

Abstract
Background:
It has recently been recognized that serum vimentin is elevated in infectious diseases, and that vimentin plays a role in regulating neutrophils and macrophages associated inflammation. However, the mechanisms are unclear. This study was designed to explore the role of vimentin in regulating monocyte survival or apoptosis as well as inflammatory cytokine secretion in response to lipopolysaccharides (LPSs).
Methods:
A human monocytic leukemia cell line (THP-1) was transfected with vimentin-specific small interfering RNA (siRNA) or vimentin over-expressing plasmid. Apoptosis was assessed by TdT-mediated dUTP Nick-End Labeling (TUNEL) and DNA content assay. Immunoblotting was performed to detect apoptosis-associated proteins. Cytokines (interleukin [IL]-6, IL-10, and tumor necrosis factor α [TNF-α]) were measured by enzyme-linked immuno sorbent assay. Two-way analysis of variance followed by Student's t test was used to compare means between different groups.
Results:
Suppression of vimentin in THP-1 cells resulted in increased apoptotic response in the presence of LPS, while over-expression of vimentin could prevent the cells from apoptosis in response to LPS. LPS alone or suppression of vimentin resulted in significant up-regulation of caspase-3 (1.42 ± 0.20 of LPS alone and 1.68 ± 0.10 of vimentin suppression vs. control, t = 5.21 and 10.28, respectively, P < 0.05). In addition, pro-inflammatory cytokines (IL-6 and TNF-α) was significantly increased (IL-6: 577.90 ± 159.90 pg/day/105 cells vs. 283.80 ± 124.60 pg/day/105 cells of control, t = 14.76, P < 0.05; TNF-α: 54.10 ± 5.80 vs. 17.10 ± 0.10 pg/day/105 cells of control, t = 6.71, P < 0.05), while anti-inflammatory cytokine (IL-10) was significantly up-regulated in the THP-1 cells that over-expressed vimentin (140.9 ± 17.2 pg/day/105 cells vs. undetectable in control cells).
Conclusions:
In summary, the vimentin may regulate innate immunity through modulating monocytes viability as well as inflammatory response in sepsis through shifting the balance of pro-inflammatory and anti-inflammatory cytokines.
Keywords: Vimentin, Sepsis, Monocyte, Apoptosis, Inflammatory cytokine
Introduction
Sepsis refers to the presence of a serious infection that correlates with organ dysfunction, which still contributes to the higher mortality in the world. Traditionally, the host immune response to sepsis was thought to be characterized by two sequential stages: The first stage is an initial hyper-inflammatory response, sometimes referred to as a cytokine storm, where the innate immune system releases pro-inflammatory cytokines to combat infection, while also recruiting members of the adaptive system to mount an intense immune response. This initial response is then thought to be followed by compensatory anti-inflammatory response syndrome (CARS), which is defined as a systemic deactivation of the immune system tasked with restoring homeostasis from an inflammatory state. Recent data suggest that both aspects of the pro-inflammatory and anti-inflammatory stages of the host immune response to severe injury and/or sepsis often occur concurrently.[1] However, specific immunologic mechanisms during sepsis are unclear.
Vimentin is a type III intermediate filament (IF) protein that is predominantly expressed in mesenchymal cells.[2] Recently, vimentin has been reported to involve in inflammatory diseases. Mor-Vaknin et al challenged wild-type and vimentin knockout mice with Escherichia coli intraperitoneally to induce colitis and found macrophages of vimentin knockout mice show significantly increased capacity to mediate bacterial killing by abundant production of ROS and nitric oxides.[2] Thiagarajan et al.'s study reported that vimentin could be an endogenous, activating ligand for Dectin-1, the non-toll pattern recognition receptor (PRR), on monocytes. The activation of monocytes contributes to the chronic inflammation such as atherosclerosis.[3] Toda et al reported that vimentin could bind with phosphorylated p38 MAPK and mediate CCL2 production in mast cells, which is a mechanism for allergic inflammation.[4] dos Santos et al demonstrated that lung inflammation and fibrosis were attenuated in the lungs of vimentin knockout mice compared to wild-type controls in the lipopolysaccharide (LPS)-induced acute lung injury mouse model; and this effect may be due to decreased activation of NLRP3 inflammasome.[5] However, so far, the role of vimentin in the pathogenesis or disease progression of sepsis is largely unknown.
In this present study, the role of vimentin in regulating monocyte survival or apoptosis and inflammatory cytokine secretion in response to LPS stimulation was investigated.
