

Abstract
Background:
Rituximab (RTX) is a chimeric monoclonal anti-CD20 antibody used off-label in the treatment of membranous nephropathy (MN). Unfortunately, limited information is available on the pharmacokinetics of therapeutic proteins such as RTX in patients with glomerular kidney diseases.
Objective:
The current study evaluated RTX pharmacokinetics in patients with MN (n=20) who received 4 RTX weekly intravenous infusions (375 mg/m2) over a month, with a repeat of the identical treatment at 6 months. Baseline patient characteristics were gender (17M/3F), age (49±13 years), and body surface area (2.2±0.24 m2).
Methods:
Compartmental pharmacokinetic analyses were conducted using Phoenix® and comparisons of these parameters were made between the MN patients and published data from two reference populations without kidney diseases (follicular lymphoma and autoimmune disorders).
Results:
Patients with MN exhibited a shorter half-life, reduced volume of central compartment, decreased area under the serum concentration-time curve (exposure), and increased RTX clearance from central compartment vs. previous reports in the reference patient populations.
Conclusions and Relevance:
These results suggest that shorter half-life and lower exposures to RTX in patients with MN may necessitate higher doses and/or changes to dosing frequency in order to optimize the relationships between serum concentrations and therapeutic effects.
Keywords: rituximab, monoclonal antibody, pharmacokinetics, membranous nephropathy, clearance
Introduction
Monoclonal antibodies are a rapidly expanding class of high molecular weight therapeutic agents approved for a variety of diseases (1). These therapies have been increasingly used for the treatment of glomerular kidney diseases (2, 3). Previous publications have reported increased systemic clearance of the anti-tumor necrosis factor-alpha (TNF-α) therapeutic antibody adalimumab in patients with focal segmental glomerulosclerosis (FSGS) and nephrotic range proteinuria. It was reported that enhancement of renal clearance, in addition to enhancement of non-renal clearance, contributes to the increased systemic clearance in patients with glomerular kidney diseases (4, 5). However, despite these findings, the dosing of therapeutic proteins in glomerular kidney diseases remains similar to doses approved by the Federal Food and Drug Administration (FDA) for other indications. The FDA does not require pharmacokinetic studies of therapeutic antibodies in kidney diseases since enhanced clearance of these agents in glomerular kidney diseases was previously not appreciated (6).
Rituximab (RTX) is a chimeric monoclonal antibody directed against the CD20 expressed on B cells and is used in the treatment of diseases characterized by B cell proliferation (7). RTX was initially FDA approved in 1997 for the treatment of non-Hodgkin’s lymphoma. Subsequently, RTX was granted FDA approval for rheumatoid arthritis, and granulomatosis with polyangiitis (GPA), which includes anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (8–10), chronic lymphocytic leukemia (11) and multiple sclerosis (12). In the kidney disease population, RTX has been used in the treatment of FSGS (13), membranous nephropathy (14), systemic lupus erythematosus (SLE) (15) and minimal change disease (16). Patients with some of these kidney diseases, including membranous nephropathy, exhibit nephrotic syndrome, with urinary protein excretion greater than 3.5 g/day.
Membranous Nephropathy (MN) is a disease characterized by basement membrane thickening and proliferation in the B cell count (17). It is a common cause of nephrotic syndrome in adults (18). It is the second most common cause of end-stage renal disease among the glomerular kidney diseases (17, 18). Since there has been an increase in the use of RTX in patients with glomerular kidney diseases secondary to MN, the current study evaluated RTX pharmacokinetics in this patient population.
Methods
RTX serum concentrations were obtained from patients (n=20) diagnosed with MN who were enrolled in a previously published study (19). The original study was approved by the Institutional Review Boards at the Mayo Clinic, the University of North Carolina at Chapel Hill, the University Health Network, University of Toronto, and was registered on www.clinicaltrials.gov (identifier NCT00405340). All patients provided written informed consent. Patients who had been receiving treatment with prednisone, cyclosporine, or mycophenolate mofetil within the previous 4 months, or alkylating agents within the previous 6 months, were not included in the study. Inclusion criteria included biopsy proven MN, creatinine clearance ≥30 mL/min per 1.73 m2, and persistent proteinuria greater than 5 g/24 h despite treatment with 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, angiotensin-converting enzyme inhibitors (ACEI), and/or angiotensin receptor blockers (ARB) at a maximum tolerated dose for at least 4 months. Patients with active infection, diabetes mellitus, or a secondary cause of MN (e.g., hepatitis B, SLE, medications, malignancies) were excluded from study participation. Further details concerning the study were previously published (19).
