

Abstract
Background:
Acute lung injury (ALI) is characterized by an acute inflammatory process, and oxidative stress in the lung tissue leads to a lack of effective therapeutics. This study aimed to identify whether the overexpression of transcription factor EB (TFEB) regulates mitophagy to protect against lipopolysaccharide (LPS)-induced ALI.
Methods:
We detected the expression of inflammatory factors, cytochrome c (Cyt.c) and nicotinamide adenine dinucleotide phosphate (NADPH), and autophagy-related proteins and observed the changes in lung histopathology induced by ALI in rats and the changes in the cell ultrastructure of primary alveolar type II epithelial cells induced by changing the expression of TFEB in the context of ALI.
Results:
The overexpression of TFEB could reduce the expression of proinflammatory factors, such as IL-1 and IL-6, and increase the expression of anti-inflammatory factors, such as IL-10, both in vitro and in vivo. In addition, the overexpression of TFEB could reduce the Cyt.c and NADPH levels both in vivo and in vitro. The overexpression of TFEB could upregulate the expression of autophagy-related proteins, such as lysosomal-associated membrane protein 1 (LAMP1), microtubule-associated protein light chain 3B (LC3B), and Beclin both in vivo and in vitro, and promote mitochondrial autophagy. The overexpression of TFEB significantly improved the histopathologic changes induced by LPS-induced ALI in rats. However, low TFEB expression produced the opposite results.
Conclusion:
TFEB overexpression can decrease inflammation and mitochondrial damage in the lung tissue and alveolar epithelial cells through regulating mitochondrial autophagy to protect against LPS-induced ALI. Therefore, TFEB is likely a potential therapeutic target in LPS-induced ALI.
Keywords: Transcription factor EB, Mitochondrial autophagy, Acute lung injury, Inflammation, Mitochondrial damage
Introduction
Acute lung injury (ALI) is a life-threatening disease with high mortality and morbidity, which is characterized by an acute inflammatory process and oxidative stress in the pulmonary parenchyma and interstitial tissue, alveolar epithelial cell dysfunction,[1] pulmonary oxidative damage, and edema.[2] ALI will progress to Acute Respiratory Distress Syndrome (ARDS) if it is not effectively controlled in the early stage.[3] ARDS is a form of respiratory failure that exhibits higher mortality.[4] Thus, how to effectively treat ALI in the early stage is an important problem.
Autophagy plays a complex role in human diseases, as it is both protective and injurious.[5] Autophagy can protect against ALI through restricting injury to the lung microvascular barrier.[6] Mitochondrial damage can release vast amounts of oxidases that aggravate lung tissue injury in sepsis-induced diseases, such as lipopolysaccharide (LPS)-induced ALI and renal injury.[7] Mitochondrial autophagy is an important way to remove damaged mitochondria and a mechanism for self-preservation.[8] It is not known whether the upregulation of mitochondrial autophagy can protect against ALI.
Transcription factor EB (TFEB) is an important transcription factor in autophagy, which can directly regulate the expression of autophagy-associated proteins.[9] Therefore, we hypothesized that TFEB can regulate mitochondrial autophagy to protect against LPS-induced ALI. In the study, we demonstrated that TFEB regulates inflammatory factors and mitochondrial damage in ALI by regulating mitochondrial autophagy and the expression of autophagy-associated proteins. These findings suggest that TFEB might be a potential therapeutic target in ALI.
Methods
Animal and materials
Adult male Sprague-Dawley rats (180–220 g) were obtained from the Animal Center of the Air Force Military Medical University (Xi’an, China). Animal experiments were approved by the Animal Care and Use Committee of the Air Force Military Medical University and were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85-23, revised 1985). Anti-lysosomal-associated membrane protein 1 (LAMP1, ab24170), anti-Beclin (ab62557), anti-microtubule-associated protein light chain 3B (LC3B, ab63817), and anti-GAPDH (ab37168) antibodies were purchased from Abcam (Cambridge, UK). An anti-TFEB antibody (13372-1-AP) was purchased from Proteintech (Rosemont, IL, USA). Anti-SP-A (sc-13977) was purchased from Santa Cruz (Dallas, TX, USA). Enzyme-linked immunosorbent assay (ELISA) kits for IL-1, IL-6, and IL-10 were purchased from R&D Systems (Minneapolis, MN, USA). Nicotinamide adenine dinucleotide phosphate (NADPH) assay kits were purchased from Sigma Chemical Company (St. Louis, MO, USA). Cytochrome c (Cyt.c) assay (ELISA) kits were purchased from Solarbio Life Sciences (Beijing, China).
