

Abstract
Background:
The increased permeability of the blood-brain barrier (BBB) induced by ischemia/hypoxia is generally correlated with alteration of tight junctions (TJs). DL-3-n-butylphthalide (NBP) has been shown to exert neuroprotective effects after ischemic injury. However, few studies have assessed the correlation between NBP and TJs. This study aimed to investigate the potential effect of NBP on the TJ proteins claudin-5, zonula occludens-1 (ZO-1), and occludin during brain ischemia.
Methods:
A chronic cerebral hypoperfusion (CCH) Sprague-Dawley rat model was established, and NBP (20, 40, or 80 mg/kg, gavage, once a day) treatment was performed for 14 days. NBP (0.1 or 1.0 μmol/L) pre-treatment was applied to an in vitro hypoxia microvascular endothelial cell model (1% O2, 24 h). BBB permeability was assessed by performing the Evans blue assay. The expressions and localization of claudin-5, ZO-1, occludin, phosphorylated/total protein kinase B (p-Akt/Akt), phosphorylated/total glycogen synthase kinase 3β (GSK-3β)/GSK-3β, and β-catenin/β-actin were evaluated by Western blotting or immunofluorescence. Reactive oxygen species (ROS) generation was measured by flow cytometry analysis. TJ ultrastructure was observed by transmission electron microscopy.
Results:
In CCH rats, treatment with 40 and 80 mg/kg NBP decreased the Evans blue content in brain tissue (9.0 ± 0.9 μg/g vs. 12.3 ± 1.9 μg/g, P = 0.005; 6.7 ± 0.6 μg/g vs. 12.3 ± 1.9 μg/g, P < 0.01), increased the expression of claudin-5 (0.79 ± 0.08 vs. 0.41 ± 0.06, P < 0.01; 0.97 ± 0.07 vs. 0.41 ± 0.06, P < 0.01), and elevated the ZO-1 protein level (P < 0.05) in brain microvascular segments in a dose-dependent manner in comparison with the corresponding values in the model group. There was no significant difference in occludin expression (P > 0.05). In the hypoxia cell model, NBP pre-treatment improved TJ ultrastructure, decreased intracellular ROS level, and increased the expression of claudin-5 (P < 0.01) and ZO-1 (P < 0.01) in comparison with the corresponding values in the hypoxia group. NBP treatment also elevated the relative expression levels of p-Akt/Akt, p-GSK-3β/GSK-3β, and β-catenin/β-actin in comparison with the corresponding values in the hypoxia group (all P < 0.05).
Conclusion:
NBP improves the barrier function of BBB against ischemic injury by upregulating the expression of TJ proteins, possibly by reducing oxidative stress and activating the Akt/GSK-3β/β-catenin signaling pathway.
Keywords: DL-3-n-butylphthalide, Blood-brain barrier, Tight junctions, Ischemia, Claudin-5
Introduction
Brain ischemia is one of the leading causes of human neurological disability. In addition to causing neuron damage, brain ischemia usually increases the permeability of the blood-brain barrier (BBB). The BBB is a diffusion barrier between capillaries and neurons that restricts the blood-brain passage of ions, water, solutes, and large molecular proteins, thus contributing to brain homeostasis. The passage of molecules through BBB depends on their structure, surface properties, and chemical composition, allowing only low molecular weight (MW) (<400) and small lipophilic molecules to enter the brain with several folds of greater competence than large molecules.[1] During the period of brain ischemia and the subsequent reperfusion, disruptions in the BBB result in an increase in vascular-derived substances in the brain and aggravate neuronal injury. As such, drugs preventing the breakdown of BBB tend to improve the outcomes of brain ischemia. The BBB is composed of specialized brain microvascular endothelial cells (BMECs), astrocyte endfeet, pericytes, and the extracellular matrix, and the first defense between the systemic circulation and the central nervous system is BMECs. One characteristic distinguishing BMECs from peripheral cells is the presence of tight junctions (TJs).
TJs are intercellular molecular binding systems that restrict paracellular flux of hydrophilic molecules across the capillary endothelial cells. The TJs between capillary endothelial cells are maintained by cytoplasmic proteins such as zonula occludens (ZO)-1, ZO-2, and ZO-3, and transmembrane proteins such as claudins, occludin, and junction adhesion molecules. Claudin-5, ZO-1, and occludin are the most important proteins in TJ complexes. Several studies have shown that the increased permeability of BBB induced by hypoxia generally correlates with alterations of claudin-5, ZO-1, and occludin.[2–4] Many signaling mediators may be involved in BBB disruption by modulating the expression of TJ proteins, but the exact mechanism remains unclear. Zuo et al[5] suggested that the expression of claudins and occludin seems to be influenced by glycogen synthase kinase 3β (GSK-3β). Lengfeld et al[6] demonstrated that the Wnt/β-catenin signaling pathway controlled the development of BBB and enhanced the BBB phenotype by increasing the expression of claudins and ZO-1. Activation of the Wnt/β-catenin signaling pathway by an inhibitor of GSK-3β could increase the expression of β-catenin, claudin-3, and ZO-1, attenuating BBB disruption.[7]
DL-3-n-butylphthalide (NBP), which was first extracted from the seeds of Apium graveolens Linn., Chinese celery, is a lipophilic compound with low MW (190.24) and also a powerful natural free radical scavenger. Multicenter clinical trials have shown that NBP is effective in improving neurological function in patients with brain ischemia and exhibits good safety. In several animal models, NBP has been proven to protect neuronal function against ischemic stroke. The underlying mechanisms may include inhibition of the inflammatory response, reduction of oxidative impairment by activation of the phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) signaling pathway, amelioration of mitochondrial function, and improvement in energy metabolism.[8–10] Our previous studies proved that NBP improved cognitive impairment in rats with chronic cerebral hypoperfusion (CCH) and in mice with repeated cerebral ischemia-reperfusion.[9,11] However, the previous studies mainly focused on the direct effect of NBP on neurons or angiogenesis in the advanced stages of brain ischemia, and little is known about the effect of NBP on TJs of endothelial cells. Since disruption of the BBB is one of the most important pathophysiological events contributing to ischemia injury and TJs are the very first structures to be injured, we hypothesized that NBP may exert its protective effect by affecting the expression and distribution of TJs in brain ischemia. To test this hypothesis, we investigated the effect of NBP on the expression and localization of claudin-5, ZO-1, and occludin in a CCH rat model. An in vitro study was also conducted in primary cultured BMECs to investigate the potential role of NBP on TJ disruption induced by hypoxia or oxygen-glucose deprivation (OGD)/reoxygenation (OGD/R). We further discuss whether the effect of NBP on TJs is related to the activation of the Akt/GSK-3β/β-catenin signaling pathway.
Methods
Ethical approval
All animal experimental procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of Hebei General Hospital.
Animal experiments
Adult male Sprague-Dawley rats weighing 230 to 280 g were purchased from the Laboratory Animal Center of Hebei Medical University. All animals were housed in an environmentally controlled room at a temperature between 20 and 23°C with a 12-h/12-h light-dark cycle and provided with free access to food and water.
