

Abstract
Fabry disease is an X-linked lysosomal storage disease caused by α-galactosidase A (α-Gal A) deficiency. Kidney and heart failure are frequent complications in adulthood and greatly contribute to patient morbidity and mortality. Because α-Gal A-deficient mouse models do not recapitulate cardiorenal findings observed in patients, a nonmouse model may be beneficial to our understanding of disease pathogenesis. In this study, we evaluated disease processes in a recently generated Fabry rat model. We found that male Fabry rats weighed significantly less than wild-type (WT) males, whereas female Fabry rats weighed significantly more than WT females. Whereas no difference in female survival was detected, we observed that male Fabry rats had a decreased lifespan. Skin histology revealed that inflammation and lipoatrophy may be chief disease mediators in patients. With respect to the kidney and heart, we found that both organs accumulate α-Gal A substrates, including the established biomarkers, globotriaosylceramide and globotriaosylsphingosine. Longitudinal serum and urine chemistry panels demonstrated pronounced renal tubule dysfunction, which was confirmed histologically. Mitral valve thickening was observed in Fabry rats using echocardiography. We conclude that Fabry rats recapitulate important kidney and heart phenotypes experienced by patients and can be further used to study disease mechanisms and test therapies.—Miller, J. J., Aoki, K., Mascari, C. A., Beltrame, A. K., Sokumbi, O., North, P. E., Tiemeyer, M., Kriegel, A. J., Dahms, N. M., α-Galactosidase A-deficient rats accumulate glycosphingolipids and develop cardiorenal phenotypes of Fabry disease.
Keywords: lysosomal storage disease, glycobiology, kidney, heart, animal model
Lysosomes play critical roles in nutrient sensing, plasma membrane repair, and macromolecule degradation (1, 2). This third function is accomplished by an arsenal of ∼60 different lysosomal enzymes. If any one of these lysosomal enzymes is deficient from a genetic mutation, then a lysosomal storage disease may develop [reviewed in Platt (3)]. One such disease, Fabry disease, is a consequence of α-galactosidase A (α-Gal A) deficiency from a galactosidase α (GLA) gene mutation. As a result, glycosphingolipids (GSLs), terminated with α-galactose, accumulate within lysosomes, ultimately causing cell and organ dysfunction. The main α-Gal A substrates include globotriaosylceramide (Gb3; also known as Gb3Cer), globotriaosylsphingosine (lyso-Gb3), digalactosylceramide (Gal2Cer), and blood group B GSLs. Like other lysosomal storage diseases, Fabry disease is progressive, with manifestations in multiple organ systems. Patients may initially experience substantial neuropathic pain, anhidrosis, and angiokeratomas. As patients age, they are at significant risk of developing cerebrovascular, heart, and kidney diseases, which often result in premature death. Fabry disease is X-linked, so males tend to be disproportionately affected. Hemizygous males and homozygous females usually, but not always, experience greater symptom severity than heterozygous (HET) females. Furthermore, patients with mutations that completely abolish α-Gal A activity generally have more severe symptoms than those with some residual activity [reviewed in Germain (4)].
Fabry mouse models [Gla knockout (KO)] have been developed and have been critical in the testing of therapies (5, 6). Whereas these mice accumulate α-galactosyl GSLs in tissues and develop microscopic lesions characteristic of Fabry disease, they live a normal lifespan and do not display altered blood and urine findings suggestive of organ dysfunction (7). Although subtle differences in cardiovascular function have been reported (8), the lack of litter- and strain-matched control mice makes interpretation of these studies difficult. In an attempt to elicit a more robust phenotype, 1 group overexpressed Gb3 synthase in Gla KO mice (9). However, the resulting transgenic mouse model developed severe neurologic and musculoskeletal phenotypes not seen in humans. Thus, a Fabry animal model that more closely mimics disease phenotypes is needed.
Previously, we generated a rat model of Fabry disease (Gla KO) and showed that Fabry rats develop somatosensory disturbances suggestive of neuropathic pain (10). In this report, we characterized Fabry rats longitudinally with specific emphasis on kidney and heart function. We found sex differences in growth, where Fabry males weigh less than wild-type (WT) males, and Fabry females weigh more than WT females. Median survival was reduced in Fabry male rats, and we showed that macrophages significantly infiltrate aged Fabry rat skin. Pronounced accumulation of the biomarkers, Gb3 and lyso-Gb3, was observed in Fabry rat kidney and heart using nanospray ionization-mass spectrometry. We performed serum and urine chemistry panels and found that Fabry rats naturally develop renal tubule disease with age. Finally, echocardiography measurements showed that mitral valve thickening, but not hypertrophic cardiomyopathy, is present in aged Fabry rats. We conclude that Fabry rats partially recapitulate kidney and heart phenotypes experienced by patients and will be useful in testing therapies and studying the pathogenic mechanisms of these phenotypes.
MATERIALS AND METHODS
Animals
The Fabry rat model (Dark Agouti background) was generated using clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeats-associated protein 9 technology, where a 47 bp deletion was introduced in exon 2 of the rat Gla gene, which results in a truncated, inactive enzyme product, as previously described (10). The existing Fabry rat colony at the Medical College of Wisconsin was used for all data collection, and cage- and littermates were used as WT controls. Rats were provided with free access to chow and drinking water. All animal procedures were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
Histology and electron microscopy
For light microscopy studies, tissues were fixed with 10% neutral-buffered formalin and paraffin-embedded. Paraffin blocks were sectioned at 4 µm, deparaffinized, and stained with hematoxylin and eosin (H&E) or Masson’s trichrome. Additional sections were antigen retrieved and stained immunohistochemically using standard-labeled streptavidin-biotin detection. Mouse anti-CD68 antibody (ED-1 clone, MAB1435, 1:100; MilliporeSigma, Burlington, MA, USA) and rabbit anti-CD3 antibody (A0452, 1:200; Dako, Santa Clara, CA, USA) were used to detect macrophages and T cells, respectively. Incubation with biotinylated anti-mouse and anti-rabbit secondary antibodies (715-066-151 and 711-066-152, respectively, both 1:500; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was performed before streptavidin-horseradish peroxidase and 3,3′-diaminobenzidine detection. A Hamamatsu (Hamamatsu, Japan) NanoZoomer 2.0-HT slide scanner was used to obtain slide images. Electron microscopy was performed as previously described (10). A board-certified pathologist assisted in the interpretation of all histology and electron microscopy images.