Alveolar type II epithelial cell isolation and culture
Primary alveolar type II epithelial cells were isolated from Sprague-Dawley rats as described in a previous article.[10] The isolated cells are resuspended to 106 cells/mL in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), then cultured for 12 h in noncoated cell culture plate in a humidified, 5% CO2 incubator at 37°C to remove residual mesenchymal cells attached to the plate. The unattached cells (mainly alveolar type II epithelial cells) are gently collected and centrifuged at 300 × g for 10 min at 4°C. The pellet is resuspended in DMEM with 10% FBS and cultured on a coated plate in 5% CO2–95% air at 37°C. The cells were identified by immunofluorescence staining. Steps were as follows. Slides of cells were fixed with 4% formaldehyde (15 min). After washed with PBS, cells were incubated with 10% normal goat serum and 5% Bull Serum Albumin (1 h), and then incubated with anti-SP-A overnight at 4°C in moist box. Cells were incubated with 594-Goat-anti-Rat for 30 min at indoor temperature. After washed with PBS, diisopropanolamine was added (5 min) and cells were subsequently examined under a fluorescence microscope.
Plasmids and lentivirus preparation
The TFEB gene product was cloned into the pCDH-CMV-MCS-EF1a-copGFP vector. A TFEB shRNA sequence was also cloned into the pCDH-CMV-MCS-EF1a-copGFP vector. The pLenti/EGFP transgene vector and the packaging plasmids pLP1 and pLP2 as well as the pLP and VSVG plasmids encoding the viral proteins pCDH-CMV-TFEB-EF1-copGFP and pCDH-CMV-shTFEB-copGFP, respectively, were prepared and transfected as previously described.[11]
Cell transfection and treatment
Primary alveolar type II epithelial cells were cultured in six-well culture dishes. There were four groups: the control group, LPS group, LPS + TFEB group (LPS + TFEB lentivirus), and LPS + shTFEB group (LPS + shTFEB lentivirus). After being challenged with 10 μg/mL LPS for 24 h, the cells were incubated in serum-free medium for 12 h. Then, the cells were infected with 20 μL TFEB lentivirus (virus titer: 7.5 × 108) or shTFEB lentivirus (virus titer: 3.4 × 108) for 48 h in DMEM supplemented with 10% FBS in 5% CO2–95% air at 37°C. Then, the cell supernatant and cell protein of each group was collected.
Cell ultrastructure
After challenge and infection, the primary alveolar type II epithelial cells from each group were collected into the AGAR empty slot in a centrifuge tube. Then, 2.5% glutaraldehyde was used to fix the fluid cells. Semithin (1-mm-thick) sections were cut from the AGAR empty slot, which included the fixed cells. The fixed cell sections were postfixed for 1 h in 1% osmium tetroxide and dehydrated in alcohol. The cells were examined under a TEM-100CX electron microscope (Japan Electron Optical Laboratory, Tokyo, Japan).
Animal model and grouping
There were four groups of rats: the control group, LPS group (LPS-induced ALI group), LPS + TFEB group (LPS-induced ALI + TFEB lentivirus), and LPS + shTFEB group (LPS-induced ALI + shTFEB lentivirus). There were five rats in each group. We established an ALI model through a tail vein injection (LPS, 10 mg/kg). The same volume of normal saline was injected via the tail vein in the control group. After 6 h the ALI model was established, rats were injected with 3% pentobarbital sodium 0.3 mL intraperitoneally with mild anesthesia. Then lentivirus suspension drops (5.0 × 108 transduction units per rat) were instilled into the rat lungs through the trachea intermittently. The control group was inhaled with the same volume of 0.9% normal saline. The rats were sacrificed by aortic transection at 48 h after lentivirus instillation. We demonstrated transfection efficiency via observing the expression of GFP in lung tissue under fluorescence microscope.
