-FTIR for A) 1:1 DC-Cs, B) 1:1 DC-Cp, C) 1:2 DC-Cs and D) 1:2 DC-Cp, i) as-made and immersed in SBF for ii) 3 and iii) 7 days.
The mass of freeze-dried DC hydrogels with different ratios of Cs and Cp (1:2 and 1:1 relative to collagen) before and after immersion in SBF was measured to determine if the addition of FDP led to a change in mass due to HA nucleation over time.
Previously [27], it was reported that DC and DC-Cs hydrogels with 1:10 Cs to collagen should result in a hydrogel that is 5 wt.% and ~60 wt.% HA, respectively, by day 7 after immersion in SBF. However, the present results obtained show that there is no significant difference between plastically compressed DC gels and DC gels with Cs incorporated (see Fig 10). Comparison of the difference in the masses of the DC, DC-Cs and DC-Cp hydrogels to the theoretical value of an as-made (day 0) gel with the same amount of additive incorporated is reported in Table 4. The mass analysis of the DC gels after PC matched up closely (-3.6%) with the theoretical mass of a gel of the same volume. Results demonstrated that the DC-Cs hydrogels have a significantly lower mass than their theoretical value, while the mass of the DC-Cp gels is close to its theoretical value. Furthermore, the difference of mass in the DC-Cs was similar to the amount of Cs added to the solution, and statistically, the difference in mass between the DC and DC-Cs gels is not significant. This indicates that Cs is expelled with the water inside the gel during the plastic compression of the highly hydrated hydrogel, and that the remaining scaffold is only collagen.
Fig 10.

Mass of DC, DC-Cs and DC-Cp hydrogels immersed in SBF for A) 0 (as-made), B) 3 and C) 7 days (*—statistically significant, one-way ANOVA, SD, n = 4, p < 0.05).
Table 4. Mass of hydrogels at day 0 compared to the theoretical values.
| Sample | Theoretical mass (mg) |
Initial mass (mg) | Difference in mass (%) | Difference in mass from control (mg) |
|---|---|---|---|---|
| DC (ctrl) | 4.7 | 4.9±0.5 | 3.6 | 0.0 |
| 1:2 DC-Cs | 7.0 | 5.1±0.5 | -27.4 | 0.2 |
| 1:1 DC-Cs | 9.4 | 4.2±0.5 | -55.7 | -0.7 |
| 1:2 DC-Cp | 7.0 | 7.2±0.4 | 3.0 | 2.3 |
| 1:1 DC-Cp | 9.4 | 10.3±0.7 | 9.7 | 5.4 |
Conclusions and perspectives
We examined the interaction in vitro between silk fibroin-derived polypeptides (FDPs) and plastically compressed collagen hydrogels developed as scaffolds for bone tissue engineering. Results in vitro show that immersing FDPs (both Cs and Cp) in simulated body fluids lead to the nucleation of hydroxyapatite (HA), yet incorporating these same FDPs within a dense collagen (DC) hydrogel does not contribute to mineralizing the collagen, as was previously reported [27]. Characterization of the polypeptides using Liquid Chromatography–Mass Spectrometry showed the presence of glutamic acid which is important to promoting HA nucleation within the protein. Glutamic acid was present in a lower quantity than expected, especially for Cs, though this may be attributed to the different experimental techniques used to quantify the amino acid composition or its processing. The results indicate that it is less suitable for promoting biomineralization than was initially thought, but it is possible that this may vary depending on the source and processing of the silk. Furthermore, mass analysis of DC hydrogels containing Cs (DC-Cs) over time indicated that the Cs fragments are immediately expelled during plastic compression (PC). The silk fibroin-derived polypeptides incorporated into a collagen hydrogel show little difference compared to a pure collagen hydrogel for promoting the nucleation of hydroxyapatite, and it cannot serve as a replacement for non-collagenous proteins (NCPs) in bone tissue engineering (BTE) without first refining the method of linking the polypeptide to the collagen fibril.
Supporting information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
An uncompressed (highly hydrated) hydrogel is placed between two pieces of nylon mesh, and placed overtop a steel mesh and a paper towel (blot paper). A weight (glass plate) is placed on top the hydrogel to produce a constant force (1 kPa) and held for 5 minutes. Water is expelled from the bottom, through the nylon and steel meshes, and soaked up by the blot paper. The hydrogel is plastically compressed into a dense collagen hydrogel, and then is removed from between the nylon meshes.