Patients received 4 weekly intravenous infusions of RTX (375 mg/m2) for an average infusion time of 3.2 ± 0.3 hours (for the first course of treatment at month 1), with a repeat of the same treatment at six months for an average infusion time of 2.8 ± 1.0 hours (for month 6). RTX serum concentrations were obtained 30 minutes after the completion of the dose (for peak concentrations) and 30 minutes prior to the subsequent infusion (for trough concentrations). Peak concentrations were obtained on days 1, 8, 15, 22, 28, 42, 60, 90, and 120 and trough concentrations were obtained on days 1, 8, 15, and 22. All samples were quantified by a proprietary ELISA (Genentech Inc., San Francisco, CA), with assay sensitivity of 500 ng/mL (19). Clinical and demographic data were recorded. Urinary protein excretion, CD19 positive B cell counts, cholesterol, triglycerides, and IgG levels were recorded for up to 12 months after initiation of RTX treatment. Two populations without kidney disease (follicular lymphoma (n=22) and autoimmune disorders (n=14)) were included as reference populations for comparison with the MN population (20). They were similarly treated with 4 weekly infusions of RTX (375 mg/m2) and their pharmacokinetics profiles were described by 2- compartment models.
RTX serum concentration vs. time graphs were constructed for each patient. The area under the serum concentration vs. time curve (AUC) was determined from RTX dose divided by clearance from the central compartment. The exposure is based on RTX AUC. Elimination rate constant and serum half-life were determined by Non Compartmental Analysis (NCA) using Phoenix® (Certara, USA, Princeton, NJ). Compartmental models (1-, 2-, and 3-) were investigated to characterize the pharmacokinetics of RTX using Phoenix®. Additive, multiplicative, and combined additive and multiplicative error models were evaluated to describe inter-patient and residual variability. Selection of a structural pharmacokinetic model and residual error model was based on the objective function, goodness of fit plots, and coefficient of variation (CV) values.
Statistical Analysis
Descriptive parameters from pharmacokinetic analyses, demographic variables, and laboratory tests included mean, median, standard deviation, and ranges. Regression relationships between pharmacokinetic parameters and clinical measures (urinary protein excretion, CD19 positive B cell counts, IgG levels, total serum cholesterol and triglyceride concentrations) at various time points after RTX initiation were evaluated using Graph Pad Instat® (La Jolla, CA). The validity of pharmacokinetic models were checked by boot strap analysis and Visual Predictive Checks (VPC).
Results
The adult patients (n=20) enrolled in this study were predominantly male (n=17) and had an age of 49 ± 13 years, gender (17 males/3 females), and body surface area 2.2 ± 0.2 m2. Other baseline laboratories were creatinine clearance 72.4 ± 33.2 mL/min, serum creatinine 1.5 ± 0.5 mg/dL, urinary protein excretion 11.3 ± 4 g/day, and serum albumin 2.7 ± 0.6 g/dL. Additional demographical data were previously published (19). The results from the time course of serum RTX concentrations at the 1 month and 6 month courses are depicted in Figure 1.
Figure 1.
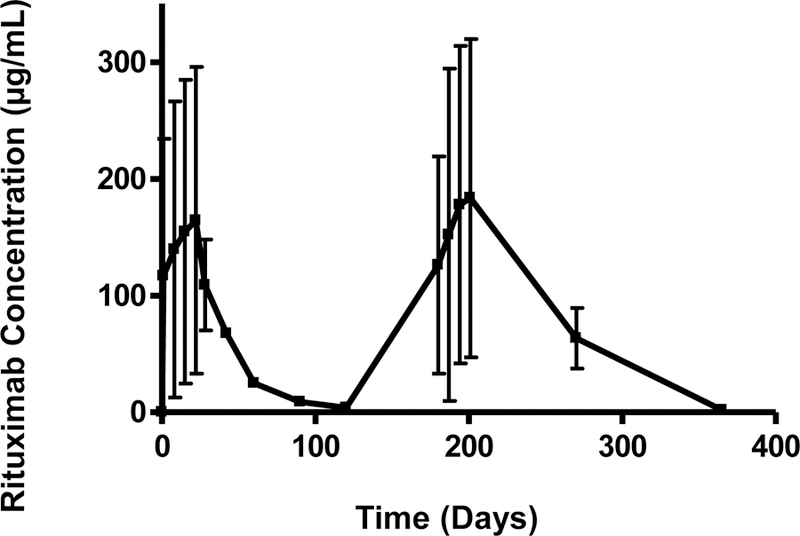
Rituximab serum concentration (µg/mL) vs. time (days) curve. The serum rituximab concentration at each time point is represented as mean ± standard deviation for 20 patients over 360 days.