The global cerebral ischemia model of CCH was induced by permanent occlusion of the bilateral common carotid arteries (BCCAO), as previously described.[12] Briefly, after anesthesia with pentobarbital sodium (50 mg/kg, intraperitoneal injection), both common carotid arteries were ligated and cut after being separated carefully from the vagus nerve through an incision in the middle of the neck. The rats were divided into five groups (n = 12 for each group) using a random number table: rats that underwent threading without actual ligation served as the sham group; rats that underwent threading and ligation of the carotid arteries served as the model groups, with rats treated with NBP 20, 40, and 80 mg/kg after threading and ligation for 14 days serving as the NBP-L (20 mg/kg), NBP-M (40 mg/kg), and NBP-H (80 mg/kg) groups. NBP (lot number: 518141001) was provided by Shijiazhuang Pharmaceutical Group NBP Pharmaceutical Co., Ltd (Shijiazhuang, Hebei, China). The obtained NBP was dissolved in 1.5 mL corn oil and administered by oral gavage once a day. Rats in the model and sham groups received the same volume of corn oil as placebo at the same time. The NBP/corn oil treatments were administered for 14 consecutive days until the rats were sacrificed.
Evans blue dye leakage assay
The integrity of the BBB was evaluated with a modified Evans blue extravasation method as previously reported.[13] After anesthesia with pentobarbital sodium (50 mg/kg, intraperitoneal injection), 37°C warmed Evans blue dye (2%, 4 mL/kg) was injected into the femoral vein and allowed to circulate for 2 h. This was followed by perfusion with phosphate-buffered saline (PBS) via the aorta to wash out any remaining dye in the blood vessels. Briefly, brains were extracted and the hemispheres of the brain were separated along the sagittal suture. Then, both hemispheres were weighed and homogenized. After the brain tissues were homogenized and centrifuged, the Evans blue dye content in the supernatant was measured by spectrophotometry at 620 nm (Thermo Multiskan FC, Thermo Fisher Scientific, Waltham, MA, USA). The concentration of Evans blue dye was expressed as microgram per gram of brain tissue weight (μg/g).
Immunohistochemical assay
The brain hemisphere tissues were fixed with 4% formalin and embedded in paraffin with standard techniques. Paraffin specimens were cut at a thickness of 5 μm. Immunohistochemical detection of TJ proteins was performed. In brief, after dewaxing, antigen retrieval, and endogenous peroxidase blockage, sections were incubated with diluted claudin-5 (1:100, 35-2500, Invitrogen Inc., Rockford, IL, USA), ZO-1 (1:100, 40-2200, Invitrogen Inc.), and occludin (1:200, 33-1500, Invitrogen Inc.) antibodies overnight at 4°C. Negative controls were obtained by incubating samples with PBS instead of the primary antibodies. After staining with diaminobenzidine and counterstaining with hematoxylin, images were observed and photographed in randomly chosen microscopic fields (ECLIPSE Ni-U, Nikon, Tokyo, Japan).
Primary culture of the rat BMECs
Rat BMECs were isolated from the brain cortex of newborn Sprague-Dawley rats (7–10 days) as previously described, with some modifications.[14] The whole brain was dissected and put into precooled D-Hanks’ solution, and the meninges and blood vessels were carefully removed on ice. The isolated brain tissue was digested in 15% dispase II (Roche, 04942078001, Indianapolis, IN, USA) at 37°C for 10 min, centrifuged at 800 × g at 4°C for 5 min, and the supernatant was discarded. Next, 15% dextran was added and mixed thoroughly, and the suspension was centrifuged at 4500 × g at 4°C for 10 min. The nerve tissue and blood vessels in the upper layer were removed into a new centrifuge tube, after which 1 mL of collagenase and disperse enzyme solution was added and the tissue was digested at 37°C for 20 min. Next, 10 μL of the digestive juice was placed on glass slides and the bead-like vascular segment was observed under a microscope. An equal amount of complete DMEM/F12 medium was added to terminate digestion and the solution was centrifuged. The isolated primary BMECs were seeded into 6-well culture plates in DMEM/F12 medium (GIBCO, Beijing, China) with 20% fetal bovine serum, 15 μg/mL endothelial cell growth supplement (Millipore, 02-102, Temecula, CA, USA), and 1% L-glutamine at 5% CO2 and 37°C. Rat BMECs within 3 to 5 passages were used in this study.
BMECs hypoxia or OGD/R models and NBP treatments
We established two BMEC injury models in vitro: the hypoxia model and the OGD/R model. In the hypoxia model, as previously reported,[15] confluent BMECs were transferred into a three-gas (94% N2, 5% CO2, 1% O2) incubator (Thermo 3131, Marietta, CH, USA) in glucose-containing DMEM and exposed to this hypoxic condition for 24 h. In the OGD/R model, confluent rat BMECs were rinsed twice with PBS and then placed into a three-gas (94% N2, 5% CO2, 1% O2) incubator in glucose-free DMEM (GIBCO, Grand Island, NY, USA) for 6 h. After 6 h of OGD, the BMECs regained oxygen and glucose supplies when they were shifted to a normal aerobic environment with fresh DMEM for 18 h.
BMECs in the control group were treated with the same procedure used for the two different models but in glucose-containing DMEM and a normoxic incubator. In the NBP group, BMECs were pre-treated with 0.1 or 1.0 μmol/L NBP for 2 h before 24 h of hypoxia or OGD/R, which was then followed by the same procedure as in the hypoxia or in OGD/R group with the same concentration of NBP. NBP was dissolved in dimethyl sulfoxide (DMSO), and the final concentration of DMSO in the medium was 0.01%.
Reactive oxygen species (ROS) detection
ROS generation was observed and measured by using a 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime Institute of Biotechnology, S0033, Shanghai, China) fluorescence probe. Flow cytometry analysis was carried out on a flow cytometer (Beckman Coulter Epics XL, Fullerton, CA, USA) equipped with 488-nm lasers and an appropriate set of detectors and filters. The level of intracellular ROS generation was determined after incubation with 1 mL DCFH-DA (1:1000) in basal medium without serum and phenol red for 30 min at 37°C. At least 10,000 gated events were collected for each sample. Data were analyzed using EXPO032ADC Analysis and CXP flow cytometry analysis programs.
In addition, cells were loaded with DCFH-DA at 10 μmol/L in the dark for 10 min, and then washed with PBS. Cell fluorescence was observed under a fluorescence microscope at an excitation wavelength of 488 nm and emission wavelength of 525 nm (DMI3000 B, Leica Microsystems, Wetzlar, Germany). Images were observed and photographed in randomly chosen microscopic fields by an inverted microscope.
Transmission electron microscopy
The confluent rat BMECs were subjected to hypoxia for 24 h with or without NBP treatment. Then, BMECs were harvested by using a cell scraper at designated time points and fixed with 4% glutaraldehyde at room temperature overnight. For transmission electron microscopy, the specimens were prepared in accordance with the conventional methods. The specimens were observed under a transmission electron microscope (Hitachi H-7500, Tokyo, Japan).