(TIF)
FTIR spectra shows that bovine bone has the characteristic peaks of collagen from the bands amide I, II and II groups at 1630, 1550 and 1240 cm-1 [58,76], while the presence of HA is seen in the large phosphate band at 1030 and 1080 cm-1 [58,74,75]. The results are supported by those seen in literature [85–87] of FTIR conducted on bone and bone samples that have had collagen removed.
(TIF)
iii) ICDD file 00-046-0905. XRD analysis show that the samples of bone match that seen in literature [88,89]. Heat treatment reveals the band pattern of bone and calcined bone, with the peaks of the latter matching that of calcium-deficient hydroxyapatite (CDHA) (ICDD file 00-046-0905).
(TIF)
Ion Chromatography of Kokubo’s SBF shows that the composition is nearly identical to the calculated theoretical values, as well as the composition of commercial SBF (Hank’s Balance Salt Solution, HBSS) [67]. In addition, it is also similar to the composition of ECF [42], with the exception of the amount carbonate, which is taken to be much lower than that of ECF given the theoretical value.
(TIF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funding was provided by the Institut national de la recherche scientifique to ID; and by the Canada Research Chairs program to FR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Aparicio C, Ginebra MP. Biomineralization and Biomaterials: Fundamentals and Applications. Woodhead Publishing; 2015. [Google Scholar]
- 2.Tomoaia G, Pasca RD. On the Collagen Mineralization. A Review. Clujul Med. 2015;88: 15–22. 10.15386/cjmed-359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nudelman F, Pieterse K, George A, Bomans PHH, Friedrich H, Brylka LJ, et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat Mater. 2010;9: 1004–1009. 10.1038/nmat2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhee S-H, Lee JD, Tanaka J. Nucleation of Hydroxyapatite Crystal through Chemical Interaction with Collagen. J Am Ceram Soc. 2000;83: 2890–2892. 10.1111/j.1151-2916.2000.tb01656.x [DOI] [Google Scholar]
- 5.Hunter GK, Kyle CL, Goldberg HA. Modulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem J. 1994;300: 723–728. 10.1042/bj3000723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmer LC, Newcomb CJ, Kaltz SR, Spoerke ED, Stupp SI. Biomimetic Systems for Hydroxyapatite Mineralization Inspired By Bone and Enamel. Chem Rev. 2008;108: 4754–4783. 10.1021/cr8004422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter GK, Hauschka PV, Poole AR, Rosenberg LC, Goldberg HA. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem J. 1996;317: 59–64. 10.1042/bj3170059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberg HA, Warner KJ, Li MC, Hunter GK. Binding of bone sialoprotein, osteopontin and synthetic polypeptides to hydroxyapatite. Connect Tissue Res. 2001;42: 25–37. [DOI] [PubMed] [Google Scholar]
- 9.Tavafoghi M, Cerruti M. The role of amino acids in hydroxyapatite mineralization. J R Soc Interface. 2016;13 10.1098/rsif.2016.0462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landi E, Celotti G, Logroscino G, Tampieri A. Carbonated hydroxyapatite as bone substitute. J Eur Ceram Soc. 2003;23: 2931–2937. 10.1016/S0955-2219(03)00304-2 [DOI] [Google Scholar]
- 11.Rhee SH, Tanaka J. Hydroxyapatite formation on cellulose cloth induced by citric acid. J Mater Sci Mater Med. 2000;11: 449–452. [DOI] [PubMed] [Google Scholar]
- 12.Song J, Malathong V, Bertozzi CR. Mineralization of synthetic polymer scaffolds: a bottom-up approach for the development of artificial bone. J Am Chem Soc. 2005;127: 3366–3372. 10.1021/ja043776z [DOI] [PubMed] [Google Scholar]