The results from the pharmacokinetic analysis are reported in Table 1. A 2-compartmental model with a multiplicative error best described the pharmacokinetics of RTX in patients with MN. The following RTX pharmacokinetic parameters from MN patients were computed from Phoenix®; clearance from the central compartment (Cl1, 10.2 ± 4.76 mL/h/m2, CV 13.04%), clearance from the peripheral compartment (Cl2, 416 ± 45.8 mL/h/m2, CV 10.18%), volume of the central compartment (V1, 0.58 ± 0.06 L/m2, CV 19.28%), and volume from the peripheral compartment (V2, 3.3 ± 0.54 L/m2, CV 12.04%). The 2- compartment model, with multiplicative error showed a −2LL value of 7525.04, while the best (smallest −2LL) 1- compartment and 3- compartment models resulted in a −2LL value of 7670.75 (larger −2LL than the 2- compartment model) and 7518.54 (larger percent coefficient of variation), respectively. The overall fit of the 2- compartment model is demonstrated in Figure 2.
Table 1.
Comparitive rituximab pharmacokinetics in membranous nephropathy vs. foliicular lymphoma and autoimmune disorder patients (20)
| Membranous Nephropathy (n=20) | Follicular Lymphoma (n=22) | Autoimmune Disorders (n=14) | |
|---|---|---|---|
| Half-Life (h) | 275 ± 130 (124–512) | 513 (248–705) | 485 (293–859) |
| Clearance 1 (Cl1) (mL/h/m2) | 10.2 ± 4.76 (1.95–22.37) | 5.1 (3.8–8.1) | 6.6 (3.8–11.9) |
| Clearance 2 (Cl2) (mL/h/m2) | 416 ± 45.8 (334–521) | Not reported | Not reported |
| Volume 1 (V1) (L/m2) | 0.58 ± 0.06 (0.46–0.73) | 1.75 (1.6–2.1) | 2.1 (1.0–2.6) |
| Volume 2 (V2) (L/m2) | 3.3 ± 0.54 (1.9–4.2) | Not reported | Not reported |
| AUC (mg h/mL) | 47.4 ± 36.0 (16.75–191.33) | 73.52 (46.3–98.7) | 56.8 (31.5–98.7) |
| Elimination rate constant (h−1) | 0.003 ± 0.001 (0.001–0.005) | 0.001 (0.002–0.001) | 0.001 (0.002–0.001) |
V1 and C1 denote volume and clearance from the central compartment.
V2 and C2 denote volume and clearance from the peripheral compartment
Figure 2.
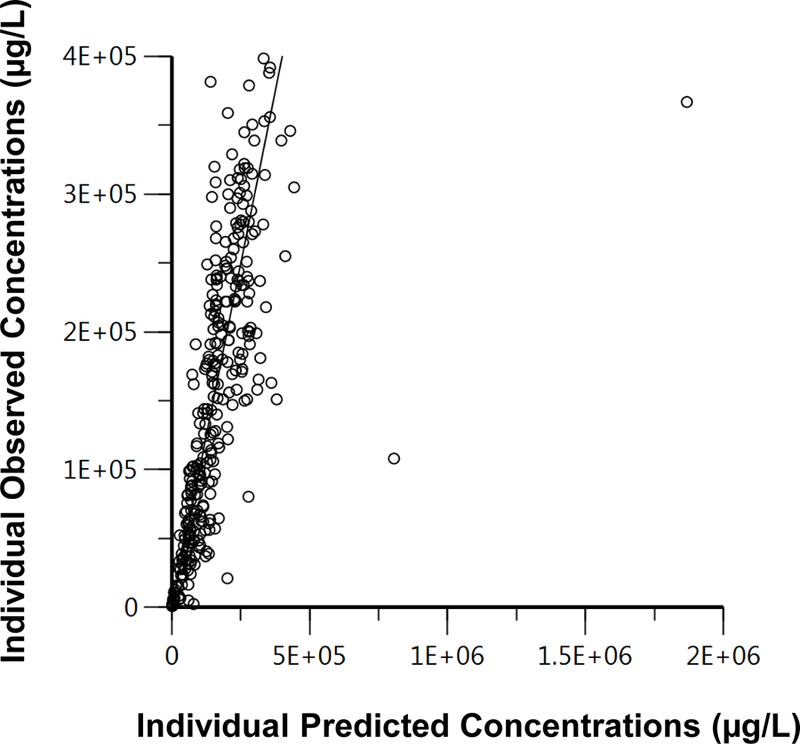

Goodness of fit graphs. 2 A) Individual observed concentrations (µg/L) vs. individual population predicted concentrations (µg/L) of rituximab using the 2- compartment model. 2B) Visual predictive check (VPC) for the final 2- compartment rituximab model. Data represents observed serum concentrations (µg/L) over time (h). Dashed line – mean concentrations vs. time; dotted line – 5th percentile concentrations vs. time; solid line – 95th percentile concentrations vs. time; dots – observed concentrations vs. time.