Immunofluorescence staining
The cells in each group were plated on coverslips, harvested at designated time points, and fixed with 4% paraformaldehyde (for ZO-1 and occludin) or methanol (for claudin-5). Samples were permeabilized with 0.5% Triton X-100 in PBS for 15 min and blocked with goat serum for 15 min at room temperature. The primary antibody (ZO-1 1:200, claudin-5 1:100, and occludin 1:100) was applied at 4°C for 24 h, followed by incubation with Cy3-conjugated secondary antibody (Abbkine, Redlands, CA, USA) at a concentration of 1:100 for 2 h at room temperature in the dark. Cell nuclei were stained with 4′, 6-diamidino-2-phenylindole. Images were observed and photographed in randomly chosen microscopic fields by an inverted microscope.
Western blotting analysis
Brain microvascular segments were isolated from brain tissues by density gradient centrifugation with 15% dextran and stored at −80°C until needed. TGN lysis buffer (HEPES 50.0 mmol/L, NP-40 0.5%, Tween-20 1.0%, PMSF 1.0 mmol/L, NaF 1.0 mmol/L, NaVO3 1.0 mmol/L) was used for extraction of proteins from cultured cells and microvascular tissues of the rat brain. After blocking with 5% milk, blotting membranes were incubated overnight at 4°C with the primary antibodies for ZO-1 (1:1000), claudin-5 (1:1000), occludin (1:500), Akt (1:2000, 4685, Cell Signaling, Danvers, MA, USA), p-Akt (Ser473) (1:2000, 9271S, Cell Signaling), GSK-3β (1:1000, 9315, Cell Signaling), p-GSK-3β (Ser9) (1:1000, 9336S, Cell Signaling), β-catenin (1:1000, 8480T, Cell Signaling), and β-actin (1:1000, 4970S, Cell Signaling). The membranes were then washed, incubated with secondary antibodies, and processed with an enhanced chemiluminescent reagent kit (Thermo Fisher Scientific). The relative levels of the target proteins were analyzed by a densitometric method. The relative levels of claudin-5, ZO-1, occludin, and β-catenin were normalized to that of β-actin. The p-Akt and p-GSK-3β levels were normalized to those of total Akt and GSK-3β, respectively.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 Statistics software (SPSS Inc., Chicago, IL, USA). Measurement data were presented as mean ± standard deviation values. Differences between groups were evaluated using one-way analysis of variance, and the Tukey post hoc test was performed for comparisons between groups. A value of P < 0.05 was considered statistically significant.
Results
NBP decreased BBB permeability in rat models of CCH
The permeability of the BBB was quantitatively determined by considering extravasation of Evans blue dye as a marker of albumin extravasation. Rats that underwent BCCAO surgery in the model group showed significantly increased leakage of Evans blue dye in comparison with the sham group (12.3 ± 1.9 μg/g vs. 4.7 ± 0.8 μg/g, P < 0.01) [Figure 1A]. This result indicated that after 14 days of CCH, BBB permeability was significantly increased in the model group. However, with NBP treatment, decreased Evans blue dye contents were observed in the NBP-M group (9.0 ± 0.9 μg/g vs. 12.3 ± 1.9 μg/g, P < 0.01) and NBP-H groups (6.7 ± 0.6 μg/g vs. 12.3 ± 1.9 μg/g, P < 0.01) in comparison to that of the model group. There were significant differences between the NBP-M and NBP-L groups (9.0 ± 0.9 μg/g vs. 6.7 ± 0.6 μg/g, P = 0.03), which indicated that NBP could improve the barrier function of BBB in a dose-dependent manner. The result suggests that NBP attenuates ischemia-induced BBB disruption.
Figure 1.
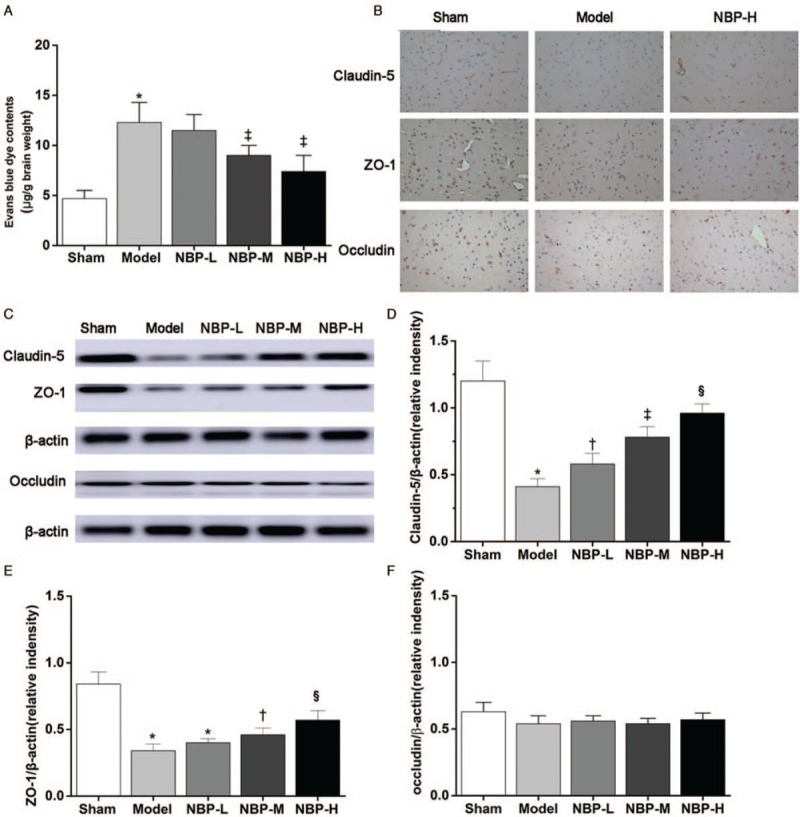
NBP decreased BBB disruption and upregulated the expression of TJ proteins. (A) Evans blue dye leakage (n = 6). (B) Representative immunohistochemistry staining graphs of claudin-5, ZO-1, and occludin (original magnification, ×400). (C) Western blotting images. (D, E, F) Densitometric analysis of protein bands (n = 6). β-actin served as the loading control. ∗P < 0.01 vs. the sham group; †P < 0.05 vs. the model group; ‡P < 0.01 vs. the model group; §P < 0.05 vs. the NBP-M group. BBB: Blood-brain barrier; NBP: DL-3-n-butylphthalide; TJ: Tight junction; ZO-1: Zonula occludens-1.
NBP increased claudin-5 and ZO-1 expression in ischemic brain
TJ proteins are essential for the restrictiveness of the BBB. Claudin-5, ZO-1, and occludin are the most important proteins in TJ complexes. Immunohistochemical staining showed that claudin-5-positive brownish cells were mainly expressed in blood vessels, while ZO-1- and occludin-positive cells could be detected in blood vessels as well as other brain tissues in the sham group [Figure 1B]. There were more claudin-5- or ZO-1-positive cells in the NBP-H group than in the model group.