Activity assay for α-Gal A
The activity of α-Gal A was determined in kidney and heart homogenates, similar to as previously described (10). In brief, organs were homogenized, and protein concentrations were determined using a commercial bicinchoninic acid assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Homogenates were diluted in reaction buffer (27 mM citric acid, 47 mM sodium phosphate dibasic, pH 4.6) and incubated with 4 mM 4-methylumbelliferyl α-d-galactopyranoside (MilliporeSigma), a synthetic α-Gal A substrate. N-Acetyl-d-galactosamine (MilliporeSigma), at 100 mM, was used as an inhibitor of α-N-acetylgalactosaminidase, an endogenous enzyme that also hydrolyzes the 4-methylumbelliferyl substrate (11, 12). The reaction was incubated at 37°C and terminated with 0.2 M glycine (pH 10.7). Fluorescence intensity was measured (excitation 365 nm, emission 448 nm) and converted to nanomole 4-methylumbelliferone product by interpolating from a linear standard curve. Activity is reported as nanomoles per hour per milligram total protein.
GSL mass spectrometry
GSLs were isolated and quantified as previously described (10). In brief, GSLs were extracted and purified from kidney and heart homogenates and were permethylated with 12C-methyliodide. As quantification reference standards, known amounts of maltotetraose were permethylated with [13C]methyliodide and spiked into the GSL samples. GSLs were analyzed by nanospray ionization-mass spectrometry in positive ion mode by direct infusion into a linear ion trap mass spectrometer (linear trap quadrupole; Thermo Fisher Scientific). Detection and absolute quantification of each GSL were accomplished using the Xcaliber software package version 2.0 (Thermo Fisher Scientific; total ion mapping and neutral loss scan functionalities). Both kidney and heart GSLs were normalized per dried protein powder (picomole GSL per milligram protein). Glycan structures are depicted using the symbol nomenclature for glycans (13).
Blood and urine chemistries
Rats were placed in metal metabolic cages overnight (∼18 h) with free access to chow and water. The collected urine was centrifuged at 3210 g for 10 min (4°C) to remove particulates and aliquoted. Rats were then anesthetized with isoflurane, and blood was collected from the ventral tail artery. Blood was placed in serum separator microtainers (Becton Dickinson, San Diego, CA, USA), allowed to clot between 30 min and 2 h, and centrifuged at 13,000 g for 90 s to yield the serum supernatant. Urine (code: VUCHM) and blood (code: ANP1) chemistry panels were performed by Marshfield Labs Veterinary Services (Waukesha, WI, USA). Urine osmolarity was measured using a VAPRO vapor pressure osmometer (ELITechGroup, Logan, UT, USA). Estimated glomerular filtration rate (eGFR) was calculated from creatinine clearance:
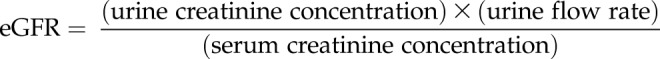 |
Echocardiography
Transthoracic echocardiography (Vivid 7; GE Healthcare, Waukesha, WI, USA) was performed on isoflurane-anesthetized rats at indicated ages. Indices of ventricular wall thickness, chamber dimensions, and function were measured from midpapillary short-axis views of the left ventricle displayed in M mode, as previously described (14, 15). Mitral flow was measured in 4-chamber views of the heart by using pulsed-wave Doppler echocardiography. The peak velocity of early E and late A waves, as well as the duration and slope of deceleration, was measured and reported as the mean of 4 cardiac cycles.
Statistics
GraphPad Prism 7 software was used for all statistical analyses. A 2-way ANOVA was used to analyze growth curves, serum, and urine chemistry longitudinal data and echocardiography data with Bonferroni’s multiple comparisons test to calculate genotype differences. Survival analyses were performed using the log-rank (Mantel-Cox) test. A 2-tailed, unpaired t test was used to compare WT and KO male enzyme activity means, and a 1-way ANOVA with Dunnett’s multiple comparisons was used to compare HET and KO female enzyme activity means to WT. Unpaired, 2-tailed t tests were used to compare WT and KO means for each GSL species. All plots show means ± sem. The set level of significance was P < 0.05, and P values are described in figure legends.
RESULTS
Fabry rats develop gross morphology, skin, and tongue changes compared with WT rats
We performed a longitudinal natural history study to determine the pathologic consequences of α-Gal A deficiency in rats. At weaning, WT and Fabry rats had similar appearances, but gross morphology differences developed with age. Starting at 17 wk, significant differences in weight were observed in male rats, with KO rats weighing less than WT (Fig. 1A). This weight difference increased with age, and at 80 wk, KO males weighed 20% less than WT. In contrast, aging Fabry (HET and KO) females weighed significantly more than WT females (Fig. 1B). Survival analysis revealed that KO males had a reduced median survival of 90 wk compared with 106 wk in WT males (Fig. 1C). There was no genotype difference in female rat survival (Fig. 1D). However, we found that all female rats had an overall reduced life expectancy compared with all male rats. In summary, Fabry male rats weigh less than and have a reduced median survival compared with WT male rats. Fabry female rats weigh more than WT female rats, but no differences in survival are observed.
Figure 1.

Fabry rat appearance and survival. A) WT and KO male rat weights are plotted with age. Representative ventral body and face images are shown (inset). Body images are from 39-wk-old rats, and face images are from 52-wk-old rats. B) Similar to A but with WT, HET, and KO female rat weights and images shown. A, B) Means ± sem are plotted, and the exact number of biologic replicates at each week are given in Supplemental Table S1. A 2-way ANOVA was used to calculate the overall effect of genotype on rat weight, and overall significance is shown to the right of each legend. Significant differences in genotype were determined at each time point using Bonferroni’s multiple comparison test, and for simplicity, the results are only shown every 10 wk. C) Male survival curve that is representative of all rats studied in the Fabry rat colony, where WT n = 153 and KO n = 173. Rats were counted as alive if they were still alive at the time of graph generation or if they were healthy but underwent a planned necropsy for organ collection. Rats were counted as dead if they died spontaneously or reached a humane endpoint and required euthanasia, as judged by the veterinary staff. A log-rank (Mantel-Cox) test was performed to compare WT with KO survival curves. D) Same as in C but with female rats, where WT n = 64, HET n = 75, and KO n = 40. *P < 0.05, **P < 0.01, ****P < 0.0001.
Skin manifestations are common in Fabry disease. Fabry rats of both sexes developed rougher coats and an unkempt appearance at ∼9 mo of age (Fig. 1A, B, insets). As the rats continued to age, alopecia and xeroderma also became prominent, and at necropsy, we noticed that the skin of aged Fabry rats was more difficult to section. Thus, we histologically examined the skin of 91-wk-old male rats. Consistent with the decreased body weight in KO males (Fig. 1A), there was evidence of subcutis loss (i.e., lipoatrophy) in KO male skin (Fig. 2A). We also observed dilated sebaceous ducts in KO rat skin (Fig. 2B), which suggested occlusion and thereby provided an explanation for the xeroderma phenotype. In the dermal layer of KO rat skin, we observed that the connective tissue was thickened and denser with a marked band of histiocytes (Fig. 2A, B). The abundant histiocytes were CD68 positive and thus are likely to be macrophages (Fig. 2C). With the dense macrophage infiltration, we also observed scattered CD3-positive T lymphocytes (Fig. 2D). Patients with Fabry disease also have macrophage and lymphocyte infiltration within tissues [reviewed in Rozenfeld and Feriozzi (16)], suggesting a similar inflammatory phenotype between Fabry rats and humans. However, no evidence of Fabry disease-associated vascular pathology, such as angiokeratomas, was observed in Fabry rat skin.