Materials and methods
Cell line and culture
THP-1 (human acute monocytic leukemia; ATCC® TIB202™ Manassas, VA, USA) cells was maintained in Dulbecco Modified Eagle Media (DMEM; Gibco, Life Technology, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS; Gibco, Life Technology), 100 μg/mL penicillin, and 100 mg/mL streptomycin (Life Technology). LPS was purchased from Sigma (Sigma-Aldrich, L5418, St. Louis, MO, USA). For the experiment, cells were treated with or without 10 μg/mL LPS for the period as indicated below.
Transfection of siRNA and plasmid
For siRNA transfection, cells were washed once with serum-free DMEM (SF-DMEM; Life Technology) without antibiotics and amphotericin B followed by plating in 60 mm dishes (3 × 106 cells/dish) in 1 mL Optimal-Minimal Essential Medium (Opti-MEM; Life Technology). Negative control siRNA (Santa Cruz Biotechnology, Cat#: sc-37007, Dallas, TX, USA) or vimentin-specific siRNA (Santa Cruz Biotechnology, Cat#: sc-29522) were mixed with lipofecatmine 2000 (Santa Cruz Biotechnology, Cat#: sc-29528) following the manufacture's instruction in Opti-MEM. The mixture was then added into the 60 mm dish containing cells (500 μL/dish, final concentration of siRNA was 200 nmol/L in 1.5 mL/dish). After 6 h transfection, cells were further cultured for 24 h with 10% FCS-DMEM supplemented with antibiotics and amphotericin B overnight. Cells were then used for experiments as designed.
For transfection of the vimentin over-expressing plasmid, cells were washed once with SF-DMEM without antibiotics and amphotericin B followed by plating in 60 mm dishes (3 × 106 cells/dish) in 1 mL Opti-MEM. The vimentin expressing plasmid (pCMV3-VIM; Sino Biological Inc., Beijing, China) or an untagged negative control vector (pCMV3-untagged Negative Control Vector) were mixed with transfection reagent (Sinofcetion-293; Sino Biological Inc.) following the manufacture's instruction in Opti-MEM. The mixture was then added into the 60 mm dish containing cells (500 μL/dish, final concentration of the plasmid was 200 nmol/L in 1.5 mL/dish). After 6 h transfection, cells were further cultured for 24 h with 10% FCS-DMEM supplemented with antibiotics and amphotericin B overnight. Cells were then used for experiments as designed.
Immunoblotting
THP-1 cells (5 × 105 cells/mL, 2 mL/well, n = 3) were seeded in 6-well plate and stimulated with LPS for 24 h. Cells were lysed with RIPA buffer (50 mmol/L Tris-HCL, pH 7.4, 50 mmol/L NaCl, 2 mM EDTA, 0.1% DS plus freshly added proteinase inhibitor cocktail including apoprotein, leupeptin, DTT, and PMSF). The cell lysate was centrifuged at 12,000×g for 10 min at 4°C. Protein concentration in the supernatant was determined by a protein dye-binding assay (Bio-Rad, Hercules, CA, USA). Total proteins were then subjected to immunoblot analysis. Briefly, after heating for 5 min at 95°C followed by cooling on ice, 10 μg of total protein was mixed with 2× sample loading buffer (0.5 mol/L Tris-HCL, pH 6.8, 10% SDS, 0.1% bromphenol blue, 20% glycerol, 2% β-mercaptoethanol) and loaded into each lane before performing electrophoresis with the Mini-protein 3 Cell System (Bio-Rad). The proteins were transferred to PVDF membrane (Bio-Rad) in transfer buffer (20 mmol/L Tris, pH 8.0, 150 mmol/L glycine, 20% methanol) at 20 V for 45 min with the semi-dry electrophoretic transfer system (Bio-Rad). The membrane was then blocked with the blocking buffer (Li-COR, Lincoln, NE, USA) at room temperature for 1 h and then exposed to primary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) at 4°C overnight. Targeted proteins were subsequently detected using IRDye 800CW goat anti-mouse antibody (Li-COR) for 1 h at room temperature and dark. Band was visualized with an image scanner (Li-COR).
TUNEL assay
THP-1 (5 × 105 cells/mL, 2 mL/well, n = 3) were seeded in 6-well plate and stimulated with LPS for 24 h. Apoptosis was assessed by TUNEL assay and DNA content assay. TUNEL assay was conducted using a commercially available kit (Roche Molecular Biochemicals, Penzberg, Germany) following the manufacture's instruction. Briefly, the cells were spun on a slide (100 μL/slide) with a cytospin device (Cytospin 2; Shandon, Canton, MA, USA) at 120g for 5 min. After air dry, the cells were fixed with 10% formalin for 1 h at room temperature followed by permeabilization with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min at 4°C and rinsed with the PBS. The cells were then reacted with the TUNEL mixture in a humidified chamber for 60 min at 37°C in the dark. After washing, the cells were counterstained with 1 μg/mL of propidium iodide for 10 min in the dark. After washing and mounting, the fluorescent incorporation into nucleotide was detected and photographed under a fluorescent microscope at 200× magnification (Leica, Wetzlar, Germany). TUNEL positive and total cell number was counted in at least five random fields for each slide.