As demonstrated in Table 1, the pharmacokinetics of RTX in patients with MN were different from published data in follicular lymphoma and autoimmune disease patients. The exposure of RTX in MN patients was lower (47.4 mg h/mL) than in patients with follicular lymphoma (73.5 mg h/mL) and autoimmune disorders (56.8 mg h/mL). The half-life of RTX in MN patients was shorter (275 h) compared to the half-life in patients with follicular lymphoma (513 h) and autoimmune disorders (485 h). Clearance of RTX from the central compartment was faster in MN patients (10.2 mL/h/m2) compared to patients with follicular lymphoma (5.1 mL/h/m2) and autoimmune disorders (6.6 mL/h/m2). Volume of RTX in the central compartment was smaller in MN patients (0.58 L/m2) compared to patients with follicular lymphoma (1.7 L/m2) and autoimmune disorders (2.1 L/m2).
The urinary protein excretion after RTX therapy initiation showed some significant regression relationships with half-life and AUC. Linear regression analyses demonstrated significant negative relationships between RTX half-life and urinary protein excretion at months 1, 3, 6, and 9 (data only shown for month 3 (Figure 3)), where the regression of the line was defined by y = −0.0215x + 434, r2 = 0.6355, p = 0.0006. There was also a negative linear regression relationship in the AUC vs. urinary protein excretion plot at 3 months, with the regression line defined as y= −0.0039x + 76.0, r2 = 0.1989, p = 0.0488 (Figure 4). No other significant relationships were noted between RTX pharmacokinetic parameters and other clinical variables (IgG, cholesterol, triglycerides, CD19 positive B cell counts).
Figure 3.
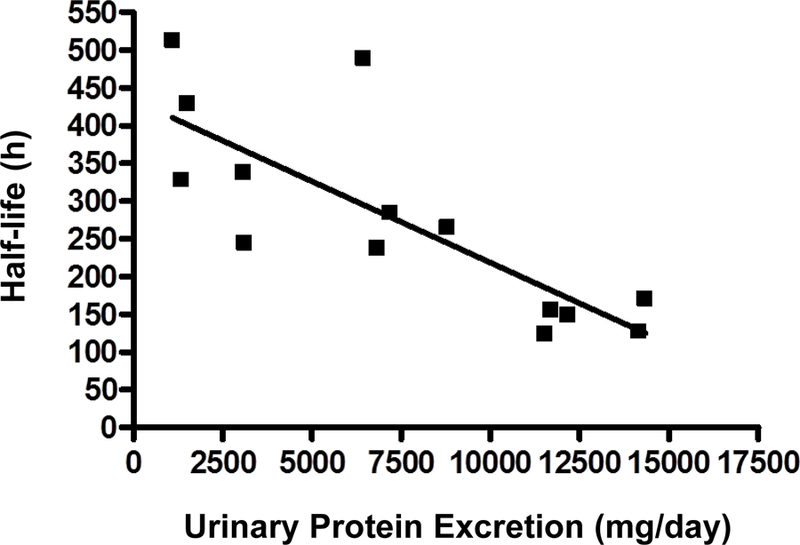
Linear regression of rituximab half-life (h) vs. urinary protein excretion (mg/day) for 20 patients at 3 months post treatment initiation. Linear regression analysis demonstrated y = −0.0215x + 434, r2 =0.6355, p = 0.0006.
Figure 4.