Figure 2.

Inflammation and lipoatrophy are evident in aged Fabry rat skin. Back-skin sections from aged (90 wk old) male rats were stained with Masson’s trichrome (A, B); anti-CD68, a marker of macrophages (C); or anti-CD3, a pan T lymphocyte marker (D). Images are representative of sections from 3 WT and 3 KO male rats. Original scale bars, 500 µm (A) and 100 µm (B–D).
We also observed enlarged tongues in Fabry male and female rats (Fig. 1A, B, insets), which is similar to findings reported in Fabry mice (17). Tongue weights (Supplemental Fig. S1A), widths (Supplemental Fig. S1B), and heights (Supplemental Fig. S1C) were all significantly increased in 13-wk-old male and female Fabry rats compared with WT controls. Histologically, we observed an expanded connective tissue area and histiocytic infiltration between the tongue muscle layers at 91 wk of age (Supplemental Fig. S1D, S1E). Similar to skin, the infiltrating cells were observed to be predominantly macrophages (Supplemental Fig. S1F), with a scattered population of T cells (Supplemental Fig. S1G). Whereas tongue enlargement is not a classic finding, Fabry patients do exhibit subtle facial dysmorphology, including midface abnormalities and lip fullness (18, 19). It is possible that the pathogenic mechanisms leading to tongue enlargement and remodeling in Fabry rodents may be similar to those contributing to facial dysmorphology in patients.
Fabry rats accumulate α-galactosyl GSLs in kidney and heart
As patients with Fabry disease are deficient in α-Gal A activity, they accumulate GSL substrates containing terminal α-galactose in virtually all tissues. These α-galactosyl GSLs include Gb3, lyso-Gb3, Gal2Cer, and blood group B (Fig. 3A). We have previously shown that Fabry rats accumulate these GSL species in serum, red blood cells, and dorsal root ganglia (10). Because kidney and heart disease have significant impacts on morbidity and mortality in Fabry disease, we next turned our Fabry rat phenotyping efforts to these 2 organs. In kidney, α-Gal A activity was undetectable in KO males and females and was significantly reduced in HET females compared with WT (Fig. 3B). Nanospray ionization-mass spectrometry analysis demonstrated that compared with WT, the KO kidney accumulated Gb3, lyso-Gb3, ceramide dihexoside (CDH; which includes lactosylceramide and Gal2Cer), and blood group B at 168-, 53-, 12-, and 11-fold increases, respectively (Fig. 3C). Additionally, poly-α-galactosylated Gb3 species were detected in KO, but not WT, kidney (Fig. 3C). As controls, we quantified GSLs that do not contain terminal α-galactose, including globotetraosylceramide (Gb4) and monosialoganglioside (GM3), and found no differences between KO and WT kidney (Fig. 3C).
Figure 3.

Fabry rat kidney and heart have undetectable α-Gal A activity and accumulate α-galactosyl GSLs. A) List of GSLs with terminal α-galactose that accumulate in Fabry disease. Carbohydrate portions of GSLs are represented using the established symbol nomenclature for glycans. B) α-Gal A activity was measured in 13-wk-old kidney homogenate using a 4-methylumbelliferyl substrate. Biologic replicates include 5 WT males, 5 KO males, 6 WT females, 6 HET females, and 3 KO females. C) Kidney GSL quantification by nanospray ionization-mass spectrometry from 3 WT and 3 KO males at 13 wk (log scale y axis). When GSL levels in KO tissue are statistically elevated above WT, the fold increase is shown above in red. GSLs detected in KO, but not WT, kidneys are highlighted with a blue box. D) Same as in B but α-Gal A activity in heart homogenate. The number of heart biologic replicates is 6 for WT and KO males and WT and HET females and 3 for KO females. E) Same as in C but GSL quantification in heart (log scale y axis). B, D) Male enzyme activity means are compared with an unpaired, 2-tailed t test, and female enzyme activity means (gray box) are compared with a 1-way ANOVA and Dunnett’s multiple comparison test. C, E) WT and KO GSL means are compared with unpaired, 2-tailed t tests. Gal-Gb3, Gb3 with 1 galactose extension; Gal-lyso-Gb3, lyso-Gb3 with 1 galactose extension; Gal2-Gb3, Gb3 with 2 galactose extensions; GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine. *P < 0.05, **P < 0.01, ***P < 0.001.
Lyso-Gb3 in Fabry rat kidney was further characterized by enzymatic digestion and total ion mapping. When incubated with a linkage-specific α-galactosidase, the signal corresponding to lyso-Gb3 shifted to lactosylsphingosine (Supplemental Fig. S2), confirming that the terminal monosaccharide of the measured lyso-Gb3 is galactose and is in an α-linkage, as predicted. We also used nanospray ionization-mass spectrometry to analyze Fabry rat urine. Although low in abundance, we detected Gb3 and lyso-Gb3 in KO urine to a greater degree than WT (Supplemental Fig. S3). In summary, α-galactosyl GSLs that are observed in patients, including Gb3 and lyso-Gb3, are also detected in Fabry rat kidney and urine.
We performed analogous enzyme activity and GSL mass spectrometry measurements in Fabry rat heart. When heart α-Gal A activity was measured, we found similar results to kidney: undetectable activity in KO males and females and significantly decreased activity in HET females compared with WT (Fig. 3D). Nanospray ionization-mass spectrometry showed that Gb3 was elevated 876-fold in KO heart relative to WT (Fig. 3E). However, unlike kidney, CDH was not significantly increased in Fabry rat heart (Fig. 3E). Several α-galactosyl GSLs that were undetectable in WT were readily detectable in KO heart, including lyso-Gb3, poly-α-galactosylated Gb3 and lyso-Gb3 species, and blood group B (Fig. 3E). There were no differences in the control, non-α-galactosyl GSLs, Gb4 and GM3 (Fig. 3E). Therefore, similar to kidney, Fabry rat hearts specifically store α-galactosyl GSLs.
Serum and urine chemistry panel analyses suggest progressive renal tubule dysfunction
A core goal of the Fabry rat natural history study was to perform serum and urine chemistry panel measurements longitudinally. Male rat samples were analyzed starting at 13 wk and continued every 13 wk until 91 wk of age. Because female rats had a reduced lifespan (Fig. 1D), we were only able to collect female chemistry data from 13 to 65 wk (in 13 wk intervals). A heat map depicting the log2 fold change (relative to WT) is shown to summarize the Fabry rat chemistry data at 2 time points: 26 and 52 wk (Fig. 4). All chemistry data are represented longitudinally in Supplemental Figs. S4–S6.
Figure 4.