We next collected the brain microvessels for Western blotting analysis. The expression of claudin-5 protein was significantly lower in the model group compared with that in the sham group (0.41 ± 0.06 vs. 1.25 ± 0.15, P < 0.01) [Figure 1C and 1D]. In comparison with the model group, the expression of claudin-5 was significantly increased in the NBP-L (0.58 ± 0.09 vs. 0.41 ± 0.06, P = 0.03), NBP-M (0.79 ± 0.08 vs. 0.41 ± 0.06, P < 0.01), and NBP-H (0.97 ± 0.07 vs. 0.41 ± 0.06, P < 0.01) groups. There were statistically significant differences among the three groups treated with NBP (0.79 ± 0.08 vs. 0.58 ± 0.09, P = 0.008; 0.97 ± 0.07 vs. 0.79 ± 0.08, P < 0.05), which indicated that the effect of NBP on claudin-5 expression was dose-dependent. The levels of ZO-1 expression in the NBP-M (P < 0.05) and NBP-H (P < 0.01) groups were also higher than those in the model group [Figure 1C and 1E]. No significant change in occludin expression was observed in different groups (F = 2.455, P = 0.075 [Figure 1C and 1F]).
NBP prevented the degradation of TJ ultrastructure after hypoxia injury
As an independent measure of TJ ultrastructure, transmission electron microscopy was performed in the hypoxic BMEC model. TJ ultrastructure showed high electronic density between adjacent cells in the control group, while it showed a lower electron-dense architecture or cellular gaps in BMECs in the hypoxia group, indicating that ultrastructural injury had occurred [Figure 2]. The alteration in TJ ultrastructure was prevented by NBP treatment, leading to an ultrastructure similar to that of the control group.
Figure 2.
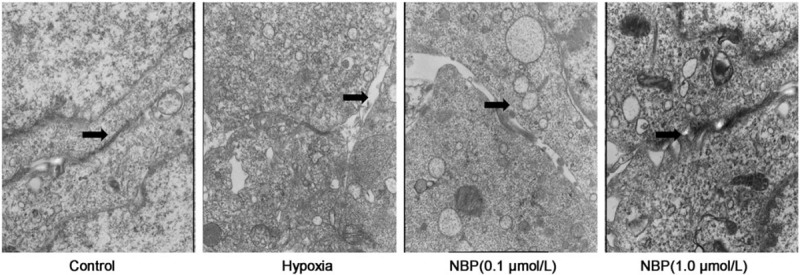
NBP improved TJ ultrastructure in BMECs. TJ ultrastructure of BMECs was observed by transmission electron microscopy. Arrows: the TJ ultrastructure between adjacent cells in the control group or cellular gap in the hypoxia group. The alteration in TJ is alleviated by NBP treatment (original magnification, ×15,000). BMECs: Brain microvascular endothelial cells; NBP: DL-3-n-butylphthalide; TJ: Tight junction.
NBP increased the expression of TJ proteins and improved the linear distribution in BMECs
BMECs in the control group showed continuous staining of claudin-5 and ZO-1 on the cell membrane by immunofluorescence [Figure 3]. After hypoxic injury, the immunofluorescence intensity of claudin-5 and ZO-1 reduced. With NBP treatment, the expression and linear distribution of TJ proteins in the membrane of BMECs improved partly. Occludin protein was observed both on the cell membrane and in the cytoplasm. There was no obvious difference in the immunofluorescence intensity of occludin in each group.
Figure 3.
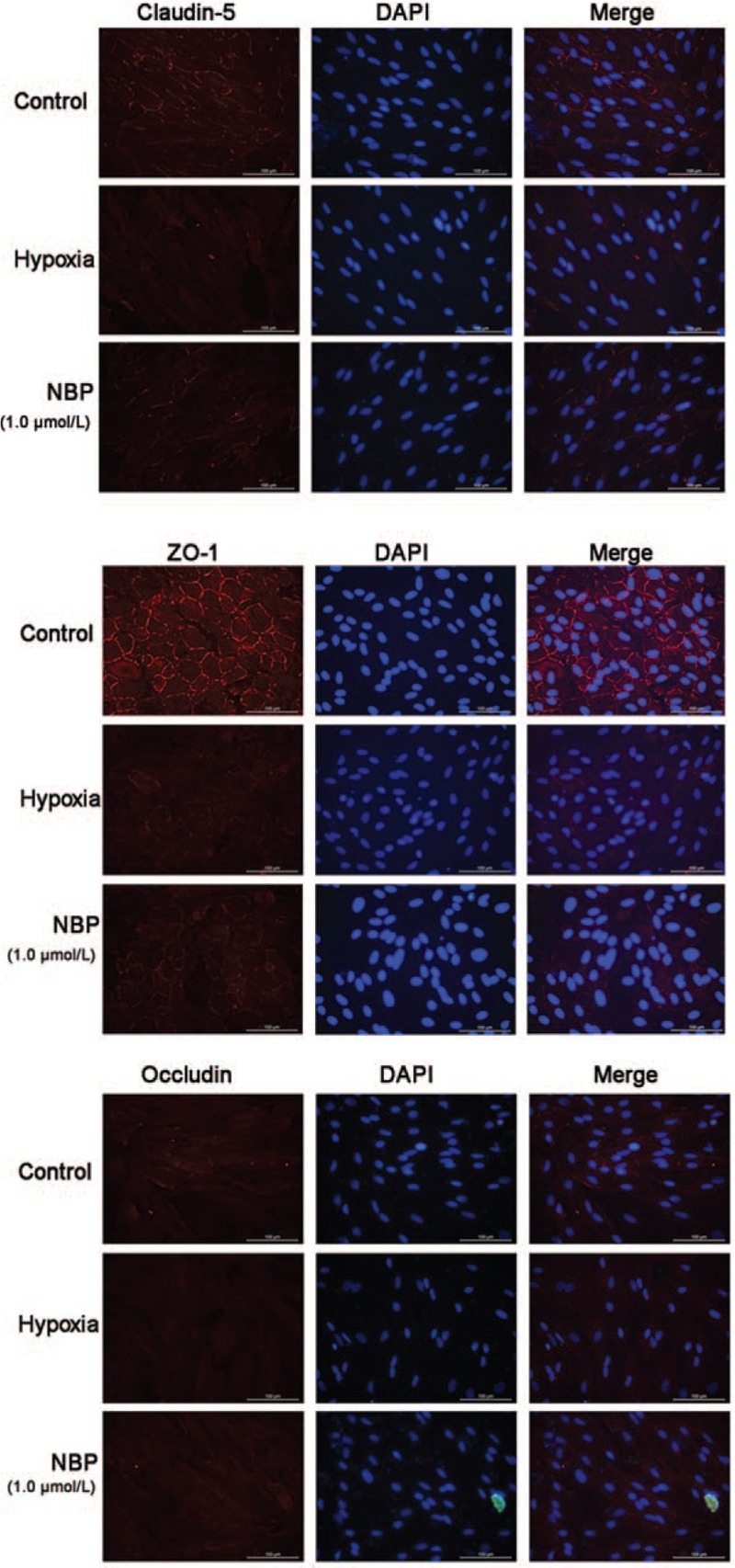
Expressions of claudin-5, ZO-1, and occludin in rat BMECs analyzed by immunofluorescence. The intensity of the red fluorescence signal represents the expression level of the corresponding protein. The nucleus was counterstained with DAPI (blue fluorescence). Scale bar = 100 μm. BMECs: Brain microvascular endothelial cells; DAPI: 4′,6-Diamidino-2-phenylindole; ZO-1: Zonula occludens-1.