Preparation of the BALF
Forty-eight hours after the lentivirus was instilled, the rats were anesthetized. The left lung was lavaged with 1-mL ice-cold phosphate-buffered saline five times in each group. We recovered 90% of the bronchoalveolar lavage fluid (BALF). The collected BALF was centrifuged at 520 × g for 20 min at 4°C, and the supernatant was used for subsequent studies.
Western blotting
The lung tissue samples from each group were collected after the rats were perfused with pH 7.4 phosphate-buffered saline to remove the blood cells from the pulmonary circulation. Total protein extracted from the primary alveolar type II epithelial cells and lung tissue was prepared according to the instructions of a Total Protein Extraction Kit. Protein concentrations were determined through the BCA method. Protein samples were separated on a denaturing 8% or 6% Sodium Dodecyl Sulfonate-polyacrylamide gel and transferred to a nitrocellulose membrane, which was incubated with rabbit monoclonal antibodies overnight. The antibody concentrations were 1:1000 anti-LAMP1 antibody, 1:1000 anti-LC3B antibody, 1:500 anti-TFEB antibody, 1:1000 anti-Beclin antibody, and 1:2500 anti-GAPDH antibody. The secondary antibody (anti-rabbit, 1:5000) was incubated for 1.5 h. Detection was performed using an enhanced chemiluminescence system (Amersham, Arlington Heights, IL, USA).
ELISA
The levels of IL-1, IL-6, IL-10, Cyt.c, and NADPH in the cell supernatant and BALF were determined by using commercially available ELISA kits according to the manufacturer's instructions.
Histopathological sampling and scoring
At 48 h after lentivirus instillation, all groups of rats were sacrificed by aortic transection. The lung tissue was removed and fixed with 4% paraformaldehyde for 24 h. The lower lobe of each right lung was embedded in paraffin and cut into 5 μm sections. Hematoxylin and eosin staining was performed according to a standard protocol.
For scoring, five samples were cut into two sections in each group so that the total number of sections per group was 10. The sections were examined by two independent pathologists who were blinded to the group assignments. Each section was evaluated for the lung injury score described in a previous standard assessment,[12] which ranged from 0 to 4 based on edema, neutrophil infiltration, hemorrhage, bronchiole epithelial desquamation, and hyaline membrane formation. The score ranged from 0 to 4 and represented the ALI severity: 0 for no injury, 1 for modest injury, 2 for intermediate injury, 3 for widespread injury, and 4 for the most prominent injury.
Statistical analysis
Data are expressed as the mean ± standard error. Statistical analysis was performed with one-way analysis of variance (ANOVA) followed by Dunnett test for multiple comparisons. A statistically significant difference was defined as P < 0.05.
Results
Effects of TFEB on inflammatory factors in LPS-induced ALI
The results showed that compared with the corresponding LPS groups, the LPS + TFEB groups showed reduced expression of IL-1 (cell supernatant level reduced from 118.47 to 0.49, P < 0.05, n = 3; BALF level reduced from 43.69 to 0.65, P < 0.05, n = 5) and IL-6 (cell supernatant level reduced from 25.75 to 4.66, P < 0.05, n = 3; BALF level reduced from 18.94 to 3.10, P < 0.05, n = 5) and increased expression of IL-10 (cell supernatant level increased from 9.76 to 32.79, P < 0.05, n = 3; BALF level increased from 9.23 to 11.41, P < 0.05, n = 5). However, compared with the corresponding LPS groups, the LPS + shTFEB groups exhibited increased expression of IL-1 (cell supernatant level increased from 118.47 to 606.41, P < 0.05, n = 3; BALF level increased from 27.66 to 323.70, P < 0.05, n = 5) and IL-6 (cell supernatant level increased from 25.75 to 46.59, P < 0.05, n = 3; BALF level increased from 18.94 to 42.28, P < 0.05, n = 5) and reduced expression of IL-10 (cell supernatant level decreased from 9.76 to 6.03, P < 0.05, n = 3; BALF level decreased from 9.23 to 7.22, P < 0.05, n = 5) [Figure 1].