- 13.Raghavan RN, Muthukumar T, Somanathan N, Sastry TP. Biomimetic mineralization of novel silane crosslinked collagen. Mater Sci Eng C. 2013;33: 1983–1988. 10.1016/j.msec.2013.01.007 [DOI] [PubMed] [Google Scholar]
- 14.Jin Y, Kundu B, Cai Y, Kundu SC, Yao J. Bio-inspired mineralization of hydroxyapatite in 3D silk fibroin hydrogel for bone tissue engineering. Colloids Surf B Biointerfaces. 2015;134: 339–345. 10.1016/j.colsurfb.2015.07.015 [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Li N, Qi Y, Dai L, Bryan TE, Mao J, et al. Intrafibrillar Collagen Mineralization Produced by Biomimetic Hierarchical Nanoapatite Assembly. Adv Mater. 2011;23: 975–980. 10.1002/adma.201003882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown CP, Rosei F, Traversa E, Licoccia S. Spider silk as a load bearing biomaterial: tailoring mechanical properties via structural modifications. Nanoscale. 2011;3: 870–876. 10.1039/c0nr00752h [DOI] [PubMed] [Google Scholar]
- 17.MacLeod J, Rosei F. Photonic crystals: Sustainable sensors from silk. Nat Mater. 2013;12: 98–100. 10.1038/nmat3552 [DOI] [PubMed] [Google Scholar]
- 18.Li JJ, Kaplan DL, Zreiqat H. Scaffold-based regeneration of skeletal tissues to meet clinical challenges. J Mater Chem B. 2014;2: 7272–7306. 10.1039/C4TB01073F [DOI] [PubMed] [Google Scholar]
- 19.Sah MK, Pramanik K. Regenerated Silk Fibroin from B. mori Silk Cocoon for Tissue Engineering Applications. Int J Environ Sci Dev. 2010;1: 404–408. 10.7763/IJESD.2010.V1.78 [DOI] [Google Scholar]
- 20.Kundu SC, Kundu B, Talukdar S, Bano S, Nayak S, Kundu J, et al. Invited review nonmulberry silk biopolymers. Biopolymers. 2012;97: 455–467. 10.1002/bip.22024 [DOI] [PubMed] [Google Scholar]
- 21.Omenetto FG, Kaplan DL. New Opportunities for an Ancient Material. Science. 2010;329: 528–531. 10.1126/science.1188936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samal SK, Dash M, Chiellini F, Kaplan DL, Chiellini E. Silk microgels formed by proteolytic enzyme activity. Acta Biomater. 2013;9: 8192–8199. 10.1016/j.actbio.2013.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freddi G, Faragò S, Maifreni T. HPLC fractionation of Cs peptides of Bombyx mori silk fibroin. Séricologia. 1989;29: 307–326. [Google Scholar]
- 24.Yang M, Shuai Y, Zhou G, Mandal N, Zhu L. Nucleation of hydroxyapatite on Antheraea pernyi (A. pernyi) silk fibroin film. Biomed Mater Eng. 2014;24: 731–740. 10.3233/BME-130861 [DOI] [PubMed] [Google Scholar]
- 25.Yang M, He W, Shuai Y, Min S, Zhu L. Nucleation of hydroxyapatite crystals by self-assembled Bombyx mori silk fibroin. J Polym Sci Part B Polym Phys. 2013;51: 742–748. 10.1002/polb.23249 [DOI] [Google Scholar]
- 26.Gulrajani ML. Degumming of silk. Rev Prog Color Relat Top. 1992;22: 79–89. 10.1111/j.1478-4408.1992.tb00091.x [DOI] [Google Scholar]
- 27.Marelli B, Ghezzi CE, Alessandrino A, Barralet JE, Freddi G, Nazhat SN. Silk fibroin derived polypeptide-induced biomineralization of collagen. Biomaterials. 2012;33: 102–108. 10.1016/j.biomaterials.2011.09.039 [DOI] [PubMed] [Google Scholar]
- 28.Termine JD. Non-collagen proteins in bone. Ciba Found Symp. 1988;136: 178–202. [DOI] [PubMed] [Google Scholar]
- 29.Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1981;26: 99–105. 10.1016/0092-8674(81)90037-4 [DOI] [PubMed] [Google Scholar]
- 30.Hunter GK, Goldberg HA. Nucleation of hydroxyapatite by bone sialoprotein. Proc Natl Acad Sci. 1993;90: 8562–8565. 10.1073/pnas.90.18.8562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujisawa R, Tamura M. Acidic bone matrix proteins and their roles in calcification. Front Biosci Landmark Ed. 2012;17: 1891–1903. [DOI] [PubMed] [Google Scholar]