Serum and urine chemistry alterations point to renal tubule dysfunction in Fabry rats. Shown are heat maps of serum and urine chemistry panel data from rat groups at 2 different ages: 26 and 52 wk. The log2 fold change is calculated by comparing HET and KO females with WT females and by comparing KO males with WT males. Each cell within the heat maps represents the log2 fold change obtained from mean chemistry values from ≥8 individual rats from each sex and genotype (WT males, KO males, WT females, HET females, KO females). The data are plotted with age in Supplemental Figs. S4–S6, and the exact numbers of biologic replicates are given in Supplemental Tables S2 and S3. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; F, female; M, male.
Several Fabry rat chemistry alterations were consistent with renal tubule disease, a phenotype observed in patients. Compared with WT controls, we observed decreased protein (Supplemental Fig. S4G), decreased albumin (Supplemental Fig. S4H), and increased potassium (Supplemental Fig. S5F) in Fabry rat serum. Serum glucose was decreased in KO males but not KO females (Supplemental Fig. S4A). Urine findings supporting renal tubule disease in KO rats include increased urine flow rate (Supplemental Fig. S6C), decreased osmolarity (Supplemental Fig. S6E), and dramatically increased urine calcium with age (Fig. 4 and Supplemental Fig. S6I). Most impressively, we found that calcium was 5.4-fold elevated in KO urine compared with WT at 91 wk. Because most calcium is reabsorbed in the proximal tubule, dysfunction of this nephron region was apparent. These chemistry alterations allowed us to conclude that Fabry rats develop renal tubule dysfunction.
As patients develop glomerular disease in addition to tubule disease, we also assessed glomerular health using the chemistry data. The eGFR was similar between WT and KO males from 13 to 78 wk but was significantly reduced in KO males at 91 wk (Supplemental Fig. S6F). eGFR was also similar among all 3 female genotypes, but at 52 wk, it was elevated in HET and KO females (Supplemental Fig. S6F). However, no differences in eGFR were observed in 65-wk-old females. We found no genotype differences in urine protein in male and female rats (Supplemental Fig. S6B). Although there were no urine protein alterations, we conclude that 91-wk-old male rats show beginning signs of glomerular decline, as assessed by eGFR.
Other striking chemistry data include significantly elevated aspartate aminotransferase and alkaline phosphatase levels with age (Supplemental Fig. S4B, D, respectively) in KO male and female rats. Alanine aminotransferase was largely similar between the genotypes (Supplemental Fig. S4C). Blood urea nitrogen (BUN) was also similar but was elevated in KO males at 78 and 91 wk and in KO females at 65 wk (Supplemental Fig. S5A). Overall, the blood and urine chemistry dataset reveals that pathology is progressive with age in Fabry rats. This is consistent with the notion that the phenotypes of Fabry disease are later onset and attenuated compared with those of other lysosomal storage diseases.
Fabry rat proximal tubule cells store inclusions progressively
To confirm the renal tubule dysfunction identified in the chemistry panel data, we next microscopically analyzed Fabry rat kidney sections. At 13 wk, the glomerular and tubule structures looked similar between WT and KO rats (Fig. 5A). However, at 90 wk, some KO glomeruli appeared atrophied (Fig. 5A). On closer magnification, the proximal tubule cells of KO rats appeared more vacuolated and accumulated several large, circular inclusions (Fig. 5B). Electron microscopy was used to evaluate glomerular and tubular ultrastructure. Similar to the light microscopy findings, 13-wk-old glomerular structures appeared similar between WT and KO rats (Fig. 5C). However, in the 90-wk-old tissues, the KO foot processes appeared less defined compared with WT (Fig. 5C). Electron microscopy of proximal tubule cells showed that inclusions of a homogeneous, medium density were present in 13-wk-old KO rats, and these inclusions remarkably increased in number and size in 90-wk-old KO rats (Fig. 5D). Similar structures are observed in chronic renal failure and are consistent with protein accumulation within secondary lysosomes (20). Furthermore, the structures are indicative of lysosomal dysfunction, as is expected in Fabry disease. As corroborated by the chemistry panel data, the histology and electron microscopy studies reveal that Fabry rat proximal tubules are more severely affected compared with other nephron structures.
Figure 5.

Inclusions accumulate in proximal tubule cells from aged Fabry rats. A) Rat kidney sections were stained with H&E, and representative images are shown from 13-wk-old rats (n = 3 each: WT males, KO males, WT females, HET females, and KO females) and 90-wk-old rats (n = 3 each: WT and KO males). Original scale bars, 100 µm. B) Magnified view of 90-wk-old kidney tubule cells with original scale bars, 50 µm. C) Representative electron microscopy images of a portion of the glomerulus. D) Representative electron microscopy images of a proximal tubule cell. C, D) Original scale bars, 2 µm. B, D) Arrows, inclusions. BM, basement membrane; DT, distal tubule; F, podocyte foot processes; G, glomerulus; N, nucleus; PT, proximal tubule.
Myocardium appears similar to WT, but some inclusions are present in aged Fabry rat heart
The impressive inclusions observed in renal tubules prompted us to examine next histologically Fabry rat hearts. The myocardium of young (13 wk) and aged (90 wk) Fabry rats appeared similar to aged-matched WT hearts on the light microscopy level (Fig. 6A). When evaluated by electron microscopy, 91-wk-old myocardium was similar between KO and WT (Fig. 6B). However, we observed the occasional presence of electron-dense structures within cardiomyocytes in KO, but not WT, samples (Fig. 6B; arrows). These inclusions are indicative of lysosomes containing undigested subcellular material. To summarize, aged Fabry rat cardiomyocytes occasionally develop inclusions but not to the striking extent observed in renal tubules.
Figure 6.

Myocardium appears similar to WT, but some electron-dense structures are observed in Fabry rat cardiomyocytes. A) H&E-stained myocardium from 13- to 90-wk-old rats. Thirteen-week-old images are representative of n = 3 each: WT males, KO males, WT females, HET females, and KO females. Ninety-week-old images are representative of n = 3 each: WT and KO males. Original scale bars, 100 µm. B) Representative electron microscopy (EM) images of 90-wk-old male myocardium. Original scale bars, 2 µm. Arrows point to inclusions.