Figure 1.
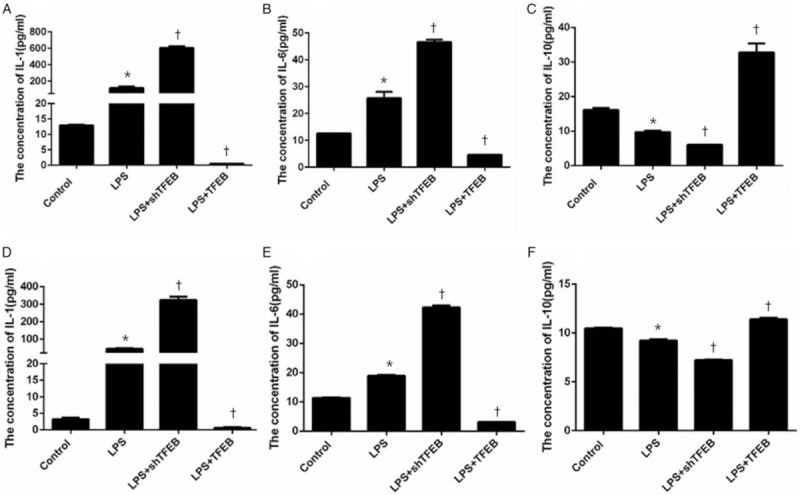
Effects of TFEB on the inflammatory factors in the cell supernatant (A–C) and in the BALF (D–F). The overexpression of TFEB could reduce the expression of IL-1 and IL-6 and increase the expression of IL-10. The knockdown of TFEB expression could increase the expression of IL-1 and IL-6 and reduce the expression of IL-10. Data are presented as the mean ± SEM (n = 3 for the cell supernatant, and n = 5 for the BALF). ∗P < 0.05 vs. control group; †P < 0.05 vs. LPS group. BALF: Bronchoalveolar lavage fluid; LPS: Lipopolysaccharide; TFEB: Transcription factor EB.
Effects of TFEB on mitochondrial damage in LPS-induced ALI
As shown in Figure 2, compared with the corresponding LPS groups, the LPS + TFEB groups exhibited decreased levels of Cyt.c (cell supernatant level decreased from 19.85 to 8.15, P < 0.05, n = 3; BALF level decreased from 21.41 to 5.79, P < 0.05, n = 5) and NADPH (cell supernatant level decreased from 129.00 to 57.78, P < 0.05, n = 3; BALF level decreased from 138.11 to 74.78, P < 0.05, n = 5), whereas the LPS + shTFEB groups displayed increased levels of Cyt.c (cell supernatant level increased from 19.85 to 37.05, P < 0.05, n = 3; BALF level increased from 21.41 to 40.40, P < 0.05, n = 5) and NADPH (cell supernatant level increased from 129.00 to 165.89, P < 0.05, n = 3; BALF level increased from 138.11 to 165.56, P < 0.05, n = 5).
Figure 2.