- 32.Neel EAA, Cheema U, Knowles JC, Brown RA, Nazhat SN. Use of multiple unconfined compression for control of collagen gel scaffold density and mechanical properties. Soft Matter. 2006;2: 986–992. 10.1039/B609784G [DOI] [PubMed] [Google Scholar]
- 33.Brown RA, Wiseman M, Chuo C-B, Cheema U, Nazhat SN. Ultrarapid Engineering of Biomimetic Materials and Tissues: Fabrication of Nano- and Microstructures by Plastic Compression. Adv Funct Mater. 2005;15: 1762–1770. 10.1002/adfm.200500042 [DOI] [Google Scholar]
- 34.Bitar M, Salih V, Brown RA, Nazhat SN. Effect of multiple unconfined compression on cellular dense collagen scaffolds for bone tissue engineering. J Mater Sci Mater Med. 2007;18: 237–244. 10.1007/s10856-006-0685-1 [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Thula TT, Jee S, Perkins SL, Aparicio C, Douglas EP, et al. Biomimetic Mineralization of Woven Bone-Like Nanocomposites: Role of Collagen Cross-Links. Biomacromolecules. 2012;13: 49–59. 10.1021/bm201070g [DOI] [PubMed] [Google Scholar]
- 36.Chicatun F, Pedraza CE, Ghezzi CE, Marelli B, Kaartinen MT, McKee MD, et al. Osteoid-mimicking dense collagen/chitosan hybrid gels. Biomacromolecules. 2011;12: 2946–2956. 10.1021/bm200528z [DOI] [PubMed] [Google Scholar]
- 37.Burg KJL, Porter S, Kellam JF. Biomaterial developments for bone tissue engineering. Biomaterials. 2000;21: 2347–2359. 10.1016/S0142-9612(00)00102-2 [DOI] [PubMed] [Google Scholar]
- 38.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27: 3413–3431. 10.1016/j.biomaterials.2006.01.039 [DOI] [PubMed] [Google Scholar]
- 39.Greenspan DC. Bioactive ceramic implant materials. Curr Opin Solid State Mater Sci. 1999;4: 389–393. 10.1016/S1359-0286(99)00021-2 [DOI] [Google Scholar]
- 40.Rong Z, Cheema U, Vadgama P. Needle enzyme electrode based glucose diffusive transport measurement in a collagen gel and validation of a simulation model. Analyst. 2006;131: 816–821. 10.1039/b600334f [DOI] [PubMed] [Google Scholar]
- 41.Cheema U, Rong Z, Kirresh O, Macrobert AJ, Vadgama P, Brown RA. Oxygen diffusion through collagen scaffolds at defined densities: implications for cell survival in tissue models. J Tissue Eng Regen Med. 2012;6: 77–84. 10.1002/term.402 [DOI] [PubMed] [Google Scholar]
- 42.Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27: 2907–2915. 10.1016/j.biomaterials.2006.01.017 [DOI] [PubMed] [Google Scholar]
- 43.Boskey AL. Noncollagenous matrix proteins and their role in mineralization. Bone Miner. 1989;6: 111–123. 10.1016/0169-6009(89)90044-5 [DOI] [PubMed] [Google Scholar]
- 44.Chicatun F, Pedraza CE, Muja N, Ghezzi CE, McKee MD, Nazhat SN. Effect of Chitosan Incorporation and Scaffold Geometry on Chondrocyte Function in Dense Collagen Type I Hydrogels. Tissue Eng Part A. 2013; 130830130010006. 10.1089/ten.tea.2013.0114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevens MM. Biomaterials for bone tissue engineering. Mater Today. 2008;11: 18–25. 10.1016/S1369-7021(08)70086-5 [DOI] [Google Scholar]
- 46.Swetha M, Sahithi K, Moorthi A, Srinivasan N, Ramasamy K, Selvamurugan N. Biocomposites containing natural polymers and hydroxyapatite for bone tissue engineering. Int J Biol Macromol. 2010;47: 1–4. 10.1016/j.ijbiomac.2010.03.015 [DOI] [PubMed] [Google Scholar]
- 47.Weiner S, Wagner HD. THE MATERIAL BONE: Structure-Mechanical Function Relations. Annu Rev Mater Sci. 1998;28: 271–298. 10.1146/annurev.matsci.28.1.271 [DOI] [Google Scholar]