For DNA content assay, cells were fixed with 70% ethanol/PBS for 30 min at 4°C followed by staining with propidium iodide (50 μg/mL). Cell cycle analysis was then performed by flow cytometry. Cells with less DNA staining than that of G1 cells (sub-G1 peak or A0 cells) were considered as apoptotic cells.
ELISA assay
Concentrations of cytokines in the supernatants of cultures were quantified using DuoSet ELISA kit (R&D System, Minneapolis, MN, USA) following the manufacture's instruction with brief modification. Briefly, 96-well plates were coated with monoclonal antibodies at 4°C overnight. Plates were washed three times, and then standards or samples were applied to individual wells and incubated at 4°C overnight, followed by washing and the application of biotinylated antibodies for 1 h at room temperature. After washing, horseradish peroxidase (HRP)-streptavidin conjugate was added for 1 h at room temperature. After a final wash, bound HRP was detected with 3,3′,5,5′-tetramethylbenzidine (TMB; Sigma, St. Louis, MO, USA). The reaction was stopped with 1 mol/L H2SO4, and quantified at 450 nm with a microplate reader (Bio-Rad). Amount of cytokines were adjusted by cell number.
Statistics analysis
Normality of data distribution was examined with Lilliefors corrected Kolmogorov-Smirnov test provided in the SPSS 16.0 software (SPSS, Chicago, IL, USA). Continuous variables with normal distribution were expressed as mean ± standard deviations (SD). One-way analysis of variance was used to compare means between different groups followed by Student's t test for paired comparison. All statistical analyses were conducted with SPSS 16.0 (SPSS), and a two-tailed P < 0.05 was considered statistically significant.
Results
Role of vimentin in modulating cell viability in response to LPS
As shown in Figure 1, vimentin was either suppressed by vimentin-specific siRNA (Vim-siRNA) or over-expressed by plasmid transfection (Vim-DNA). To investigate the role of vimentin in modulating THP-1 cell viability in response to LPS, the cells were transfected with either scramble (control) siRNA or vimentin-specific siRNA. Cells were then exposed to LPS for 24 h. As shown in Figure 2, THP-1 cells underwent apoptosis in response to LPS exposure as evidenced by significant increase of TUNEL positivity (2.50 ± 0.65% of control vs. 12.10 ± 1.29% of LPS exposure, t = 13.81, P < 0.05, Figure 2A, 2B, and 2E). Suppression of vimentin in the THP-1 cells resulted in further augmentation of LPS-induced apoptosis (27.51 ± 2.47%, t = 5.28, P < 0.05 compared with LPS alone) [Figure 2B, 2D, and 2E]. Similarly, DNA content assay also demonstrated that suppression of vimentin resulted in significant increase of apoptosis in THP-1 cells [Figure 3].
Figure 1.
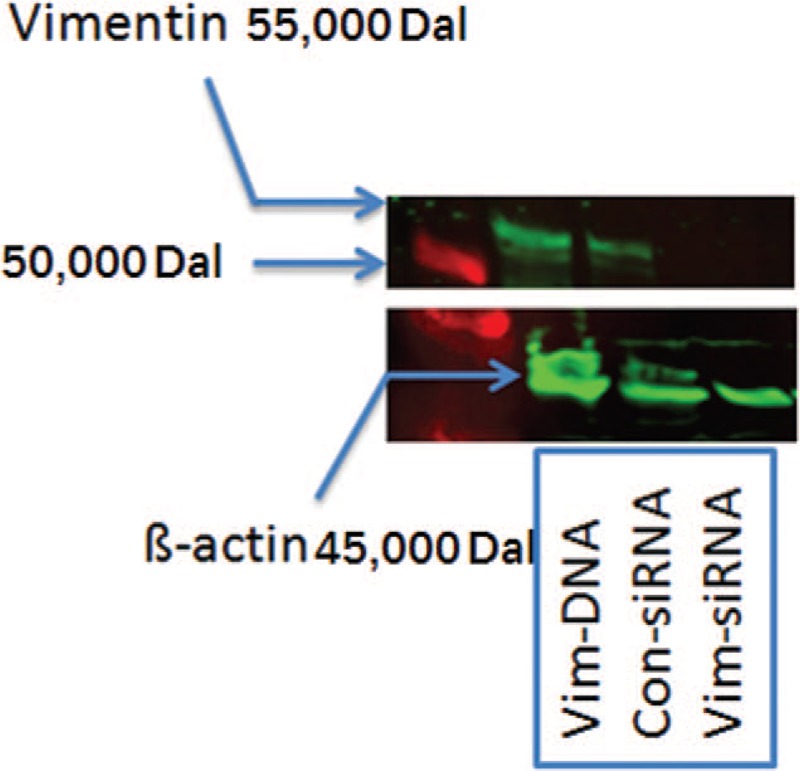
Representative immunoblots indicating suppression of vimentin by siRNA (Vim-siRNA) or over-expression of vimentin (Vim-DNA). THP-1 cells were transfected with control siRNA, vimentin siRNA or a plasmid expressing vimentin and immunoblotting was performed as described in the methods.