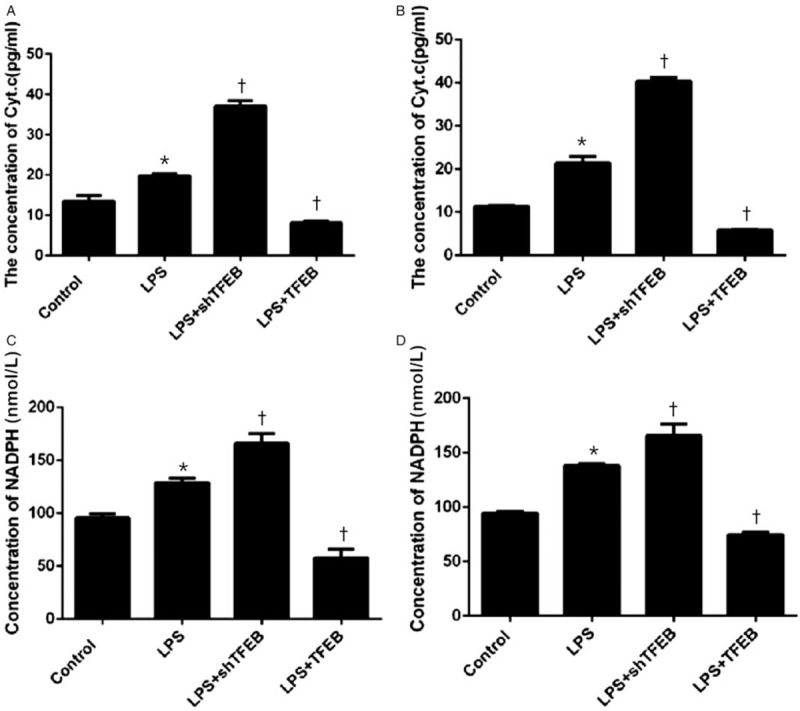
Effects of TFEB on Cyt.c (A, B) and NADPH (C, D) expression in the cell supernatant (A, C) and in the BALF (B, D). The overexpression of TFEB could reduce the expression of Cyt.c and NADPH. The knockdown of TFEB expression could increase the expression of Cyt.c and NADPH. Data are presented as the mean ± SEM (n = 3 for the cell supernatant, and n = 5 for the BALF). ∗P < 0.05 vs. control group; †P < 0.05 vs. LPS group. BALF: Bronchoalveolar lavage fluid; Cyt.c: Cytochrome c; LPS: Lipopolysaccharide; NADPH: Nicotinamide adenine dinucleotide phosphate; TFEB: Transcription factor EB.
Effects of TFEB on mitochondrial autophagy-related proteins in LPS-induced ALI
We measured the expression of mitochondrial autophagy-related proteins, such as LAMP1, Beclin, and LC3B. The results showed that the protein expression of LAMP1, Beclin, and LC3B was obviously increased in the LPS + TFEB groups but decreased in the LPS + shTFEB groups compared with the corresponding LPS groups in vitro and in vivo [Figure 3].
Figure 3.

Effects of TFEB on mitochondrial autophagy-related proteins in primary alveolar type II epithelial cells (A) and in LPS-induced acute lung injury in rats (B). LAMP-1: lysosomal-associated membrane protein 1; LC3B: Microtubule-associated protein light chain 3B; LPS: Lipopolysaccharide; TFEB: Transcription factor EB.
Effects of TFEB on histopathology in LPS-induced ALI
Histopathologic staining showed that compared with control, LPS induced lung edema, inflammatory cell infiltration in the pulmonary mesenchyme and alveoli, hemorrhage, bronchiole epithelia desquamation, and hyaline membrane appearance [Figure 4B]. Compared with the LPS group, the LPS + TFEB groups showed obvious improvements in lung edema, inflammatory cell infiltration, and hemorrhage [Figure 4C]. In the LPS + shTFEB group, the inflammatory cell infiltration was more serious than that in the LPS group [Figure 4D]. The assigned lung injury scores of every group are shown in Table 1.
Figure 4.
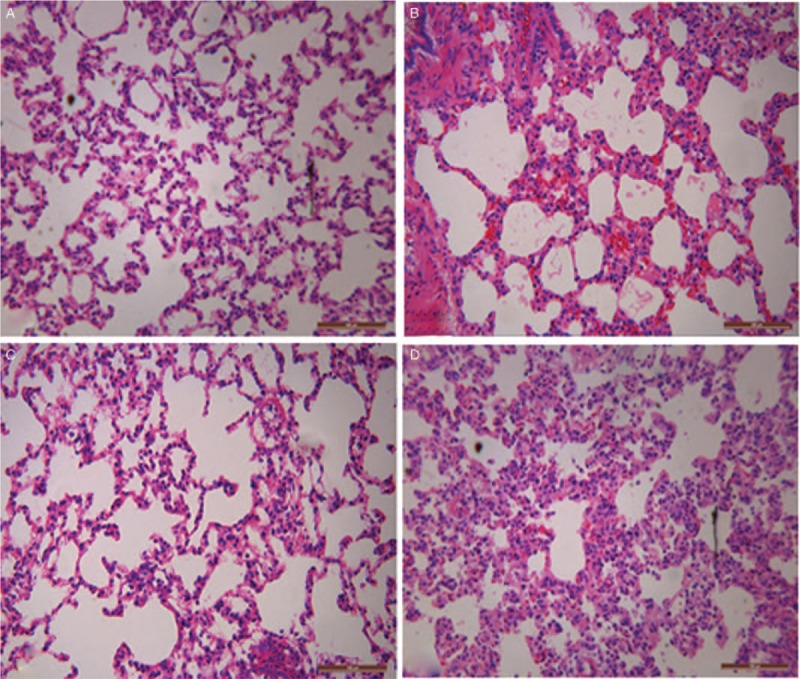
Effects of TFEB on histopathology in LPS-induced acute lung injury. (A) Control group; (B) LPS group; (C) LPS + TFEB group; (D) LPS + shTFEB group (hematoxylin and eosin staining, original magnification ×200. LPS: Lipopolysaccharide; TFEB: Transcription factor EB.