Western blotting analysis showed that hypoxia decreased the expression of claudin-5 (P < 0.01) and ZO-1 (P < 0.01) compared with the control group [Figure 4A]. NBP (0.1 μmol/L) and NBP (1.0 μmol/L) treatment increased the expression of claudin-5 (0.45 ± 0.06 vs. 0.34 ± 0.05, P < 0.05; 0.55 ± 0.06 vs. 0.34 ± 0.05, P < 0.01) and ZO-1 (0.37 ± 0.09 vs. 0.24 ± 0.05, P < 0.05; 0.42 ± 0.09 vs. 0.24 ± 0.05, P < 0.01) in comparison with the hypoxia group. There was no significant difference in occludin expression among the four groups (F = 0.064, P = 0.598) [Figure 4A].
Figure 4.
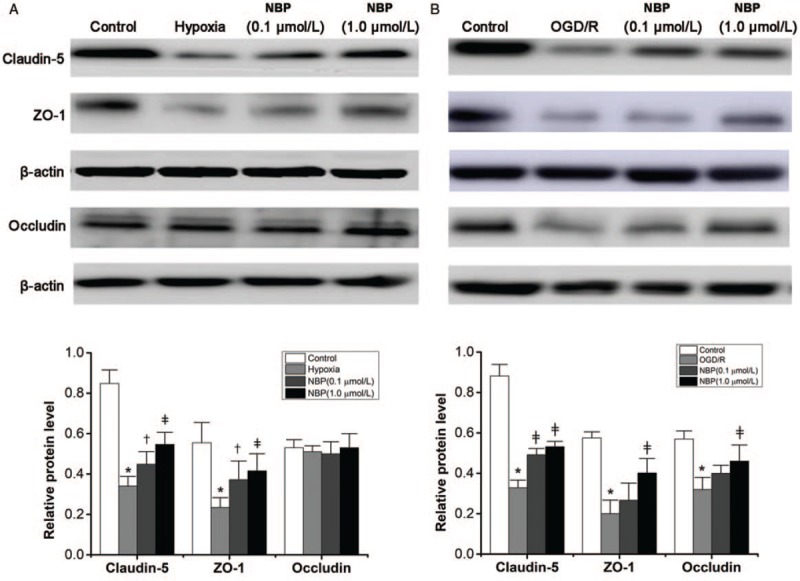
Expression of tight junction proteins in BMECs analyzed by Western blotting. (A) Protein level of claudin-5, ZO-1, and occludin detected by Western blotting in the hypoxia model and the densitometric analysis of protein expression (n = 6). (B) Protein level of claudin-5, ZO-1, and occludin detected by Western blotting in the OGD/R model and densitometric analysis of protein expression (n = 6). ∗P < 0.01 vs. the control group; †P < 0.05 vs. the hypoxia group or OGD/R group. ‡P < 0.01 vs. the hypoxia group or OGD/R group. BMECs: Brain microvascular endothelial cells; OGD/R: Oxygen-glucose deprivation/reoxygenation; ZO-1: Zonula occludens-1.
In the OGD/R model, the expression of claudin-5, ZO-1, and occludin was significantly lower than that in the control group (all P < 0.01) [Figure 4B]. NBP treatment (0.1 and 1.0 μmol/L) increased claudin-5 expression in comparison with the OGD/R group (both P < 0.01), but ZO-1 and occludin expression were increased only in the 1.0 μmol/L NBP group (both P < 0.01).
NBP attenuated ROS generation induced by hypoxia in BMECs
ROS levels were measured using DCFH-DA, a fluorescent dye that allows the visualization of ROS. As shown in Figure 5A, NBP decreased the hypoxia-induced increase in 2’-7’-dichlorofluorescein-positive cells (green). According to the results of flow cytometry analysis, hypoxia induced a significant increase in intracellular ROS generation in the hypoxia group in comparison with the control group (73.2% ± 7.4% vs. 7.1% ± 1.7%, P < 0.01). Treatment with NBP (0.1 μmol/L) (25.6% ± 3.0% vs. 73.2% ± 7.4%, P < 0.01) or NBP (1.0 μmol/L) (17.3% ± 2.6% vs. 73.2% ± 7.4%, P < 0.01) significantly decreased intracellular ROS level in a dose-dependent manner (NBP 0.1 μmol/L: 25.6% ± 3.0% vs. NBP 1.0 μmol/L: 17.3% ± 2.6%, P < 0.01) in comparison with the hypoxia group [Figure 5B]. These results indicate that NBP inhibits ROS generation induced by hypoxia in rat BMECs.
Figure 5.
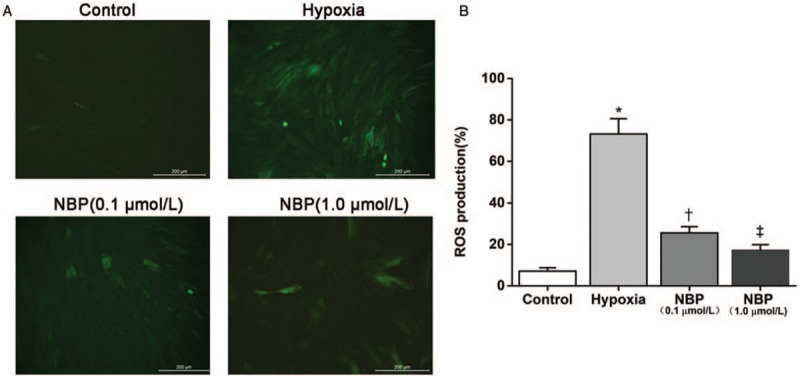
NBP attenuated ROS generation induced by hypoxia in rat BMECs. (A) Representative micrographs of ROS generation (under fluorescence microscope). Scale bar = 200 μm. (B) ROS generation measured by flow cytometry (n = 6). ∗P < 0.01 vs. the control group; †P < 0.01 vs. the hypoxia group; ‡P < 0.01 vs. NBP (0.1 μmol/L) group. BMECs: Brain microvascular endothelial cells; NBP: DL-3-n-butylphthalide; ROS: Reactive oxygen species.
NBP inhibited the hypoxia-induced depression of the Akt/GSK-3β/β-catenin pathway
We attempted to identify whether the Akt/GSK-3β/β-catenin signaling pathway was involved in the protective effect of NBP on TJs. Hypoxia injury decreased the relative expression of p-Akt/Akt (0.22 ± 0.07 vs. 0.64 ± 0.11, P < 0.01), p-GSK-3β/GSK-3β (0.36 ± 0.09 vs. 0.93 ± 0.10, P < 0.01), and β-catenin/β-actin (0.23 ± 0.03 vs. 0.54 ± 0.07, P < 0.01) in BMECs in comparison with the control group [Figure 6A and 6B]. In comparison with the hypoxia group, NBP treatment elevated the expression level of p-Akt/Akt, p-GSK-3β/GSK-3β, and β-catenin/β-actin (all P < 0.05). These results suggest that NBP could preserve BBB integrity, possibly by enhancing p-Akt activation, stabilizing β-catenin via GSK-3β inhibition, and then effectively raising the expression of TJs.