Figure 2.
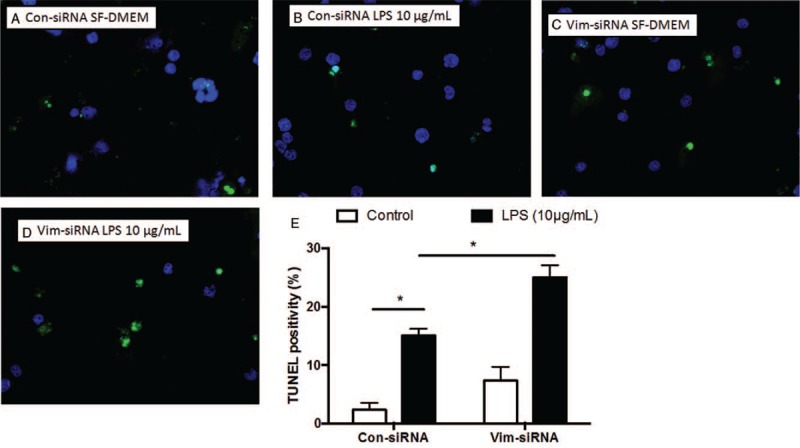
Effect of vimentin suppression by siRNA on cell survival assessed by TUNEL assay. THP-1 cells were transfected with vimentin-specific or scramble siRNA followed by exposure to LPS (10 μg/mL), and apoptosis was assessed by TUNEL assay as described in the methods. (A–D) Representative images of TUNEL assay. Green: positive TUNEL staining; blue: nuclei staining with DAPI. (E) Average of three separate TUNEL assay results. Vertical axis: TUNEL positivity (%); horizontal axis: cells transfected with control or vimentin-specific siRNA. Open bar: cells cultured in SF-DMEM only; closed bar: cells treated with LPS. ∗P < 0.05. LPS: Lipopolysaccharide; SF-DMEM: Serum-free Dulbecco modified Eagle media; TUNEL: TdT-mediated dUTP Nick-End Labeling.
Figure 3.
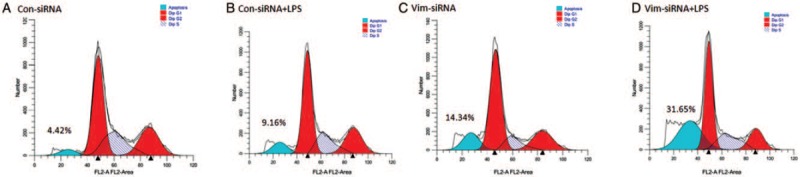
Effect of vimentin suppression by siRNA on cell survival assessed by DNA content assay. THP-1 cells were transfected with vimentin-specific or scramble siRNA followed by exposure to LPS (10 μg/mL), and apoptosis was assessed by DNA content assay as described in the methods. (A) Cells were transfected with control-siRNA and cultured in medium only. (B) Cells were transfected with control-siRNA and treated with LPS. (C) Cells were transfected with vimentin-siRNA and cultured in medium only. (D) Cells were transfected with vimentin-siRNA and treated with LPS. Blue peak indicates apoptotic cells with less DNA content. Data presented were one representative of three separate experiments. LPS: Lipopolysaccharide.