Aging Fabry rats develop left-ventricular wall thinning
Hypertrophic cardiomyopathy is a well-established phenotype in patients with Fabry disease. We next used echocardiography to evaluate the cardiac function in Fabry rats. To observe potential age-related changes, we assessed rats at 2 ages: 7 mo (31 wk, referred to as young rats) and 17 mo (74–77 wk, referred to as aged rats). Note that the measurements from 1 aged Fabry rat were excluded because of the presence of an arrhythmia. Diastolic and systolic measurements were taken from rat hearts in short-axis view (Fig. 7A). There were no significant differences in the diastolic or systolic internal dimension between genotypes in either young or aged rats (Fig. 7B, C). Although not different in young rats, left-ventricular ventral walls of aged Fabry rats were significantly thinner in both diastole (Fig. 7D) and systole (Fig. 7E) than WT rats. Likewise, left-ventricular dorsal walls were thinner in systole but not diastole in aged Fabry rats compared with WT (Fig. 7F, G). Left-ventricular mass was calculated using the American Society of Echocardiography estimation. Aged but not young Fabry rats were found to have significantly decreased left-ventricular mass in both diastole (Fig. 7H) and systole (Fig. 7I). Finally, whereas there were no differences in young rats, fractional shortening (Fig. 7J) and ejection fraction (Fig. 7K) were found to be reduced in aged Fabry rats compared with WT controls. From these echocardiography data, we conclude that the left-ventricular walls are thinner in aged Fabry rats, which may affect cardiac function. Therefore, Fabry rats do not recapitulate hypertrophic cardiomyopathy, which is a characteristic symptom in patients with Fabry disease.
Figure 7.

Aged Fabry rats develop wall thinning of the left ventricle. A) Diagram of left-ventricle (LV) echocardiographic measurements in short-axis view. Diastole indicates maximal cardiac relaxation, and systole indicates maximal cardiac contraction. B–K) Echocardiographic measurements include the following: internal dimension at diastole (LVIDd) (B), internal dimension at systole (LVIDs) (C), LV ventral-wall thickness at diastole (IVSd) (D), LV ventral-wall thickness at systole (IVSs) (E), LV dorsal-wall thickness at diastole (LVPWd) (F), LV dorsal-wall thickness at systole (LVPWs) (G), LV mass at diastole (LVd) (H), LV mass at systole (LVs) (I), fractional shortening (FS) (J), and ejection fraction (EF) (K). LV mass (H, I) was calculated using the American Society of Echocardiography estimation. Data in B–K were obtained from 4 WT and 4 KO male rats at 31 wk (7 mo) and 8 WT and 5 KO male rats at 74–77 wk (17 mo). Data were analyzed using a 2-way ANOVA, and genotype differences in echocardiography parameters were determined with Bonferroni’s multiple comparisons test. *P < 0.05, **P < 0.01.
Mitral valve thickening is evident in aged Fabry rats
We also used echocardiography to assess mitral valve function, which is often impacted in Fabry disease. We found that mitral valve deceleration time was significantly increased in aged but not young Fabry rats compared with WT (Fig. 8A). Consistently, we found that the mitral valve-deceleration slope was decreased in aged Fabry rats (Fig. 8B). These 2 findings are suggestive of impaired diastolic filling through the mitral valve and/or impaired relaxation of the left ventricle. The echocardiography exams also revealed hypertrophied papillary muscles in some Fabry rats, which may be indicative of mitral valve dysfunction. We next histologically examined the mitral valve of aged Fabry rats, and we observed substantial thickening of the valve with cellular infiltration when compared with WT (Fig. 8C). To summarize, the mitral valve of aged Fabry rats is thickened, which is a common finding in patients. Thus, α-Gal A-deficient rats partially recapitulate the cardiac phenotype of Fabry disease: mitral valve thickening without hypertrophic cardiomyopathy.
Figure 8.

Aged Fabry rats show evidence of mitral valve thickening. A, B) Mitral valve (MV) deceleration time (A) and MV deceleration slope (B) were obtained from echocardiography measurements. Data were obtained from 4 WT and 4 KO male rats at 31 wk (7 mo) and 7 WT and 5 KO male rats at 74–77 wk (17 mo). MV echocardiography data were analyzed using a 2-way ANOVA, and genotype differences were determined with Bonferroni’s multiple comparisons test. C) Ninety-week-old (21 mo) heart sections were stained with Masson’s trichrome, and representative valve images are shown. Arrows point to the section of the valve that is magnified; original scale bars, 100 µm. *P < 0.05.
DISCUSSION
Recent newborn screening studies have provided evidence that Fabry disease is likely to be the most common lysosomal storage disease (21–24). As more countries screen newborns for Fabry disease, this patient population is expected to expand greatly. Given that the disease symptoms are extremely debilitating, and the efficacy of enzyme replacement therapy is questioned, increased research effort is desperately needed to better understand the pathogenesis of this complex disease. We recently developed a rat model of Fabry disease, and in this natural history study, we longitudinally characterized general phenotypes, including kidney and heart function in Fabry rats. We found weight differences, skin inflammation, and α-galactosyl GSL accumulation in kidney and heart. We also found evidence of renal tubule dysfunction and mitral valve thickening, 2 serious phenotypes observed in patients. Thus, the Fabry rat model serves as a valuable new tool to study various disease processes.
We found that Fabry rats recapitulate the life expectancy and weight phenotypes reported in patients with Fabry disease. In Dutch patients, the male median survival is reduced from ∼78 to 57 yr, and the female median survival is reduced from ∼80 to 72 yr (25). However, no difference in median survival is detected in Fabry mice (7). We found that Fabry male rats have a statistically reduced lifespan with a median survival of 90 wk (∼1.7 yr) compared with a median survival of 106 wk (∼2 yr) in WT male rats. Thus, unlike Fabry mouse models, we observe recapitulation of the human disease in Fabry male rats with respect to life expectancy. There was no genotype-dependent difference in female rat survival, but this may be attributed to the fact that all female rat groups in our study had an overall reduced life expectancy compared with males.
Fabry rats also better reproduce human disease-growth phenotypes compared with Fabry mice. Fabry male mice (C57BL/6, 129/SvJ mixed background) weigh more than WT C57BL/6 mice (17, 26, 27), whereas Fabry female mice weigh less than WT (17). We found the opposite trends in Fabry rats: males weigh less than WT, and females weigh more than WT. These results match the weight findings in a large group of pediatric patients with Fabry disease. In patients, mean male weight is less than the 50th percentile, whereas mean female weight is greater than the 50th percentile (28). Furthermore, symptomatic adult male patients have difficulty gaining weight [reviewed in Germain (4)]. For example, adult male patients with gastrointestinal symptoms were shown to have a reduced body mass index compared with those without gastrointestinal symptoms (29). In contrast, Wang et al. (30) examined a cohort of 44 adult females with Fabry disease and found that 68% had a body mass index greater than normal, with 44% meeting criteria for obesity. Thus, compared with α-Gal A-deficient mouse models, Fabry rats more closely recapitulate the sex-specific weight differences observed in pediatric and adult patients with Fabry disease. The question remains as to what factors contribute to the sex differences in weight seen in Fabry disease. Patients are reported to have endocrine dysfunction (31), so perhaps sex-hormone disturbances play a role in the effect of α-Gal A deficiency on weight.