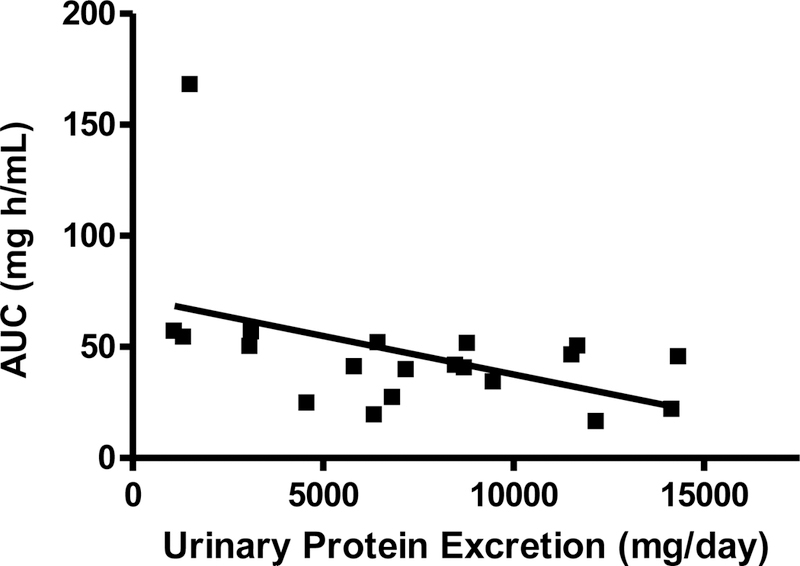
Linear regression of rituximab area under the serum concentration - time curve (mg h/mL) vs. urinary protein excretion (mg/day) for 20 patients at 3 months post treatment initiation. Linear regression analysis demonstrated y = −0.0039x + 76.0, r2 =0.1989, p = 0.0487.
Discussion
RTX is prescribed for a variety of clinical indications in patients with and without kidney diseases. RTX has been increasingly used in glomerular kidney diseases and detailed pharmacokinetic data is lacking in these patients. The current study sought to evaluate RTX pharmacokinetics in patients with MN. The results demonstrated altered RTX pharmacokinetics; shorter half-life (275 h vs. 513 h and 485 h), increased systemic clearance from the central compartment (10.2 mL/h/m2 vs. 5.1 mL/h/m2 and 6.6 mL/h/m2), decreased volume of the central compartment (0.5 L/m2 vs. 1.8 L/m2 and 2.1 L/m2), and lower exposures (47.4 mg h/mL vs. 73.5 mg h/mL and 56.8 mg h/mL) in patients with MN vs. the reference populations (follicular lymphoma and autoimmune diseases, respectively). However, the previous publications and the current study all support a 2- compartment model for describing the pharmacokinetics of RTX (20–22). These pharmacokinetic outcomes indicate that MN patients have lower than expected exposures to RTX administered at the same doses as other populations. A previous publication explored the pharmacokinetics of RTX (administered as a 375 mg/m2 dose) in children with nephrotic syndrome (23). The authors reported a similar half-life (14.6 ± 5.2 days) to the current study. However, the exposures (83.2 ± 53.1 mg h/mL) and volume (2.2 ± 0.37 L/m2) were greater, and clearance (5.83 ± 2.97 mL/h/m2) values were lower in the pediatric study. The pharmacokinetic results from the current study employing RTX are consistent with previous publications describing increased clearance and lower exposures to the monoclonal antibody adalimumab in patients with FSGS, another glomerular disease associated with heavy proteinuria (4, 5). Enhanced clearance and lower expected exposures to RTX could have clinical implications in terms of reduced efficacy to treatment. There might also be financial implications to increasing RTX doses to compensate for the enhanced clearance in patients with MN. These issues, however, need to be explored more completely in future studies.
In the current study, the volume of the central compartment in MN patients was lower than reported for the comparator populations. This could be due to leakage of RTX from vasculature into interstitial space. Two publications reported enhanced clearance of RTX in a patient with nephrotic syndrome and in a population with chronic lymphocytic leukemia (24, 25), and this data was discussed in a subsequent publication (26). The pharmacokinetic findings from the current study are important since previous dosing strategies of therapeutic monoclonal antibodies for MN were borrowed from other FDA approved indications. The current data and former publications suggests that conventional RTX doses may not directly translate to similar exposures in patients with glomerular diseases. However, the use of a single dose of RTX has been reported to induce remission of proteinuria in the majority of MN patients (27), but a more recent study has challenged these observations (28). The current study also showed longer half-lives and higher exposures for RTX with lower urinary protein excretion. As such, a proper understanding of dose and concentration relationships in glomerular kidney diseases associated with heavy proteinuria is essential in order to personalize and optimize treatment-related outcomes.
Therapeutic monoclonal antibodies are large molecules (~145 KD) that do not typically undergo renal filtration as a clearance mechanism. Hence, the FDA does not require testing in patients with impaired renal function under the Guidance for Industry Document on Pharmacokinetics (29). However, our previous study employing adalimumab in patients with FSGS (4) and a review paper published by Kamath et al. (30) indicate that therapeutic monoclonal antibodies can be cleared through kidneys that are damaged. In fact, a previously published case report in a pediatric patient demonstrated that at least 25% of RTX total body clearance occurred through the renal route (25). These findings are important since conventional information on clearance of monoclonal antibodies suggest specific and nonspecific pathways that do not implicate filtration clearance through the kidneys. The clearance pathways for antibodies traditionally include target-mediated drug disposition where the monoclonal antibody binds to the bound or unbound antigen, undergoes cellular internalization, and is subsequently degraded. The pharmacology of RTX leads to a reduction in B cells, leading to a subsequent down-modulation of target (CD20). A previous report showed reduced RTX clearance with repeated dosing (30).