Figure 6.
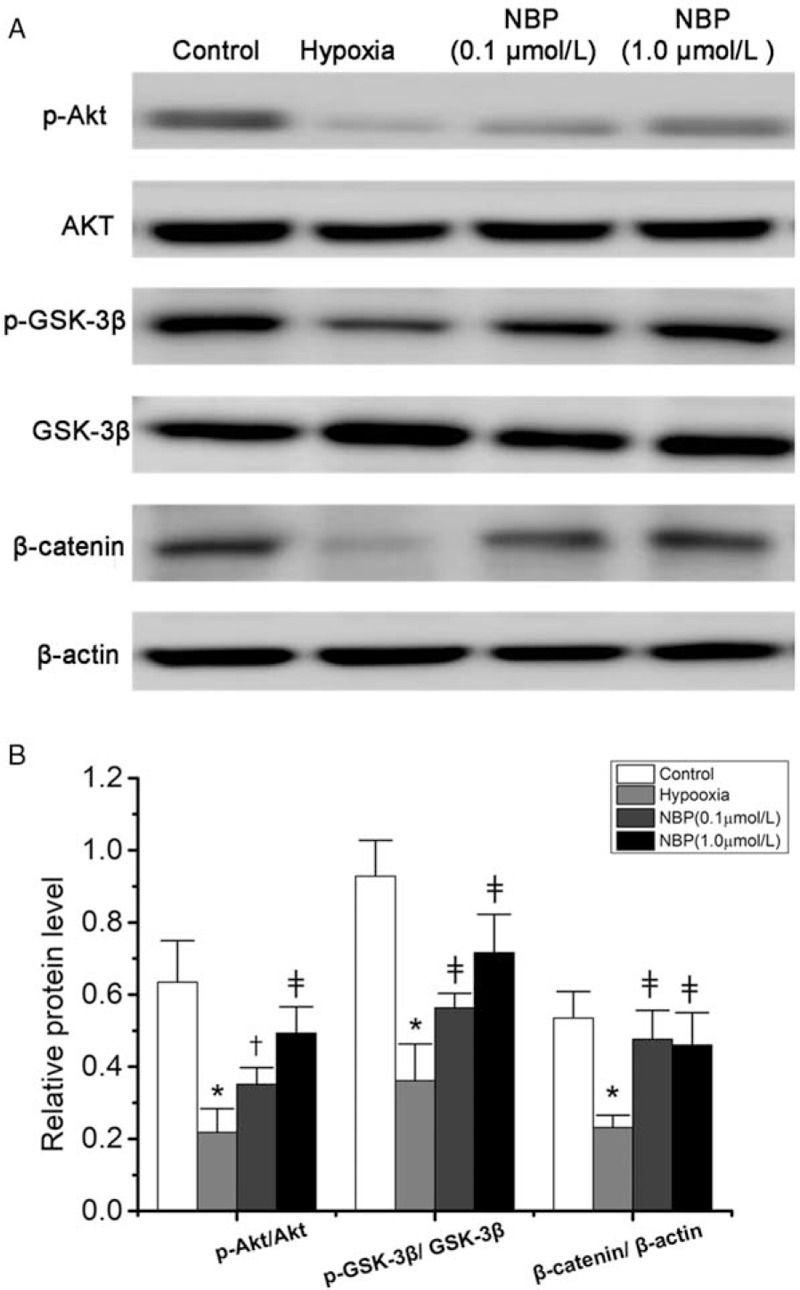
Akt/GSK-3β/β-catenin signaling pathway was activated by NBP in BMECs. (A) Protein level of p-Akt, Akt, p-GSK-3β, GSK-3β, β-catenin proteins detected by Western blotting. (B) Densitometric analysis of protein expression (n = 6). ∗P < 0.01 vs. the control group; †P < 0.05, ‡P < 0.01 vs. the hypoxia group. Akt: Protein kinase B; BMECs: brain microvascular endothelial cells; GSK-3β: Glycogen synthase kinase 3β; NBP: DL-3-n-butylphthalide.
Discussion
NBP is a compound extracted from the seeds of Chinese celery and was approved by the China Food and Drug Administration for the treatment of ischemic stroke in 2002. Numerous studies have demonstrated that NBP is a multi-target drug for the treatment of ischemic stroke and some other animal models of cerebral diseases. Meanwhile, many studies also confirmed that NBP could decrease the permeability of the BBB and improve the BBB function. Hu et al[16] demonstrated that NBP could reduce the brain water content and protect the BBB in a rat model of focal cerebral infarction. A previous study in a mouse model of concussive head injury showed that NBP was effective in reducing brain edema formation, as shown by the lower leakage of radioiodine and Evans blue albumin.[17] Nevertheless, the mechanism by which NBP maintains the functional stability of the BBB and its contribution to the ultrastructure of the BBB remains unclear. In the present study, we established a CCH rat model by BCCAO. In rats, BCCAO results in a loss of blood supply in the brain tissue and the resultant ischemia/hypoxia triggers a cascade of events, including hyperpermeability of BBB. Similarly, our present results showed that NBP significantly reduced the permeability of BBB, indicating that NBP may protect the BBB against brain hypoxic injury.
The BBB is composed of specialized BMECs, and the barrier property of BBB primarily depends on the integrity of BMECs and the TJs located between BMECs. TJs are responsible for restricting paracellular permeability at the BBB. Under normal physiological conditions, TJs are dynamic complexes of multiple protein constituents. Among these proteins, ZO-1, claudin-5, and occludin are the most important components for the barrier integrity of BBB. Therefore, our research focused on these three TJ proteins. In the rat model of CCH, our results showed that NBP increased the expression of claudin-5 and ZO-1 in rat brain tissue under continuous hypoperfusion. To further explore the effect of NBP on TJ expression, we established two BMEC models in vitro: hypoxia model (1% O2, 24 h) and OGD/R model (OGD 6 h/R 18 h). The hypoxic BMECs injury model was consistent with the CCH rat model used in this study, in which hypoxia/ischemia is the main cause of injury. The OGD/R injury model is known as an appropriate model of stoke in vitro, in which the reperfusion after occlusion results in more serious brain injury. The present study mainly focused on hypoxia/ischemia injury. In both cell injury models, we found that the expressions of claudin-5 and ZO-1 decreased and the NBP treatment was effective in preventing the degradation of the two TJ proteins. The results from the cell model in our study were consistent with those obtained using the animal model.