Next, to confirm the role of vimentin in regulating apoptosis, THP-1 cells were transfected with a plasmid containing either a negative control vector (pCMV3-untagged negative control vector) or vimentin gene ORF cDNA clone expression plasmid. Cells were then exposed to LPS for 24 h. As shown in Figure 4, over-expression of vimentin in THP-1 cells lead to significant reduction of apoptosis in response to LPS exposure (10.4 ± 1.2% of LPS exposure to control cells vs. 2.4 ± 0.4% of LPS exposure to the cells over-expressing vimentin, Figure 4B, 4D, and 4E, t = 5.75, P < 0.05).
Figure 4.
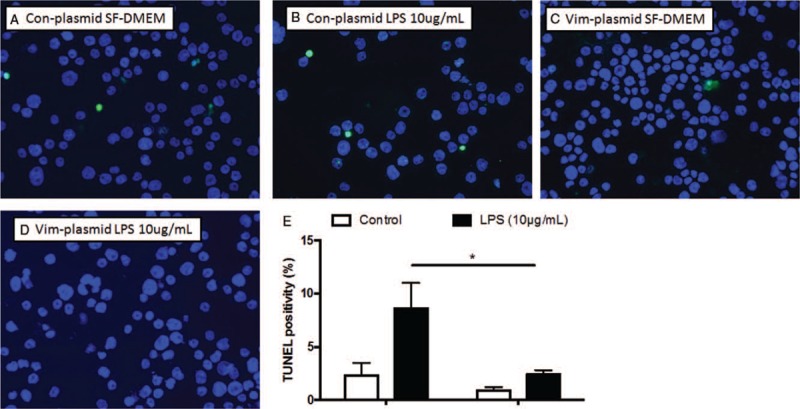
Effect of vimentin over-expression on cell survival assessed by TUNEL assay. THP-1 cells were transfected with a plasmid containing either a negative control vector or vimentin gene followed by exposure to LPS (10 μg/mL), and apoptosis was assessed by TUNEL assay as described in the methods. (A–D) Representative images of TUNEL assay. Green: positive TUNEL staining; blue: nuclei staining with DAPI. (E) average of three separate TUNEL assay results. Vertical axis: TUNEL positivity (%); horizontal axis: cells transfected with control or vimentin-specific siRNA. Open bar: cells cultured in SF-DMEM only; closed bar: cells treated with LPS. ∗P < 0.05. LPS: Lipopolysaccharide; SF-DMEM: Serum-free Dulbecco modified Eagle media; TUNEL: TdT-mediated dUTP Nick-End Labeling.
Apoptosis associated protein expression
Next, expression of apoptosis-associated proteins was examined. LPS alone or suppression of vimentin resulted in significant up-regulation of caspase-3 compared with control (1.42 ± 0.20 of LPS alone and 1.68 ± 0.10 of vimentin suppression vs. control, Figure 5A and 5B, t = 5.21 and 10.28, respectively, P < 0.05). Moreover, caspase-3 level was even more increased in the cells lacking vimentin and exposed to LPS (2.15 ± 0.12, Figure 5A and 5B, t = 17.95, P < 0.05 compared with control). Other caspases including caspase-8 and -9 were not altered (data not shown).
Figure 5.
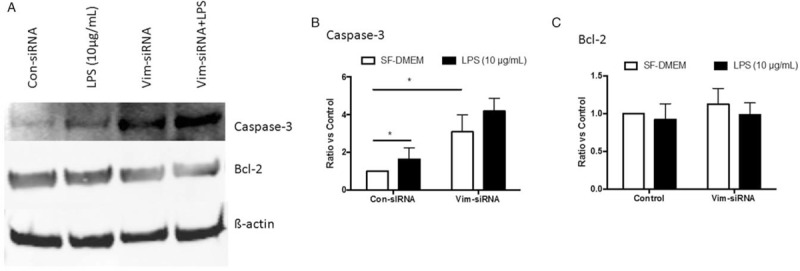
Effect of vimentin suppression on caspase-3 and Bcl-2. THP-1 cells were transfected with vimentin-specific or scramble siRNA followed by exposure to LPS (10 μg/mL), and caspase-3 and Bcl-2 were assessed by immunoblotting as described in the methods. (A) Representative images of immunoblotting. (B, C) Average of three separate immunoblottings for caspase-3 (B) and Bcl-2 (C). Vertical axis: ratio to control; horizontal axis: cells transfected with control or vimentin-specific siRNA. Open bar: cells cultured in SF-DMEM only; closed bar: cells treated with LPS. ∗P < 0.05. LPS: Lipopolysaccharide; SF-DMEM: Serum-free Dulbecco modified Eagle media.
As shown in Figure 5A and 5C, neither LPS exposure nor vimentin suppression altered the protein level of Bcl-2 in THP-1 cells (P > 0.05). In addition, Bax and Bim were not significantly altered, either, in the cells lacking vimentin regardless of LPS exposure (data not shown). In addition, neither caspase-3 nor Bcl-2 was altered in the cells over-expressing vimentin [Figure 6A–6C].