Nonspecific clearance of therapeutic antibodies occurs through neonatal Fc receptor (FcRn) expression and function. FcRn is a 52 kD heterodimeric protein comprised of light chain β2-microglobulin (β2M) and a heavy chain (31). FcRn is widely expressed in many tissues, vascular endothelium, and renal locations including proximal tubule cells, glomerular podocytes, and endothelial cells. FcRn is also responsible for the extended half-life of albumin (21 days) and IgG antibodies (7–21 days) and these proteins bind to separate binding sites (32). The Fc (constant region) on IgG is known to bind to intracellular FcRn within the acidic endosome (pH <6.8). This binding serves to salvage IgG (and albumin) from lysosomal degradation (32). There is dissociation of the IgG-FcRn complex at neutral pH (or higher), with transcytosis of the IgG back to the cell membrane and subsequent release into the circulation. Balthasar et al., previously reported that monoclonal IgG antibody clearance is substantially increased in animals lacking FcRn (33). β2M and FcRn α-chain knockout mice, respectively, are unable to express functional FcRn and are reported to have a 10- to 15- fold higher IgG clearance (33, 34). Given the role of FcRn in influencing the half-life of IgG, mechanisms such as decreased binding affinity of therapeutic antibodies, reduced functionality, or decreased function of FcRn secondary to genetic polymorphisms in FCGRT could all be plausible mechanisms for the increased RTX clearance demonstrated in patients with MN (35, 36).
Immunogenicity is known to contribute to enhanced clearance of monoclonal antibodies. Immunogenicity implies formation of anti-drug antibodies (ADAs) in response to an administered exogenous antibody and enhanced therapeutic antibody clearance has been demonstrated with ADA formation (37). RTX is a chimeric antibody (77% human, 33% mouse) and reported rates of immunogenicity are between 1 to 11% (37, 38). In addition to neutralization of monoclonal antibody therapeutics, aggregation of therapeutic proteins can also lead to formation of ADAs that cross-react with endogenous proteins (39). Intrinsic factors (protein sequence, oxidation, glycosylation, and aggregation) (31) and extrinsic factors (subcutaneous vs. intravenous route (40), lower doses (41, 42), longer duration and intermittent therapy (37), and protein aggregation (43)) can contribute to immunogenicity. The charge on the therapeutic antibody has also been related to clearance, whereby an enhancement in positive charge has been shown to increase clearance (30). These former factors are all responsible for variability in ADA responses observed across the biologic therapeutic modalities, species, and disease states and can affect therapeutic antibody pharmacokinetics and pharmacodynamics (37).
The current publication has demonstrated through a 2- compartment pharmacokinetic analysis that RTX has an altered pharmacokinetic profile in patients with MN as compared to patient populations without kidney diseases. Enhanced clearance, shorter half-life, and lower exposures were demonstrated in patients with MN. However, the current study has several limitations. The pharmacokinetic study was limited in size to 20 patients who were predominantly male. Additionally, all patients were adults, so applicability to pediatric patients receiving RTX cannot be predicted by the current study. The applicability of these findings across patient demographics is unknown. Future assessments will incorporate a population pharmacokinetic approach with covariate analyses and pharmacodynamic modeling. Simulations will be performed to describe the best dosing strategy for patients with MN. Physiologically-based pharmacokinetic (PBPK) modeling will be utilized to ascertain predictions related to therapeutic antibody disposition. PBPK modeling can also probe proposed mechanisms for alterations in FcRn and influence on RTX clearance. The current study did not have urine data available, so the contribution of renal clearance to total body clearance of RTX was not determined. It would also be beneficial to evaluate other kidney diseases that do not exhibit significant urinary protein excretion in order to evaluate how proteinuria directly influences the pharmacokinetics of RTX. Furthermore, while we have shown enhanced clearance of adalimumab in FSGS and RTX in MN, it is currently unknown whether other glomerular diseases and therapeutic monoclonal antibody combinations are associated with these pharmacokinetic alterations.