Claudin-5 belongs to the claudin family, which has two extracellular loops that interlink with claudins, forming the primary seal of TJs. As indicated in the immunochemistry images, claudin-5 protein was only expressed along the cerebral vessels, suggesting that claudin-5 is closely related to the integrity of BBB and may be a sensitive indicator of the normal and disturbed functional states of BBB. The lack of claudin-5 can directly lead to the increased BBB permeability and selective permeability of proteins with MW less than 800. Claudin-5 is essential for the formation of BBB and is involved in the selective permeability of TJs and in cell polarization. A number of studies have reported that claudin-5 plays a key role in regulating the integrity and permeability of BBB. In a BBB model established by using BMECs in vitro, the expression of claudin-5 was reduced by the siRNA technique, which was followed by increased cell permeability.[18] Studies on animal models of ischemic stroke have confirmed that neurons will benefit indirectly from the increased expression of claudin-5, and claudin-5 knockout mice all died in 10 h after birth.[19]
ZO-1 is a cytoplasmic membrane-associated accessory protein that interacts with occludin and claudins to anchor the TJ to the cytoskeletal scaffold and actin proteins of endothelial cells. In our study, similar to claudin-5, hypoxia and ODG/R also resulted in the downregulation of ZO-1, and NBP restored the expression in a dose-dependent manner. Our observations were in good agreement with the findings of a previous investigation in which ZO-1 was expressed both in the BBB and the non-vessel barrier.[20] Although ZO-1 is not a specific TJ marker, ZO-1 is an important component protein of TJs and an important TJ regulator, which plays an important role in the maintenance of cell polarity, cytoskeleton formation, TJ localization, and paracellular barrier. ZO-1 is closely related to BBB permeability. Unlike claudin-5 and ZO-1, we found decreased expression of occludin only in the OGD/R cell model, and the NBP treatment elevated occludin expression. Decreased expression of occludin has been reported to be associated with increased BBB permeability, as shown in animal models of hypoxia/reoxygenation stress. Lochhead et al[21] revealed that pathological stressors, such as oxidative stress, could cause the movement of occludin away from TJ complexes, and free radical scavengers could prevent such localization. Sub-cellular fractionation of cerebral microvessels revealed alterations in occludin oligomeric assemblies in TJs associated with plasma membrane lipid rafts. The phosphorylation state of occludin has been proposed to regulate its association within the cell membrane at the Ohtake et al[22] and Comerford et al[23] observed that serine and threonine phosphorylation was necessary for occludin localization within the membrane, while tyrosine phosphorylation promoted occludin disassociation from intercellular proteins, which led to increased BBB permeability. In our present study, we only measured the expression level of occludin protein. The phosphorylation state and localization of occludin in hypoxia and OGD/R deserve further investigation.
Taken together, the present results highlight that NBP improves the barrier function of the BBB against ischemia injury by upregulating the expression of TJ proteins. However, the signaling pathways by which NBP regulates TJs have not yet been fully elucidated. Oxidative stress during CCH has been shown to result in serious brain damage in multiple studies. In the present study, we also found that hypoxia-induced increased ROS generation in BMECs, while NBP decreased intracellular ROS generation. Many studies have demonstrated clearly that ROS play an important role in BBB disruption during ischemia and reperfusion. Although the exact mechanism by which ROS induce BBB breakdown has not fully clarified, one harmful effect on BBB could be mediated by modifications of TJs. The protective effect of NBP on TJ proteins can be explained, at least in part, by reduced ROS generation. The PI3K/Akt signaling pathway is considered to be one of the cell survival pathways. Many reports have demonstrated that Akt plays a major role in protection from cell death under oxidative stress and attenuates ROS production. The PI3K/Akt signaling pathway has been demonstrated to be involved in the neuroprotective effect of NBP during CCH or oxidative stress. Previous studies have shown that NBP could activate the PI3K/Akt pathway to protect stem cells, promote angiogenesis, inhibit neuronal apoptosis, regulate autophagy, and improve cognitive impairment, etc.[8–11] Therefore, PI3K/Akt is a very important downstream effector of NBP. Phosphorylation of Akt plays a vital role in activating the PI3K/Akt signaling pathway. Qi et al[11] reported that NBP could enhance the phosphorylation of Akt due to its antioxidant property. Phosphorylation of Akt is followed by the phosphorylation on Ser9 of GSK-3β, which belongs to the PI3K/Akt family, and the subsequent phosphorylation of GSK-3β could stabilize β-catenin.[24] The Wnt/β-catenin signaling pathway is crucial to a number of physiological processes, including the barrier function of the BBB. β-catenin is involved in the formation of the BBB during the fetal period and enhances BBB function by increasing the expression of claudins and occludin. Under pathologic conditions, a dysfunctional Wnt/β-catenin signaling pathway has been reported to contribute to BBB breakdown. Lengfeld et al[6] reported that the Wnt/β-catenin pathway in the blood vessels of the central nervous system during the progression of experimental autoimmune encephalomyelitis was associated with disruption of TJs. Taddei et al[25] reported that PI3K/Akt activation could limit the translocation of β-catenin to the nucleus, which finally increased the expression of claudin-5. Several studies also indicated that activation of PI3K/Akt could raise the expression of claudin-3 and claudin-5 through GSK-3β and β-catenin. In the present study, our data demonstrated that phosphorylation of Akt was increased by NBP, leading to high phosphorylation of GSK-3β/GSK-3β and elevated expression of β-catenin. Taken together, these findings suggest that the effect of NBP on TJ may be related to activation of the Akt/GSK-3β/β-catenin signaling pathway.
Our study had some limitations. First, TJs are complexes of multiple protein constituents and we only investigated the effect of NBP on claudin-5, ZO-1, and occludin. The effects of NBP on other components of TJ, such as claudin-1 and ZO-2, deserve further investigation. Second, the evidence for the signaling mechanisms involved in the effect of NBP on TJ, to some extent, is only correlative, and direct evidence is still needed.
In summary, NBP improves the barrier function of the BBB by upregulating the expression of TJ proteins against ischemic injury. The effect of NBP on TJs during hypoxia may be related to activation of the Akt/GSK-3β/β-catenin signaling pathway. Thus, these findings may provide the basic data for the clinical use of NBP in cases of brain ischemia and stroke.
Acknowledgements
The authors thank Dr. Bo Feng (Animal Science and Technology College, Beijing University of Agriculture, Beijing, China) for the primary cell culture. The authors wish to express the deepest thanks and appreciation to the staff of Department of Hebei Key Laboratory of Metabolic Disease for their support of this research.
Funding
This work was supported by a grant from the National Natural Science Funds of China (No. 81241037).
Conflicts of interest
None.
Footnotes
How to cite this article: Ye ZY, Xing HY, Wang B, Liu M, Lv PY. DL-3-n-butylphthalide protects the blood-brain barrier against ischemia/hypoxia injury via upregulation of tight junction proteins. Chin Med J 2019;00:00–00. doi:10.1097/CM9.0000000000000232
Zhan-Ying Ye and Han-Ying Xing contributed equally to this work.