Table 1.
Effects of TFEB on the assigned lung injury scores in LPS-induced ALI.

Effects of TFEB on cell ultrastructure
To further clarify the effects of TFEB on mitochondrial autophagy, we observed the cell ultrastructure of primary alveolar type II epithelial cells using electron microscopy. In the control group, the cells showed normal ultrastructure. In the LPS group, the autophagosome reactivity was stronger than that of the control group. There were more autophagosomes in the LPS + TFEB group than those in the LPS group. Compared with the LPS group, the LPS + shTFEB group exhibited decreased autophagosome accumulation [Figure 5].
Figure 5.
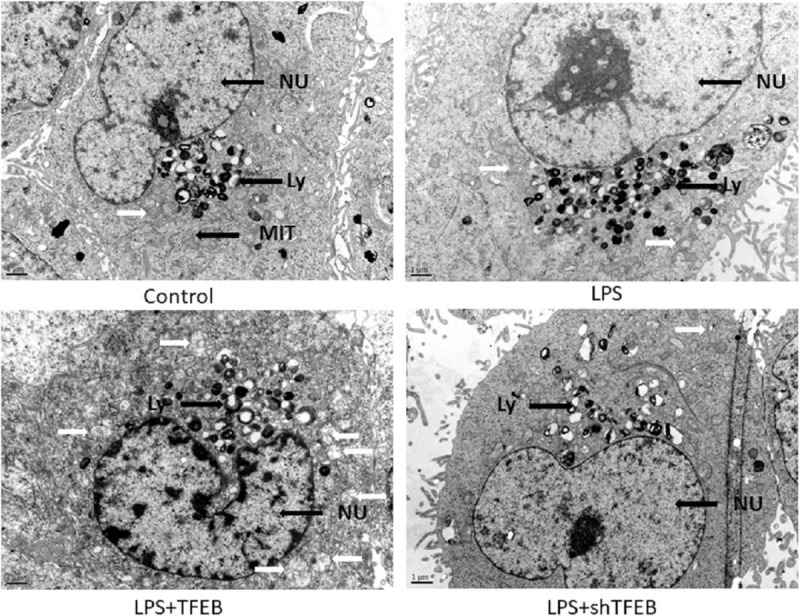
Effects of TFEB on the cell ultrastructure of primary alveolar type II epithelial cells. White arrows indicate autophagosomes (×1000). Ly: Lysosome; MIT: Mitochondria; NU: Cell nucleus; TFEB: Transcription factor EB.
Discussion
In this study, we demonstrated that TFEB regulated mitochondrial autophagy to protect against LPS-induced ALI in rats. The overexpression of TFEB could reduce the expression of proinflammatory factors, such as IL-1 and IL-6, and increase the expression of anti-inflammatory factors, such as IL-10, both in vitro and in vivo. The overexpression of TFEB could upregulate the expression of autophagy-related proteins, such as LAMP1, LC3B, and Beclin; reduce the expression of Cyt.c and NADPH both in vitro and in vivo; and significantly increase the accumulation of autophagosomes in alveolar type II epithelial cells. The overexpression of TFEB significantly improved the histopathologic changes associated with LPS-induced ALI in rats. However, knocking down the expression of TFEB had opposite results.