Conclusions and Relevance
The MN glomerular kidney disease population with significant proteinuria demonstrated marked differences in the pharmacokinetics of the therapeutic antibody RTX. The findings of shortened half-life, lower volume of distribution, and increased clearance and decreased exposures to RTX are worrisome and suggest that fixed dosing protocols may not be adequate for all patients with proteinuric kidney diseases. Higher RTX doses and/or more frequent dosing needs to be weighed against the enhanced risk of immunogenicity, as well as for increased financial burden of treatment. Future work will focus on investigating the mechanism(s) for enhancement of RTX clearance in MN and potentially other glomerular diseases.
References
- 1.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs 2015;7(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitchlu A, Fingrut W, Avila-Casado C, Chan CT, Crump M, Hogg D, et al. Nephrotic Syndrome With Cancer Immunotherapies: A Report of 2 Cases. Am J Kidney Dis 2017;70(4):581–5. [DOI] [PubMed] [Google Scholar]
- 3.Manrique J, Cravedi P. Role of monoclonal antibodies in the treatment of immune-mediated glomerular diseases. Nefrologia 2014;34(3):388–97. [DOI] [PubMed] [Google Scholar]
- 4.Roberts BV, Susano I, Gipson DS, Trachtman H, Joy MS. Contribution of renal and non-renal clearance on increased total clearance of adalimumab in glomerular disease. J Clin Pharmacol 2013;53(9):919–24. [DOI] [PubMed] [Google Scholar]
- 5.Joy MS, Gipson DS, Powell L, MacHardy J, Jennette JC, Vento S, et al. Phase 1 trial of adalimumab in Focal Segmental Glomerulosclerosis (FSGS): II. Report of the FONT (Novel Therapies for Resistant FSGS) study group. Am J Kidney Dis 2010;55(1):50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chebib FT, Torres VE. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am J Kidney Dis 2016;67(5):792–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motta G, Cea M, Moran E, Carbone F, Augusti V, Patrone F, et al. Monoclonal antibodies for non-Hodgkin’s lymphoma: state of the art and perspectives. Clin Dev Immunol 2010;2010:428253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen Tervaert JW. Rituximab in ANCA-associated vasculitis: a revolution? Nephrol Dial Transplant 2011;26(10):3077–9. [DOI] [PubMed] [Google Scholar]
- 9.Randall KL. Rituximab in autoimmune diseases. Aust Prescr 2016;39(4):131–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenert A, Lenert P. Current and emerging treatment options for ANCA-associated vasculitis: potential role of belimumab and other BAFF/APRIL targeting agents. Drug Des Devel Ther 2015;9:333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaglowski SM, Byrd JC. Rituximab in chronic lymphocytic leukemia. Semin Hematol 2010;47(2):156–69. [DOI] [PubMed] [Google Scholar]
- 12.Salzer J, Svenningsson R, Alping P, Novakova L, Bjorck A, Fink K, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology 2016;87(20):2074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roccatello D, Sciascia S, Rossi D, Alpa M, Naretto C, Radin M, et al. High-Dose Rituximab Ineffective for Focal Segmental Glomerulosclerosis: A Long-Term Observation Study. Am J Nephrol 2017;46(2):108–13. [DOI] [PubMed] [Google Scholar]
- 14.Fervenza FC, Canetta PA, Barbour SJ, Lafayette RA, Rovin BH, Aslam N, et al. A Multicenter Randomized Controlled Trial of Rituximab versus Cyclosporine in the Treatment of Idiopathic Membranous Nephropathy (MENTOR). Nephron 2015;130(3):159–68. [DOI] [PubMed] [Google Scholar]
- 15.Beckwith H, Lightstone L. Rituximab in systemic lupus erythematosus and lupus nephritis. Nephron Clin Pract 2014;128(3–4):250–4. [DOI] [PubMed] [Google Scholar]
- 16.Madanchi N, Bitzan M, Takano T. Rituximab in Minimal Change Disease: Mechanisms of Action and Hypotheses for Future Studies. Can J Kidney Health Dis 2017;4:2054358117698667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y, Butkowski R, Burke B, Kleppel MM, Crosson J, Katz A, et al. Differential expression of basement membrane collagen in membranous nephropathy. Am J Pathol 1991;139(6):1381–8. [PMC free article] [PubMed] [Google Scholar]
- 18.Fervenza FC, Sethi S, Specks U. Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol 2008;3(3):905–19. [DOI] [PubMed] [Google Scholar]
- 19.Fervenza FC, Abraham RS, Erickson SB, Irazabal MV, Eirin A, Specks U, et al. Rituximab therapy in idiopathic membranous nephropathy: a 2-year study. Clin J Am Soc Nephrol 2010;5(12):2188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Regazzi MB, Iacona I, Avanzini MA, Arcaini L, Merlini G, Perfetti V, et al. Pharmacokinetic behavior of rituximab: a study of different schedules of administration for heterogeneous clinical settings. Ther Drug Monit 2005;27(6):785–92. [DOI] [PubMed] [Google Scholar]