References
- 1.Sharma G, Sharma AR, Lee SS, Bhattacharya M, Nam JS, Chakraborty C. Advances in nanocarriers enabled brain targeted drug delivery across blood brain barrier. Int J Pharm 2019; 559:360–372. doi: 10.1016/j.ijpharm.2019.01.056. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal A, Singh I, Sandhir R. Protective effect of S-nitrosoglutathione administration against hyperglycemia induced disruption of blood brain barrier is mediated by modulation of tight junction proteins and cell adhesion molecules. Neurochem Int 2018; 118:205–216. doi: 10.1016/j.neuint.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Koto T, Takubo K, Ishida S, Shinoda H, Inoue M, Tsubota K, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol 2007; 170:1389–1397. doi: 10.2353/ajpath.2007.060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang YF, Gu YT, Qin GH, Zhong L, Meng YN. Curcumin ameliorates the permeability of the blood-brain barrier during hypoxia by upregulating heme oxygenase-1 expression in brain microvascular endothelial cells. J Mol Neurosci 2013; 51:344–351. doi: 10.1007/s12031-013-9989-4. [DOI] [PubMed] [Google Scholar]
- 5.Zuo S, Ge H, Li Q, Zhang X, Hu R, Hu SL, et al. Artesunate protected blood-brain barrier via sphingosine 1 phosphate receptor 1/phosphatidylinositol 3 kinase pathway after subarachnoid hemorrhage in rats. Mol Neurobiol 2017; 54:1213–1228. doi: 10.1007/s12035-016-9732-6. [DOI] [PubMed] [Google Scholar]
- 6.Lengfeld JE, Lutz SE, Smith JR, Diaconu C, Scott C, Kofman SB, et al. Endothelial Wnt/beta-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci U S A 2017; 114:E1168–E1177. doi: 10.1073/pnas.1609905114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang W, Li M, Wang Y, Li Q, Deng G, Wan JR, et al. GSK-3β inhibitor TWS119 attenuates rtPA-induced hemorrhagic transformation and activates the Wnt/β-catenin signaling pathway after acute ischemic stroke in rats. Mol Neurobiol 2016; 53:7028–7036. doi: 10.1007/s12035-015-9607-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen N, Zhou Z, Li J, Li BC, Feng JH, He D, et al. 3-n-butylphthalide exerts neuroprotective effects by enhancing anti-oxidation and attenuating mitochondrial dysfunction in an in vitro model of ischemic stroke. Drug Des Devel Ther 2018; 12:4261–4271. doi: 10.2147/DDDT.S189472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Huai YP, Meng N, Dong YH, Liu ZJ, Qi QQ, et al. L-3-n-Butylphthalide activates Akt/mTOR signaling, inhibits neuronal apoptosis and autophagy and improves cognitive impairment in mice with repeated cerebral ischemia-reperfusion injury. Neurochem Res 2017; 42:2968–2981. doi: 10.1007 /s11064-017-2328-3. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Zhang B, Tao Y, Wang Y, Wei H, Zhao J, et al. DL-3-n-butylphthalide protects endothelial cells against oxidative/nitrosative stress, mitochondrial damage and subsequent cell death after oxygen glucose deprivation in vitro. Brain Res 2009; 1290:91–101. doi: 10.1016/j.brainres.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 11.Qi QQ, Xu J, Lv PY, Dong YH, Liu ZJ, Hu M, et al. DL-3-n-butylphthalide alleviates vascular cognitive impairment induced by chronic cerebral hypoperfusion by activating the Akt/Nrf2 signaling pathway in the hippocampus of rats. Neurosci Lett 2018; 672:59–64. doi: 10.1016/j.neulet.2017.11.051. [DOI] [PubMed] [Google Scholar]
- 12.Jin W, Jia YQ, Huang LL, Wang TJ, Wang HB, Dong YH, et al. Lipoxin A4 methyl ester ameliorates cognitive deficits induced by chronic cerebral hypoperfusion through activating ERK/Nrf2 signaling pathway in rats. Pharmacol Biochem Behav 2014; 124:145–152. doi: 10.1016/j.pbb.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Alluri H, Grimsley M, Anasooya Shaji C, Varghese KP, Zhang SL, Peddaboina C, et al. Attenuation of blood-brain barrier breakdown and hyperpermeability by calpain inhibition. J Biol Chem 2016; 291:26958–26969. doi: 10.1074/jbc.M116.735365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Q, Liu X, Wang N, Zheng X, Fu J, Zheng J. Nitric oxide from brain microvascular endothelial cells may initiate the compensatory response to mild hypoxia of astrocytes in a hypoxia-inducible factor-1 alpha dependent manner. Am J Transl Res 2016; 8:4735–4749. [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X, Zhang H, Pan Q, Zhao YH, Chen J, Zhao B, et al. Hypoxia/aglycemia-induced endothelial barrier dysfunction and tight junction protein downregulation can be ameliorated by citicoline. PLoS One 2013; 8:e82604.doi: 10.1371/journal.pone.0082604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu J, Wen Q, Wu Y, Li BZ, Gao P. The effect of butylphthalide on the brain edema, blood-brain barrier of rats after focal cerebral infarction and the expression of Rho A. Cell Biochem Biophys 2014; 69:363–368. doi: 10.1007/s12013-013-9808-0. [DOI] [PubMed] [Google Scholar]
- 17.Feng L, Sharma A, Niu F, Huang Y, Lafuente JV, Muresanu DF, et al. TiO2-nanowired delivery of DL-3-n-butylphthalide (DL-NBP) attenuates blood-brain barrier disruption, brain edema formation, and neuronal damages following concussive head injury. Mol Neurobiol 2018; 55:350–358. doi: 10.1007/s12035-017-0746-5. [DOI] [PubMed] [Google Scholar]
- 18.Luissint AC, Federici C, Guillonneau F, Fabrice C, Luc C, Fabienne G, et al. Guanine nucleotide-binding protein G αi2: a new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J Cereb Blood Flow Metab 2012; 32:860–873. doi: 10.1038/jcbfm.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003; 161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bi M, Zhang M, Guo D, Bi W, Liu B, Zou Y, et al. N-Butylphthalide alleviates blood-brain barrier impairment in rats exposed to carbon monoxide. Front Pharmacol 2016; 7:394.doi: 10.3389/fphar.2016.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab 2010; 30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohtake K, Maeno T, Ueda H, Ogihara M, Natsume H, Morimoto Y. Poly-L-arginine enhances paracellular permeability via serine/threonine phosphorylation of ZO-1 and tyrosine dephosphorylation of occludin in rabbit nasal epithelium. Pharm Res 2003; 20:1838–1845. doi: 10.1023/b:pham.0000003383.86238.d1. [DOI] [PubMed] [Google Scholar]
- 23.Comerford KM, Lawrence DW, Synnestvedt K, Levi Boaz P, Colgan Sean P, Colgan Sean P. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J 2002; 16:583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 24.Wang YH, Liou KT, Tsai KC, Liu HK, Yang LM, Chern CM, et al. GSK-3 inhibition through GLP-1R allosteric activation mediates the neurogenesis promoting effect of P7C3 after cerebral ischemic/reperfusional injury in mice. Toxicol Appl Pharmacol 2018; 357:88–105. doi: 10.1016/j.taap.2018.08.023. [DOI] [PubMed] [Google Scholar]
- 25.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 2018; 10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]