ALI is characterized by noncardiogenic pulmonary edema with alveolar epithelial cell and pulmonary mesenchyme inflammation and oxidative stress.[13,14] The main treatment strategies for ALI include ventilation,[4] anti-inflammatory,[15] antioxidant,[14] and mesenchymal stem cell therapies[16]; however, none of these treatment strategies is the most effective. Therefore, in the present study, we focused on a potentially effective treatment for ALI. The mitochondria are the only organelles containing DNA other than the nucleus,[17] so they are very sensitive to oxidative stress and liable to be damaged.[18] When the mitochondria are damaged in ALI, vast amounts of oxidases are released[19]; excessive oxidase release then leads to more serious oxidative stress and mitochondrial damage.[8,20] When the mitochondria are damaged, mitochondrial respiratory chain products, such as Cyt.c and NADPH, are released.[21–23] Therefore, the increases in the levels of Cyt.c and NADPH are signs of mitochondrial damage. Mitophagy is the mechanism of mitochondrial damage self-repair.[8,24,25] Upregulating mitophagy decreases the levels of mitochondrial respiratory chain products, such as Cyt.c and NADPH.[18,26] Therefore, Cyt.c and NADPH are the biomarkers of mitophagy. In this study, we found that the Cyt.c and NADPH levels were significantly increased in the LPS group and significantly decreased in the LPS + TFEB group.
Some studies found that upregulated autophagy can decrease mitochondrial damage and lung inflammation.[27] TFEB was the first identified member of the MiTF/TFE family, which include of TFEB, TFE3, MITF, and TFEC,[28] and plays a pivotal role in the regulation of lysosomal biogenesis and autophagy.[29] However, it is not known whether upregulating TFEB expression will improve mitochondrial autophagy and protect against ALI. In our study, we observed that the overexpression of TFEB could promote autophagy in damaged mitochondria under an electron microscope. We also found that the overexpression of TFEB could upregulate the expression of autophagy-related proteins and decrease inflammation and Cyt.c and NADPH expression, whereas knocking down the expression of TFEB exerted opposite effects. Thus, TFEB likely regulates mitochondrial damage by regulating autophagy-related proteins.
In conclusion, upregulated mitochondrial autophagy can decrease inflammation and mitochondrial damage in LPS-induced ALI. The overexpression of TFEB can upregulate mitochondrial autophagy through upregulating autophagy-related protein expression to protect against LPS-induced ALI. Thus, TFEB might be a potential therapeutic target in LPS-induced ALI.
Funding
This study was supported by a grant from the Natural Science Foundation of China (No. 81600053).
Conflicts of interest
None.
Footnotes
How to cite this article: Liu W, Li CC, Lu X, Bo LY, Jin FG. Overexpression of transcription factor EB regulates mitochondrial autophagy to protect lipopolysaccharide-induced acute lung injury. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000243
References
- 1.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 2009; 77:1763–1772. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhargava M, Wendt CH. Biomarkers in acute lung injury. Transl Res 2012; 159:205–217. doi: 10.1016/j.trsl.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cross L, Matthay MA. Biomarkers in acute lung injury: insights into the pathogenesis of acute lung injury. Crit Care Clin 2011; 27:355–377. doi: 10.1016/j.ccc.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA 2018; 319:698–710. doi: 10.1001/jama.2017.21907. [DOI] [PubMed] [Google Scholar]