Patients with Fabry disease develop kidney damage that often progresses to end-stage disease, requiring renal replacement therapy [reviewed in Germain (4)]. Patients are found to have glomerular disease and proteinuria resulting from GSL-laden podocytes that eventually detach (32). Proximal and distal tubule abnormalities also develop (33–36). On the cellular level, GSL inclusions accumulate in podocytes, renal vascular endothelial cells, and distal tubule cells, with some sparing of proximal tubule cells (37). Knowledge of the renal injury mechanisms in response to GSL storage is incomplete. Studies have shown that when applied to an immortalized podocyte cell line, lyso-Gb3 activates fibrogenic, inflammatory, and dedifferentiation processes (38, 39). It has been proposed that the effects of Gb3 and lyso-Gb3 on cultured renal tubule cells lead to an epithelial-to-mesenchymal transition (40) and altered lipid-raft dynamics (41). Therefore, an animal model of Fabry disease with any renal phenotype, such as the Fabry rat, will be valuable in further evaluating these proposed injury mechanisms.
Fabry mice are poor models for kidney disease, as they do not spontaneously develop a renal phenotype. At 40 and 80 wk of age, Fabry mice are reported to show no significant deviations in blood and urine chemistries [e.g., BUN, serum creatinine, urine creatinine, urine protein (7)]. Fabry mice overexpressing Gb3 synthase are reported to be a better model of renal disease, as these mice develop decreased urine osmolarity at 5 wk, polyuria at 10 wk, and increased BUN at 15 wk (9). However, this transgenic mouse model does not develop glomerular disease, in that no structural abnormalities are seen in Bowman’s capsule, and few to no GSL inclusions are seen in glomerular (e.g., podocytes, mesangial, endothelial) cells (9). Furthermore, the Gb3 synthase-overexpressing Fabry mouse has irrelevant neuromuscular phenotypes, including spontaneous tremors, slow movements, rounded backs, gait disturbances, and premature death by 35 wk (9). In summary, neither mouse model seems to be ideal in the study of Fabry kidney disease.
We found that aging Fabry rats spontaneously develop a renal phenotype; however, Fabry renal disease is different in rats compared with humans. Fabry rats show evidence of a pronounced renal tubule phenotype, with a mild glomerular phenotype. Evidence of progressive renal disease in KO rats is observed with the following abnormalities: decreased urine osmolarity and elevated urine calcium, serum potassium, BUN, and urine flow rate. The increase in calcium excretion in Fabry rats was unexpected and warrants further study, as osteopenia and osteoporosis have been reported in patients with Fabry disease (42, 43). The increased urine calcium suggests bone demineralization in Fabry rats, a finding that may provide clues as to why many patients develop bone disease. The data obtained from the chemistry panels allow us to conclude that Fabry rats partially recapitulate the renal phenotype of Fabry disease. Furthermore, these data highlight new avenues of research, such as investigating why patients experience bone demineralization.
Similar to kidney disease, Fabry rats partially recapitulate heart phenotypes, in that mitral valve thickening but not ventricular hypertrophy is observed. It should be noted that we did observe papillary muscle hypertrophy in some Fabry rats, a finding reported in patients and suggested to be a diagnostic marker (44). The cause of ventricular hypertrophy in Fabry patients is unknown, but it has been reported not to be directly from GSL deposits or pressure overloading as a result of hypertension (45–47). Biologic differences in tissue types may explain why the rat mitral valve is susceptible to damage, but the myocardium is relatively spared in the context of α-Gal A deficiency. For example, similar to connective tissue in skin, it appears that mitral valve-connective tissue is vulnerable to cellular infiltration. It is also possible that prolonged mitral valve dysfunction could contribute to ventricular hypertrophy but is not extensive enough to elicit a hypertrophic remodeling in this model within the study period. Nevertheless, mitral valve dysfunction is a cardiac manifestation often observed in pediatric patients (48). Thus, the Fabry rat valvular phenotype could lead to a better understanding of and therapies for mitral valve disease in patients.
In summary, a longitudinal natural history study was performed in the newly generated Fabry rat model to inform future mechanistic studies. Fabry rats show weight differences that recapitulate findings in patients, and male Fabry rats have a decreased life expectancy. Although minimal phenotypic differences are apparent in young rats, aging Fabry rats slowly develop renal tubule disease and mitral valve thickening, consistent with Fabry disease being a late-onset and attenuated lysosomal storage disease. We anticipate that these progressive phenotypes in the Fabry rat will be informative in future studies to enhance therapies and improve mechanistic understanding of Fabry disease pathogenesis.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Dr. David Mattson for guidance, especially during the early stages of this project. The authors also thank Christine Duris, Stephanie Wirsbinski, Tatunya Bufford, and Qiuhui Yang of the Children’s Hospital of Wisconsin Children’s Research Institute Histology Core for histology and immunohistochemistry services. The electron microscopy services and interpretation by Clive Wells are greatly appreciated. This work was supported by U.S. National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK042667 and NIH National Institute of Neurological Disorders and Stroke Grant R21NS095627 (to N.M.D.), NIH National Institute of Allergy and Infectious Diseases Grant R21AI129873 (to K.A.), NIH National Institute of General Medical Sciences Grant P41GM103490 (to M.T.), and NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant F30DK113641 (to J.J.M.). J.J.M. is a trainee of the Medical College of Wisconsin Medical Scientist Training Program, which is partially supported by T32GM080202. The Fabry rat model was generated under R24HL114474, a resource grant from NIH National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors declare no conflicts of interest.
Glossary
- α-Gal A
α-galactosidase A
- BUN
blood urea nitrogen
- CDH
ceramide dihexoside
- eGFR
estimated glomerular filtration rate
- Gal2Cer
digalactosylceramide
- Gb3
globotriaosylceramide
- Gb4
globotetraosylceramide
- Gla
galactosidase α
- GM3
monosialoganglioside
- GSL
glycosphingolipid
- H&E
hematoxylin and eosin
- HET
heterozygous
- KO
knockout
- lyso-Gb3
globotriaosylsphingosine
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
J. J. Miller, K. Aoki, M. Tiemeyer, A. J. Kriegel, and N. M. Dahms designed research; J. J. Miller, K. Aoki, C. A. Mascari, A. K. Beltrame, and A. J. Kriegel performed research; J. J. Miller, K. Aoki, C. A. Mascari, A. K. Beltrame, O. Sokumbi, P. E. North, M. Tiemeyer, A. J. Kriegel, and N. M. Dahms analyzed data; J. J. Miller wrote the original manuscript draft; and J. J. Miller, K. Aoki, C. A. Mascari, A. K. Beltrame, M. Tiemeyer, A. J. Kriegel, and N. M. Dahms revised and edited the manuscript.