Figure 6.
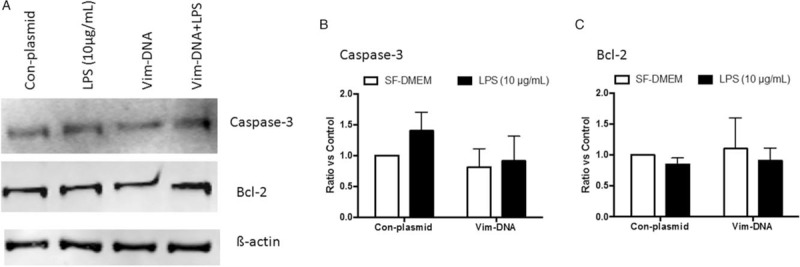
Effect of vimentin over-expression on caspase-3 and Bcl-2. THP-1 cells were transfected with a plasmid containing negative control vector or vimentin gene followed by exposure to LPS (10 μg/mL), and caspase-3 and Bcl-2 were assessed by immunoblotting as described in the methods. (A) Representative images of immunoblotting. (B, C) Average of three separate immunoblotting for caspase-3 (B) and Bcl-2 (C). Vertical axis: ratio to control; horizontal axis: cells transfected with plasmids containing negative control vector or vimentin gene. Open bar: cells cultured in SF-DMEM only; closed bar: cells treated with LPS. ∗P < 0.05. LPS: Lipopolysaccharide; SF-DMEM: Serum-free Dulbecco modified Eagle media; Vim-DNA: Over-expression of vimentin.
Role of vimentin in regulating cytokine release in response to LPS
To assess potential role of vimentin in regulating THP-1-mediated inflammation, effect of vimentin suppression or over-expression on inflammation-associated cytokines and matrix metalloproteinases were examined. As shown in Figure 7, pro-inflammatory cytokines, interleukin [IL]-6 and tumor necrosis factor α (TNF-α), were significantly up-regulated in the cells lacking vimentin (IL-6: 577.9 ± 159.9 pg/day/105 cells vs. 283.8 ± 124.6 pg/day/105 cells of control, t = 14.76, P < 0.05; TNF-α: 54.1 ± 5.8 vs. 17.1 ± 0.1 pg/day/105 cells of control, t = 6.71, P < 0.05) [Figure 7A and 7B]. LPS exposure resulted in up-regulation of IL-6 and TNF-α in the cells transfected with scramble siRNA (IL-6: 935.2 ± 68.6 pg/day/105 cells; TNF-α: 65.1 ± 12.2 pg/day/105 cells, Figure 7A and 7B, t = 24.67 and 5.28, P < 0.05 compared with control), and even more significant up-regulation of IL-6 in the cells lacking vimentin (2887.7 ± 831.0 pg/day/105 cells, t = 9.48, P < 0.05 compared to LPS-exposed control cells) [Figure 7A]. In contrast, over-expression of vimentin in the THP-1 cells significantly blocked TNF-α release under control (undetectable) as well as in the presence of LPS (40.6 ± 3.6 pg/day/105 cells, Figure 7B, t = 5.40, P < 0.05 compared to the cells lacking vimentin and exposed to LPS), while IL-6 release was not significantly altered in the cells over-expressing vimentin compared with control cells (Figure 7A, t = 1.99, P > 0.05).
Figure 7.
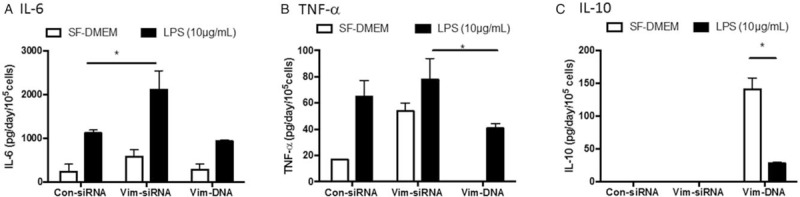
Effect of vimentin suppression or over-expression on cytokine release. Vimentin was either suppressed by siRNA or over-expressed by transfecting a plasmid containing vimentin promoter followed by exposure to LPS (10 μg/mL), and cytokines were quantified by ELISA as described in the methods. (A) IL-6. (B) TNF-α. (C) IL-10. Vertical axis: cytokine concentration (pg/day/105 cells); horizontal axis: cells transfected with control-siRNA, vimentin-specific siRNA or a plasmid containing vimentin promoter. Open bar: cells cultured in SF-DMEM only; closed bar: cells treated with LPS. Data presented were an average of three separate experiments. ∗P < 0.05. IL: Interleukin; LPS: Lipopolysaccharide; SF-DMEM: Serum-free Dulbecco modified Eagle media.