- 5.Mizumura K, Cloonan SM, Haspel JA, Choi A. The emerging importance of autophagy in pulmonary diseases. Chest 2012; 142:1289–1299. doi: 10.1378/chest.12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D, Zhou J, Ye LC, Li J, Wu Z, Li Y, et al. Autophagy maintains the integrity of endothelial barrier in LPS-induced lung injury. J Cell Physiol 2018; 233:688–698. doi: 10.1002/jcp.25928. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houten BV, Hunter SE, Meyer JN. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front Biosci (Landmark Ed) 2016; 21:42–54. doi:10.2741/4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 10.Sun F, Xiao G, Qu Z. Isolation of murine alveolar type II epithelial cells. Bio Protoc 2017; 7.doi: 10.21769/BioProtoc.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SH, Jun HJ, Jang SI, You JC. The determination of importance of sequences neighboring the Psi sequence in lentiviral vector transduction and packaging efficiency. PLoS One 2012; 7:e50148.doi: 10.1371/journal.pone.0050148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou ZH, Sun B, Lin K, Zhu LW. Prevention of rabbit acute lung injury by surfactant, inhaled nitric oxide, and pressure support ventilation. Am J Respir Crit Care Med 2000; 161:581–588. doi:10.1164/ajrccm.161.2.9901048. [DOI] [PubMed] [Google Scholar]
- 13.Dias-Freitas F, Metelo-Coimbra C, Roncon-Albuquerque RJ. Molecular mechanisms underlying hyperoxia acute lung injury. Respir Med 2016; 119:23–28. doi: 10.1016/j.rmed.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal 2007; 9:1991–2002. doi: 10.1089/ars.2007.1785. [DOI] [PubMed] [Google Scholar]
- 15.Huang X, Xiu H, Zhang S, Zhang G. The role of macrophages in the pathogenesis of ALI/ARDS. Mediators Inflamm 2018; 2018:1264913.doi: 10.1155/2018/1264913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antebi B, Mohammadipoor A, Batchinsky AI, Cancio LC. The promise of mesenchymal stem cell therapy for acute respiratory distress syndrome. J Trauma Acute Care Surg 2018; 84:183–191. doi: 10.1097/TA.0000000000001713. [DOI] [PubMed] [Google Scholar]
- 17.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 2014; 15:634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su SH, Wu YF, Wang DP, Hai J. Inhibition of excessive autophagy and mitophagy mediates neuroprotective effects of URB597 against chronic cerebral hypoperfusion. Cell Death Dis 2018; 9:733.doi: 10.1038/s41419-018-0755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J, Shi J, Wang D, Dong S, Zhang Y, Wang M, et al. Heme oxygenase-1/carbon monoxide-regulated mitochondrial dynamic equilibrium contributes to the attenuation of endotoxin-induced acute lung injury in rats and in lipopolysaccharide-activated macrophages. Anesthesiology 2016; 125:1190–1201. doi: 10.1097/ALN.0000000000001333. [DOI] [PubMed] [Google Scholar]
- 20.Kuck JL, Obiako BO, Gorodnya OM, Pastukh VM, Kua J, Simmons JD, et al. Mitochondrial DNA damage-associated molecular patterns mediate a feed-forward cycle of bacteria-induced vascular injury in perfused rat lungs. Am J Physiol Lung Cell Mol Physiol 2015; 308:L1078–L1085. doi: 10.1152/ajplung.00015.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng H, Li T, Tian J, Zhang LL. TUBP1 protein lead to mitochondria-mediated apoptotic cell death in Verticillium dahliae. Int J Biochem Cell Biol 2018; 103:35–44. doi: 10.1016/j.biocel.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med 2018; 117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Yan X, Yang D, Zhou J, Song J, Yang D. Rapamycin attenuates mitochondrial injury and renal tubular cell apoptosis in experimental contrast-induced acute kidney injury in rats. Biosci Rep 2018; 38:pii: BSR20180876.doi: 10.1042/BSR20180876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang D, Li C, Zhou J, Song Y, Fang X, Ou J, et al. Autophagy protects against ischemia/reperfusion-induced lung injury through alleviating blood-air barrier damage. J Heart Lung Transplant 2015; 34:746–755. doi: 10.1016/j.healun.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 2016; 311:L433–L452. doi: 10.1152/ajplung.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng J, Chen X, Lu S, Li W, Yang D, Su W, et al. Naringin attenuates cerebral ischemia-reperfusion injury through inhibiting peroxynitrite-mediated mitophagy activation. Molecular Neurobiology 2018; 55:9029–9042. doi: 10.1007/s12035-018-1027-7. [DOI] [PubMed] [Google Scholar]
- 27.Jurkuvenaite A, Benavides GA, Komarova S, Doran SF, Johnson M, Aggarwal S, et al. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med 2015; 85:83–94. doi: 10.1016/j.freeradbiomed.2015.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 2016; 32:255–278. doi: 10.1146/annurev-cellbio-111315-125407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci 2016; 129:2475–2481. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]