- 48.Rockwood DN, Preda RC, Yücel T, Wang X, Lovett ML, Kaplan DL. Materials fabrication from Bombyx mori silk fibroin. Nat Protoc. 2011;6: 1612–1631. 10.1038/nprot.2011.379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zahn H, Schade W, Ziegler K. Fractionation of the chymotryptic precipitate of Bombyx mori silk fibroin. Biochem J. 1967;104: 1019–1026. 10.1042/bj1041019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi Y, Wang H, Wei K, Yang Y, Zheng R-Y, Kim IS, et al. A Review of Structure Construction of Silk Fibroin Biomaterials from Single Structures to Multi-Level Structures. Int J Mol Sci. 2017;18 10.3390/ijms18030237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sashina ES, Bochek AM, Novoselov NP, Kirichenko DA. Structure and solubility of natural silk fibroin. Russ J Appl Chem. 2006;79: 869–876. 10.1134/S1070427206060012 [DOI] [Google Scholar]
- 52.Salvi G, De Los Rios P, Vendruscolo M. Effective interactions between chaotropic agents and proteins. Proteins. 2005;61: 492–499. 10.1002/prot.20626 [DOI] [PubMed] [Google Scholar]
- 53.Sawyer WH, Puckridge J. The dissociation of proteins by chaotropic salts. J Biol Chem. 1973;248: 8429–8433. [PubMed] [Google Scholar]
- 54.Parker ST, Domachuk P, Amsden J, Bressner J, Lewis JA, Kaplan DL, et al. Biocompatible Silk Printed Optical Waveguides. Adv Mater. 2009;21: 2411–2415. 10.1002/adma.200801580 [DOI] [Google Scholar]
- 55.Freddi G, Gotoh Y, Mori T, Tsutsui I, Tsukada M. Chemical structure and physical properties of antheraea assama silk. J Appl Polym Sci. 1994;52: 775–781. 10.1002/app.1994.070520608 [DOI] [Google Scholar]
- 56.Gou Z, Chang J. Synthesis and in vitro bioactivity of dicalcium silicate powders. J Eur Ceram Soc. 2004;24: 93–99. 10.1016/S0955-2219(03)00320-0 [DOI] [Google Scholar]
- 57.Stoch A, Jastrzbski W, Brożek A, Trybalska B, Cichocińska M, Szarawara E. FTIR monitoring of the growth of the carbonate containing apatite layers from simulated and natural body fluids. J Mol Struct. 1999;511–512: 287–294. 10.1016/S0022-2860(99)00170-2 [DOI] [Google Scholar]
- 58.Roessler S, Born R, Scharnweber D, Worch H, Sewing A, Dard M. Biomimetic coatings functionalized with adhesion peptides for dental implants. J Mater Sci Mater Med. 2001;12: 871–877. [DOI] [PubMed] [Google Scholar]
- 59.Cao Z, Chen X, Yao J, Huang L, Shao Z. The preparation of regenerated silk fibroin microspheres. Soft Matter. 2007;3: 910 10.1039/b703139d [DOI] [PubMed] [Google Scholar]
- 60.Zhang H, Li L, Dai F, Zhang H, Ni B, Zhou W, et al. Preparation and characterization of silk fibroin as a biomaterial with potential for drug delivery. J Transl Med. 2012;10: 117 10.1186/1479-5876-10-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Zhang K-Q. Silk Fiber—Molecular Formation Mechanism, Structure- Property Relationship and Advanced Applications In: Lesieur C, editor. Oligomerization of Chemical and Biological Compounds. 1st ed Rijeka: IntechOpen; 2014. pp. 69–102. 10.5772/57611 [DOI] [Google Scholar]
- 62.Huang Y, Bailey K, Wang S, Feng X. Silk fibroin films for potential applications in controlled release. React Funct Polym. 2017;116: 57–68. 10.1016/j.reactfunctpolym.2017.05.007 [DOI] [Google Scholar]
- 63.Morin A, Pahlevan M, Alam P. Silk Biocomposites: Structure and Chemistry. In: Thakur VK, Thakur MK, Kessler MR, editors. Handbook of Composites from Renewable Materials. 2017. pp. 189–220. 10.1002/9781119441632.ch8 [DOI] [Google Scholar]
- 64.Warwicker JO. The crystal structure of silk fibroin. Acta Crystallogr. 1954;7: 565–573. 10.1107/S0365110X54001867 [DOI] [Google Scholar]
- 65.Wongpinyochit T, Johnston BF, Seib FP. Degradation Behavior of Silk Nanoparticles—Enzyme Responsiveness. ACS Biomater Sci Eng. 2018;4: 942–951. 10.1021/acsbiomaterials.7b01021 [DOI] [PubMed] [Google Scholar]