Interestingly, anti-inflammatory cytokine, IL-10, was not detectable in the THP-1 cells transfected with control-siRNA or vimentin-specific siRNA regardless of LPS stimulation [Figure 7C]. However, IL-10 secretion was dramatically increased in the cells over-expressing vimentin (140.9 ± 17.2 pg/day/105 cells), which was significantly blocked by LPS (28.1 ± 1.2 pg/day/105 cells, t = 12.99, P < 0.05) [Figure 7C].
Discussion
Vimentin is the most abundant IF protein and it is important for stabilizing the architecture of the cytoplasm. In this study, we investigated the role of vimentin in modulating monocytes viability and cytokine release. We found that suppression of vimentin by siRNA resulted in apoptotic cell death, especially in the presence of LPS, while over-expression of vimentin could significantly reduce apoptosis of THP-1 cells in response to LPS exposure. Consistently, caspase-3 was up-regulated in the cells lacking vimentin. Additionally, suppression of vimentin led to up-regulation of pro-inflammatory cytokines (IL-6 and TNF-α) but down-regulation of anti-inflammatory cytokine (IL-10). These findings suggested that vimentin plays an important role in the immunomodulatory mechanism of sepsis through modulating monocytes viability as well as balance of inflammation-associated cytokines.
Vimentin has been more focus in oncology research. However, vimentin is also highly abundant in human monocytes and activated macrophages,[6] and increased expression of vimentin has been shown to be a late event in the differentiation of human monocytes,[7,8] while suppression of vimentin inhibited formation of macrophage foam cells in response to oxidized LDL.[9] Consistently, a growing number of studies suggest that the infection caused by different pathogens can lead to increased vimentin expression, such as necrotizing fasciitis and myonecrosis caused by invasive infection with group A streptococci,[10] neonatal meningitis caused by E. coli,[11] acute gastroenteritis caused by Salmonella typhimurium,[12] or even Chlamydia and virus infections.[13,14] Mak and Bruggemann[15] reviewed the relationship between vimentin and infection, and pointed out two main aspects of bacteria-vimentin interactions: the role of vimentin in pathogen binding on the cell surface and subsequent bacterial invasion; the interaction of cytosolic vimentin and intracellular pathogens with regards to innate immune signaling. These findings suggest that vimentin may be one of the key molecules that mediate inflammation and immune responses in the infections. However, the mechanism by which pathogenic bacteria bind and function with vimentin is not well understood at present.
The current study demonstrated that vimentin was associated with THP-1 cell apoptosis. In this content, we found that THP-1 cells lacking vimentin were prone to undergoing apoptosis, especially in the presence of LPS, while vimentin over-expression prevented THP-1 cells from apoptosis in response to LPS. Our finding was consistent with the previously reported study, that is, assembly of normal vimentin was disturbed and contributed to amplify the cell death signal.[16] Furthermore, Moisan and Girard reported that vimentin expressed on the cell surfaces of activated human neutrophils.[17] Studies have also shown that IFs control the intracellular distribution of caspases during apoptosis,[18] that apoptosis clearance was related with O-GlcNAc proteins interaction,[19] and that vimentin was involved in peptidylarginine deiminase 2 process during the apoptosis.[20] These findings, together with our findings, suggested that vimentin play a role in regulating innate immunity in sepsis through modulating monocytes viability. The mechanisms of vimentin modulation on monocytes apoptosis, however, are complicated and remain to be further defined. In this content, the current study demonstrated that caspase-3 was increased in the cells lacking vimentin but was not altered in the cells over-expressing vimentin regardless of LPS presence or absence, suggesting amount and activity of caspase-3 may be dependent not only on the interactive binding of vimentin and caspase-3 or other apoptosis-associated proteins, but also on the amount of vimentin and other cytoskeletal proteins expressed by the monocytes. This remains to be further invested in the future.