- 21.Candelaria M, Gonzalez D, Fernandez Gomez FJ, Paravisini A, Del Campo Garcia A, Perez L, et al. Comparative assessment of pharmacokinetics, and pharmacodynamics between RTXM83, a rituximab biosimilar, and rituximab in diffuse large B-cell lymphoma patients: a population PK model approach. Cancer Chemother Pharmacol 2018;81(3):515–27. [DOI] [PubMed] [Google Scholar]
- 22.Golay J, Semenzato G, Rambaldi A, Foa R, Gaidano G, Gamba E, et al. Lessons for the clinic from rituximab pharmacokinetics and pharmacodynamics. MAbs 2013;5(6):826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zachwieja J, Silska M, Ostalska-Nowicka D, Soltysiak J, Lipkowska K, Blumczynski A, et al. Efficacy and safety of rituximab treatment in children with primary glomerulonephritis. J Nephrol 2012;25(6):1060–6. [DOI] [PubMed] [Google Scholar]
- 24.Mo CC, Njuguna N, Beum PV, Lindorfer MA, Vire B, Lee E, et al. Rapid clearance of rituximab may contribute to the continued high incidence of autoimmune hematologic complications of chemoimmunotherapy for chronic lymphocytic leukemia. Haematologica 2013;98(8):1259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Counsilman CE, Jol-van der Zijde CM, Stevens J, Cransberg K, Bredius RG, Sukhai RN. Pharmacokinetics of rituximab in a pediatric patient with therapy-resistant nephrotic syndrome. Pediatr Nephrol 2015;30(8):1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivas NR. Commonality of rituximab pharmacokinetic disposition in nephrotic syndrome and autoimmune cytopenias in chronic lymphocytic leukemia patients. Pediatr Nephrol 2016;31(2):335–6. [DOI] [PubMed] [Google Scholar]
- 27.Cravedi P, Ruggenenti P, Sghirlanzoni MC, Remuzzi G. Titrating rituximab to circulating B cells to optimize lymphocytolytic therapy in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 2007;2(5):932–7. [DOI] [PubMed] [Google Scholar]
- 28.Moroni G, Depetri F, Del Vecchio L, Gallelli B, Raffiotta F, Giglio E, et al. Low-dose rituximab is poorly effective in patients with primary membranous nephropathy. Nephrol Dial Transplant 2017;32(10):1691–6. [DOI] [PubMed] [Google Scholar]
- 29.Administration USFaD. Guidance for Industry. Pharmacokinetics in patients with impaired renal function. Study design, data analysis, and impact on dosing and labeling In: U.S. Department of Health and Human Services CfDEaR, Center for Biologics Evaluation and Research, editor. 1998.
- 30.Kamath AV. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov Today Technol 2016;21–22:75–83. [DOI] [PubMed] [Google Scholar]
- 31.Vugmeyster Y, Xu X, Theil FP, Khawli LA, Leach MW. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J Biol Chem 2012;3(4):73–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Challa DK, Velmurugan R, Ober RJ, Sally Ward E. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr Top Microbiol Immunol 2014;382:249–72. [DOI] [PubMed] [Google Scholar]
- 33.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn 2007;34(5):687–709. [DOI] [PubMed] [Google Scholar]
- 34.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology 1996;89(4):573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 2009;157(2):220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Passot C, Azzopardi N, Renault S, Baroukh N, Arnoult C, Ohresser M, et al. Influence of FCGRT gene polymorphisms on pharmacokinetics of therapeutic antibodies. MAbs 2013;5(4):614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J 2012;14(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mok CC. Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther 2013;8:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishna M, Nadler SG. Immunogenicity to Biotherapeutics - The Role of Anti-drug Immune Complexes. Front Immunol 2016;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fathallah AM, Bankert RB, Balu-Iyer SV. Immunogenicity of subcutaneously administered therapeutic proteins--a mechanistic perspective. AAPS J 2013;15(4):897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuriakose A, Chirmule N, Nair P. Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J Immunol Res 2016;2016:1298473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryman JT, Meibohm B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol 2017;6(9):576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, et al. Immunogenicity of Therapeutic Protein Aggregates. J Pharm Sci 2016;105(2):417–30. [DOI] [PubMed] [Google Scholar]