- 66.Lotz B, Colonna Cesari F. The chemical structure and the crystalline structures of Bombyx mori silk fibroin. Biochimie. 1979;61: 205–214. 10.1016/S0300-9084(79)80067-X [DOI] [PubMed] [Google Scholar]
- 67.Thermo Fisher Scientific. 14025—HBSS, calcium, magnesium, no phenol red. In: Thermo Fisher Scientific [Internet]. 2016 [cited 14 Sep 2016]. Available: http://www.thermofisher.com/ca/en/home/technical-resources/media-formulation.153.html
- 68.Guyton AC, Hall JE. Textbook of Medical Physiology. 11th ed Philadelphia: Elsevier Saunders; 2006. [Google Scholar]
- 69.Chen C, Huang Z, Yuan W, Li J, Cheng X, Chi R. Pressure effecting on morphology of hydroxyapatite crystals in homogeneous system. CrystEngComm. 2011;13: 1632–1637. 10.1039/C0CE00090F [DOI] [Google Scholar]
- 70.Rodríguez-Clemente R, López-Macipe A, Gómez-Morales J, Torrent-Burgués J, Castaño VM. Hydroxyapatite precipitation: A case of nucleation-aggregation-agglomeration-growth mechanism. J Eur Ceram Soc. 1998;18: 1351–1356. 10.1016/S0955-2219(98)00064-8 [DOI] [Google Scholar]
- 71.Addadi L, Weiner S. Interactions between acidic proteins and crystals: stereochemical requirements in biomineralization. Proc Natl Acad Sci U S A. 1985;82: 4110–4114. 10.1073/pnas.82.12.4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Veis A, Perry A. The Phosphoprotein of the Dentin Matrix*. Biochemistry. 1967;6: 2409–2416. 10.1021/bi00860a017 [DOI] [PubMed] [Google Scholar]
- 73.Baht GS, Hunter GK, Goldberg HA. Bone sialoprotein–collagen interaction promotes hydroxyapatite nucleation. Matrix Biol. 2008;27: 600–608. 10.1016/j.matbio.2008.06.004 [DOI] [PubMed] [Google Scholar]
- 74.Ślósarczyk A, Paluszkiewicz C, Gawlicki M, Paszkiewicz Z. The FTIR spectroscopy and QXRD studies of calcium phosphate based materials produced from the powder precursors with different CaP ratios. Ceram Int. 1997;23: 297–304. 10.1016/S0272-8842(96)00016-8 [DOI] [Google Scholar]
- 75.Rapacz-Kmita A, Paluszkiewicz C, Ślósarczyk A, Paszkiewicz Z. FTIR and XRD investigations on the thermal stability of hydroxyapatite during hot pressing and pressureless sintering processes. J Mol Struct. 2005;744–747: 653–656. 10.1016/j.molstruc.2004.11.070 [DOI] [Google Scholar]
- 76.Nagai T, Suzuki N, Tanoue Y, Kai N. Collagen from Tendon of Yezo Sika Deer (Cervus nippon yesoensis) as By-Product. Food Nutr Sci. 2012;03: 72–79. 10.4236/fns.2012.31012 [DOI] [Google Scholar]
- 77.Yang Y, Cui Q, Sahai N. How Does Bone Sialoprotein Promote the Nucleation of Hydroxyapatite? A Molecular Dynamics Study Using Model Peptides of Different Conformations. Langmuir. 2010;26: 9848–9859. 10.1021/la100192z [DOI] [PubMed] [Google Scholar]
- 78.He G, George A. Dentin matrix protein 1 immobilized on type I collagen fibrils facilitates apatite deposition in vitro. J Biol Chem. 2004;279: 11649–11656. 10.1074/jbc.M309296200 [DOI] [PubMed] [Google Scholar]
- 79.Prokopowicz M, Zegliński J, Gandhi A, Sawicki W, Tofail SAM. Bioactive silica-based drug delivery systems containing doxorubicin hydrochloride: In vitro studies. Colloids Surf B Biointerfaces. 2012;93: 249–59. 10.1016/j.colsurfb.2012.01.020 [DOI] [PubMed] [Google Scholar]
- 80.Wilcock C J., Gentile P, Hatton P, Miller C. Rapid Mix Preparation of Bioinspired Nanoscale Hydroxyapatite for Biomedical Applications. J Vis Exp. 2016;2017 10.3791/55343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.López EO, Mello A, Farina M, Rossi AM, Rossi AL. Nanoscale analysis of calcium phosphate films obtained by RF magnetron sputtering during the initial stages of deposition. Surf Coat Technol. 2015;279: 16–24. 10.1016/j.surfcoat.2015.08.021 [DOI] [Google Scholar]