REFERENCES
- 1.De Duve C. (2005) The lysosome turns fifty. Nat. Cell Biol. 7, 847–849 10.1038/ncb0905-847 [DOI] [PubMed] [Google Scholar]
- 2.Ballabio A. (2016) The awesome lysosome. EMBO Mol. Med. 8, 73–76 10.15252/emmm.201505966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platt F. M. (2014) Sphingolipid lysosomal storage disorders. Nature 510, 68–75 10.1038/nature13476 [DOI] [PubMed] [Google Scholar]
- 4.Germain D. P. (2010) Fabry disease. Orphanet J. Rare Dis. 5, 30 10.1186/1750-1172-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ioannou Y. A., Zeidner K. M., Gordon R. E., Desnick R. J. (2001) Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. Am. J. Hum. Genet. 68, 14–25 10.1086/316953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohshima T., Murray G. J., Swaim W. D., Longenecker G., Quirk J. M., Cardarelli C. O., Sugimoto Y., Pastan I., Gottesman M. M., Brady R. O., Kulkarni A. B. (1997) alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc. Natl. Acad. Sci. USA 94, 2540–2544 10.1073/pnas.94.6.2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohshima T., Schiffmann R., Murray G. J., Kopp J., Quirk J. M., Stahl S., Chan C. C., Zerfas P., Tao-Cheng J. H., Ward J. M., Brady R. O., Kulkarni A. B. (1999) Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. Proc. Natl. Acad. Sci. USA 96, 6423–6427 10.1073/pnas.96.11.6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen Dinh Cat A., Escoubet B., Agrapart V., Griol-Charhbili V., Schoeb T., Feng W., Jaimes E., Warnock D. G., Jaisser F. (2012) Cardiomyopathy and response to enzyme replacement therapy in a male mouse model for Fabry disease. PLoS One 7, e33743 10.1371/journal.pone.0033743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taguchi A., Maruyama H., Nameta M., Yamamoto T., Matsuda J., Kulkarni A. B., Yoshioka H., Ishii S. (2013) A symptomatic Fabry disease mouse model generated by inducing globotriaosylceramide synthesis. Biochem. J. 456, 373–383 10.1042/BJ20130825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller J. J., Aoki K., Moehring F., Murphy C. A., O’Hara C. L., Tiemeyer M., Stucky C. L., Dahms N. M. (2018) Neuropathic pain in a Fabry disease rat model. JCI Insight 3, e99171 10.1172/jci.insight.99171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dean K. J., Sung S. S., Sweeley C. C. (1977) The identification of alpha-galactosidase B from human liver as an alpha-N-acetylgalactosaminidase. Biochem. Biophys. Res. Commun. 77, 1411–1417 10.1016/S0006-291X(77)80136-8 [DOI] [PubMed] [Google Scholar]
- 12.Mayes J. S., Scheerer J. B., Sifers R. N., Donaldson M. L. (1981) Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin. Chim. Acta 112, 247–251 10.1016/0009-8981(81)90384-3 [DOI] [PubMed] [Google Scholar]
- 13.Varki A., Cummings R. D., Aebi M., Packer N. H., Seeberger P. H., Esko J. D., Stanley P., Hart G., Darvill A., Kinoshita T., Prestegard J. J., Schnaar R. L., Freeze H. H., Marth J. D., Bertozzi C. R., Etzler M. E., Frank M., Vliegenthart J. F., Lütteke T., Perez S., Bolton E., Rudd P., Paulson J., Kanehisa M., Toukach P., Aoki-Kinoshita K. F., Dell A., Narimatsu H., York W., Taniguchi N., Kornfeld S. (2015) Symbol nomenclature for graphical representations of glycans. Glycobiology 25, 1323–1324 10.1093/glycob/cwv091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriegel A. J., Didier D. N., Li P., Lazar J., Greene A. S. (2012) Mechanisms of cardioprotection resulting from Brown Norway chromosome 16 substitution in the salt-sensitive Dahl rat. Physiol. Genomics 44, 819–827 10.1152/physiolgenomics.00175.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kriegel A. J., Greene A. S. (2008) Substitution of Brown Norway chromosome 16 preserves cardiac function with aging in a salt-sensitive Dahl consomic rat. Physiol. Genomics 36, 35–42 10.1152/physiolgenomics.00054.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rozenfeld P., Feriozzi S. (2017) Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 122, 19–27 10.1016/j.ymgme.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 17.Üçeyler N., Biko L., Hose D., Hofmann L., Sommer C. (2016) Comprehensive and differential long-term characterization of the alpha-galactosidase A deficient mouse model of Fabry disease focusing on the sensory system and pain development. Mol. Pain 12, 1–10 10.1177/1744806916646379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ries M., Moore D. F., Robinson C. J., Tifft C. J., Rosenbaum K. N., Brady R. O., Schiffmann R., Krasnewich D. (2006) Quantitative dysmorphology assessment in Fabry disease. Genet. Med. 8, 96–101 10.1097/01.gim.0000200950.25118.dd [DOI] [PubMed] [Google Scholar]
- 19.Cox-Brinkman J., Vedder A., Hollak C., Richfield L., Mehta A., Orteu K., Wijburg F., Hammond P. (2007) Three-dimensional face shape in Fabry disease. Eur. J. Hum. Genet. 15, 535–542 10.1038/sj.ejhg.5201798 [DOI] [PubMed] [Google Scholar]
- 20.Kretchmer N., Bernstein J. (1974) The dynamic morphology of the nephron: morphogenesis of the “protein droplet”. Kidney Int. 5, 96–105 10.1038/ki.1974.13 [DOI] [PubMed] [Google Scholar]
- 21.Elliott S., Buroker N., Cournoyer J. J., Potier A. M., Trometer J. D., Elbin C., Schermer M. J., Kantola J., Boyce A., Turecek F., Gelb M. H., Scott C. R. (2016) Pilot study of newborn screening for six lysosomal storage diseases using tandem mass spectrometry. Mol. Genet. Metab. 118, 304–309 10.1016/j.ymgme.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopkins P. V., Campbell C., Klug T., Rogers S., Raburn-Miller J., Kiesling J. (2015) Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 166, 172–177 10.1016/j.jpeds.2014.09.023 [DOI] [PubMed] [Google Scholar]
- 23.Spada M., Pagliardini S., Yasuda M., Tukel T., Thiagarajan G., Sakuraba H., Ponzone A., Desnick R. J. (2006) High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 79, 31–40 10.1086/504601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burton B. K., Charrow J., Hoganson G. E., Waggoner D., Tinkle B., Braddock S. R., Schneider M., Grange D. K., Nash C., Shryock H., Barnett R., Shao R., Basheeruddin K., Dizikes G. (2017) Newborn screening for lysosomal storage disorders in illinois: the initial 15-month experience. J. Pediatr. 190, 130–135 10.1016/j.jpeds.2017.06.048 [DOI] [PubMed] [Google Scholar]
- 25.Vedder A. C., Linthorst G. E., van Breemen M. J., Groener J. E., Bemelman F. J., Strijland A., Mannens M. M., Aerts J. M., Hollak C. E. (2007) The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J. Inherit. Metab. Dis. 30, 68–78 10.1007/s10545-006-0484-8 [DOI] [PubMed] [Google Scholar]
- 26.Rodrigues L. G., Ferraz M. J., Rodrigues D., Pais-Vieira M., Lima D., Brady R. O., Sousa M. M., Sá-Miranda M. C. (2009) Neurophysiological, behavioral and morphological abnormalities in the Fabry knockout mice. Neurobiol. Dis. 33, 48–56 10.1016/j.nbd.2008.09.001 [DOI] [PubMed] [Google Scholar]
- 27.Lakomá J., Rimondini R., Donadio V., Liguori R., Caprini M. (2014) Pain related channels are differentially expressed in neuronal and non-neuronal cells of glabrous skin of fabry knockout male mice. PLoS One 9, e108641 10.1371/journal.pone.0108641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopkin R. J., Bissler J., Banikazemi M., Clarke L., Eng C. M., Germain D. P., Lemay R., Tylki-Szymanska A., Wilcox W. R. (2008) Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr. Res. 64, 550–555 10.1203/PDR.0b013e318183f132 [DOI] [PubMed] [Google Scholar]
- 29.MacDermot K. D., Holmes A., Miners A. H. (2001) Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J. Med. Genet. 38, 750–760 10.1136/jmg.38.11.750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R. Y., Lelis A., Mirocha J., Wilcox W. R. (2007) Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet. Med. 9, 34–45 10.1097/GIM.0b013e31802d8321 [DOI] [PubMed] [Google Scholar]
- 31.Faggiano A., Pisani A., Milone F., Gaccione M., Filippella M., Santoro A., Vallone G., Tortora F., Sabbatini M., Spinelli L., Lombardi G., Cianciaruso B., Colao A. (2006) Endocrine dysfunction in patients with Fabry disease. J. Clin. Endocrinol. Metab. 91, 4319–4325 10.1210/jc.2006-0858 [DOI] [PubMed] [Google Scholar]
- 32.Fall B., Scott C. R., Mauer M., Shankland S., Pippin J., Jefferson J. A., Wallace E., Warnock D., Najafian B. (2016) Urinary podocyte loss is increased in patients with Fabry disease and correlates with clinical severity of Fabry nephropathy. PLoS One 11, e0168346 10.1371/journal.pone.0168346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colley J. R., Miller D. L., Hutt M. S., Wallace H. J., De Wardener H. E. (1958) The renal lesion in angiokeratoma corporis diffusum. BMJ 1, 1266–1268 10.1136/bmj.1.5082.1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henry E. W., Rally C. R. (1963) The renal lesion in angiokeratoma corporis diffusum (Fabry’s disease). Can. Med. Assoc. J. 89, 206–213 [PMC free article] [PubMed] [Google Scholar]
- 35.Pabico R. C., Atancio B. C., McKenna B. A., Pamukcoglu T., Yodaiken R. (1973) Renal pathologic lesions and functional alterations in a man with Fabry’s disease. Am. J. Med. 55, 415–425 10.1016/0002-9343(73)90140-X [DOI] [PubMed] [Google Scholar]
- 36.Burkholder P. M., Updike S. J., Ware R. A., Reese O. G. (1980) Clinicopathologic, enzymatic, and genetic features in a case of Fabry’s disease. Arch. Pathol. Lab. Med. 104, 17–25 [PubMed] [Google Scholar]
- 37.Alroy J., Sabnis S., Kopp J. B. (2002) Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 13(Suppl 2), S134–S138 [PubMed] [Google Scholar]
- 38.Sanchez-Niño M. D., Carpio D., Sanz A. B., Ruiz-Ortega M., Mezzano S., Ortiz A. (2015) Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 24, 5720–5732 10.1093/hmg/ddv291 [DOI] [PubMed] [Google Scholar]
- 39.Sanchez-Niño M. D., Sanz A. B., Carrasco S., Saleem M. A., Mathieson P. W., Valdivielso J. M., Ruiz-Ortega M., Egido J., Ortiz A. (2011) Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol. Dial. Transplant. 26, 1797–1802 10.1093/ndt/gfq306 [DOI] [PubMed] [Google Scholar]
- 40.Shin Y. J., Jeon Y. J., Jung N., Park J. W., Park H. Y., Jung S. C. (2015) Substrate-specific gene expression profiles in different kidney cell types are associated with Fabry disease. Mol. Med. Rep. 12, 5049–5057 10.3892/mmr.2015.4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labilloy A., Youker R. T., Bruns J. R., Kukic I., Kiselyov K., Halfter W., Finegold D., do Monte S. J., Weisz O. A. (2014) Altered dynamics of a lipid raft associated protein in a kidney model of Fabry disease. Mol. Genet. Metab. 111, 184–192 10.1016/j.ymgme.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Germain D. P., Benistan K., Boutouyrie P., Mutschler C. (2005) Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. Clin. Genet. 68, 93–95 10.1111/j.1399-0004.2005.00457.x [DOI] [PubMed] [Google Scholar]
- 43.Mersebach H., Johansson J. O., Rasmussen A. K., Bengtsson B. A., Rosenberg K., Hasholt L., Sørensen S. A., Sørensen S. S., Feldt-Rasmussen U. (2007) Osteopenia: a common aspect of Fabry disease. Predictors of bone mineral density. Genet. Med. 9, 812–818 10.1097/GIM.0b013e31815cb197 [DOI] [PubMed] [Google Scholar]
- 44.Niemann M., Liu D., Hu K., Herrmann S., Breunig F., Strotmann J., Störk S., Voelker W., Ertl G., Wanner C., Weidemann F. (2011) Prominent papillary muscles in Fabry disease: a diagnostic marker? Ultrasound Med. Biol. 37, 37–43 10.1016/j.ultrasmedbio.2010.10.017 [DOI] [PubMed] [Google Scholar]
- 45.Barbey F., Brakch N., Linhart A., Rosenblatt-Velin N., Jeanrenaud X., Qanadli S., Steinmann B., Burnier M., Palecek T., Bultas J., Hayoz D. (2006) Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. Arterioscler. Thromb. Vasc. Biol. 26, 839–844 10.1161/01.ATV.0000209649.60409.38 [DOI] [PubMed] [Google Scholar]
- 46.Elleder M., Bradová V., Smíd F., Budĕsínský M., Harzer K., Kustermann-Kuhn B., Ledvinová J., Bĕlohlávek, Král V., Dorazilová V. (1990) Cardiocyte storage and hypertrophy as a sole manifestation of Fabry’s disease. Report on a case simulating hypertrophic non-obstructive cardiomyopathy. Virchows Arch. A Pathol. Anat. Histopathol. 417, 449–455 10.1007/BF01606034 [DOI] [PubMed] [Google Scholar]
- 47.Von Scheidt W., Eng C. M., Fitzmaurice T. F., Erdmann E., Hübner G., Olsen E. G., Christomanou H., Kandolf R., Bishop D. F., Desnick R. J. (1991) An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N. Engl. J. Med. 324, 395–399 10.1056/NEJM199102073240607 [DOI] [PubMed] [Google Scholar]
- 48.Kampmann C., Wiethoff C. M., Whybra C., Baehner F. A., Mengel E., Beck M. (2008) Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 97, 463–469 10.1111/j.1651-2227.2008.00700.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.