- 82.Venkateswarlu K, Chandra Bose A, Rameshbabu N. X-ray peak broadening studies of nanocrystalline hydroxyapatite by Williamson–Hall analysis. Phys B Condens Matter. 2010;405: 4256–4261. 10.1016/j.physb.2010.07.020 [DOI] [Google Scholar]
- 83.Calasans-Maia MD, Melo BR de, Alves ATNN, Resende RF de B, Louro RS, Sartoretto SC, et al. Cytocompatibility and biocompatibility of nanostructured carbonated hydroxyapatite spheres for bone repair. J Appl Oral Sci. 2015;23: 599–608. 10.1590/1678-775720150122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lam E, Gu Q, Swedlund PJ, Marchesseau S, Hemar Y. X-ray diffraction investigation of amorphous calcium phosphate and hydroxyapatite under ultra-high hydrostatic pressure. Int J Miner Metall Mater. 2015;22: 1225–1231. 10.1007/s12613-015-1189-5 [DOI] [Google Scholar]
- 85.Querido W, Rossi AL, Campos APC, Rossi AM, Farina M. Does crystallinity of extracted bone mineral increase over storage time? Mater Res. 2013;16: 970–974. 10.1590/S1516-14392013005000096 [DOI] [Google Scholar]
- 86.Akindoyo J, Beg H, Ghazali S, Akindoyo E, Jeyaratnam N. Synthesis of Hydroxyapatite through Ultrasound and Calcination Techniques. IOP Conf Ser Mater Sci Eng. 2017;203: 012003 10.1088/1757-899X/203/1/012003 [DOI] [Google Scholar]
- 87.Bano N, Jikan SS, Basri H, Adzila S, Zago DM. XRD and FTIR study of A&B type carbonated hydroxyapatite extracted from bovine bone. AIP Conf Proc. 2019;2068: 020100 10.1063/1.5089399 [DOI] [Google Scholar]
- 88.Liu Q, Huang S, Matinlinna JP, Chen Z, Pan H. Insight into Biological Apatite: Physiochemical Properties and Preparation Approaches. BioMed Res Int. 2013;2013 10.1155/2013/929748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pramanik S, A. S. M. H, Pingguan-Murphy B, Abu Osman NA. Morphological Change of Heat Treated Bovine Bone: A Comparative Study. Materials. 2013;6: 65–75. 10.3390/Ma6010065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
An uncompressed (highly hydrated) hydrogel is placed between two pieces of nylon mesh, and placed overtop a steel mesh and a paper towel (blot paper). A weight (glass plate) is placed on top the hydrogel to produce a constant force (1 kPa) and held for 5 minutes. Water is expelled from the bottom, through the nylon and steel meshes, and soaked up by the blot paper. The hydrogel is plastically compressed into a dense collagen hydrogel, and then is removed from between the nylon meshes.
(TIF)
FTIR spectra shows that bovine bone has the characteristic peaks of collagen from the bands amide I, II and II groups at 1630, 1550 and 1240 cm-1 [58,76], while the presence of HA is seen in the large phosphate band at 1030 and 1080 cm-1 [58,74,75]. The results are supported by those seen in literature [85–87] of FTIR conducted on bone and bone samples that have had collagen removed.
(TIF)
iii) ICDD file 00-046-0905. XRD analysis show that the samples of bone match that seen in literature [88,89]. Heat treatment reveals the band pattern of bone and calcined bone, with the peaks of the latter matching that of calcium-deficient hydroxyapatite (CDHA) (ICDD file 00-046-0905).
(TIF)
Ion Chromatography of Kokubo’s SBF shows that the composition is nearly identical to the calculated theoretical values, as well as the composition of commercial SBF (Hank’s Balance Salt Solution, HBSS) [67]. In addition, it is also similar to the composition of ECF [42], with the exception of the amount carbonate, which is taken to be much lower than that of ECF given the theoretical value.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.