Vimentin is involved in and regulates the inflammatory response. Jiang et al[21] found that cardioprotective effects in the myocardial ischemia injury model are related to reductions in the inflammatory response by targeting vimentin. Dos Santos et al revealed that vimentin might be a key regulator of the NLRP3 inflammasome.[5] Consistent with these reports, the current study demonstrated suppression of vimentin expression resulted in up-regulation of pro-inflammatory cytokines (IL-6 and TNF-α) but down-regulation of anti-inflammatory cytokines (IL-10). Interestingly, however, suppression of vimentin by siRNA resulted in significant increase of IL-6 and TNF-α release under control culture condition (SF-DMEM), but the effect was mild in the presence of LPS stimulation. In contrast, over-expression of vimentin had dramatic effect IL-10 release, which was significantly reduced in the presence of LPS. While the mechanism of the vimentin regulation on inflammatory cytokines remains to be defined, it may be associated with several factors including NF-κB and NOD2 regulation,[22,23] Dectin-1 recognition and binding,[3] or PKCβ phosphorylation.[24]
In this study, we demonstrated that suppression of vimentin in THP-1 cells resulted in increased apoptotic response in the presence of LPS, while over-expression of vimentin could prevent the cells from apoptosis in response to LPS. In addition, pro-inflammatory cytokines (IL-6 and TNF-α) was increased in the THP-1 cells lacking vimentin, and in contrast, anti-inflammatory cytokine (IL-10) was increased in the cells over-expressing vimentin. These findings suggested that vimentin may regulate innate immunity through modulating monocytes viability as well as inflammatory response in sepsis through shifting the balance of pro-inflammatory and anti-inflammatory cytokines.
Funding
This study was supported by a grant from the National Science Foundation for Young Scientists of China (No. 81501707).
Conflicts of interest
None.
Footnotes
How to cite this article: Su LX, Pan P, Wang XT, Long Y, Liu DW, Zhou X. Vimentin modulates apoptosis and inflammatory cytokine release by a human monocytic cell line (THP-1) in response to lipopolysaccharides in vitro. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000187
Long-Xiang Su and Pan Pan contributed equally to this work.
References
- 1.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013; 13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest 2009; 119:1763–1771. doi: 10.1172/JCI38339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiagarajan PS, Yakubenko VP, Elsori DH, Yadav SP, Willard B, Tan CD, et al. Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc Res 2013; 99:494–504. doi: 10.1093/cvr/cvt117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toda M, Kuo CH, Borman SK, Richardson RM, Inoko A, Inagaki M, et al. Evidence that formation of vimentin mitogen-activated protein kinase (MAPK) complex mediates mast cell activation following FcepsilonRI/CC chemokine receptor 1 cross-talk. J Biol Chem 2012; 287:24516–24524. doi: 10.1074/jbc.M111.319624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 2015; 6:6574.doi: 10.1038/ncomms7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cain H, Kraus B, Krauspe R, Osborn M, Weber K. Vimentin filaments in peritoneal macrophages at various stages of differentiation and with altered function. Virchows Arch B Cell Pathol Incl Mol Pathol 1983; 42:65–81. [DOI] [PubMed] [Google Scholar]
- 7.Rius C, Cabanas C, Aller P. The induction of vimentin gene expression by sodium butyrate in human promonocytic leukemia U937 cells. Exp Cell Res 1990; 188:129–134. doi: 10.1016/0014-4827(90)90287-K. [DOI] [PubMed] [Google Scholar]
- 8.Rius C, Aller P. Vimentin expression as a late event in the in vitro differentiation of human promonocytic cells. J Cell Sci 1992; 101:395–401. [DOI] [PubMed] [Google Scholar]
- 9.Yao W, Huang L, Sun Q, Yang L, Tang L, Meng G, et al. The inhibition of macrophage foam cell formation by tetrahydroxystilbene glucoside is driven by suppressing vimentin cytoskeleton. Biomed Pharmacother 2016; 83:1132–1140. doi: 10.1016/j.biopha.2016.08.032. [DOI] [PubMed] [Google Scholar]
- 10.Bryant AE, Bayer CR, Huntington JD, Stevens DL. Group A streptococcal myonecrosis: increased vimentin expression after skeletal-muscle injury mediates the binding of Streptococcus pyogenes. J Infect Dis 2006; 193:1685–1692. doi: 10.1086/504261. [DOI] [PubMed] [Google Scholar]
- 11.Zou Y, He L, Huang SH. Identification of a surface protein on human brain microvascular endothelial cells as vimentin interacting with Escherichia coli invasion protein IbeA. Biochem Biophys Res Commun 2006; 351:625–630. doi: 10.1016/j.bbrc.2006.10.091. [DOI] [PubMed] [Google Scholar]
- 12.Murli S, Watson RO, Galan JE. Role of tyrosine kinases and the tyrosine phosphatase SptP in the interaction of Salmonella with host cells. Cell Microbiol 2001; 3:795–810. doi: 10.1046/j.1462-5822.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 13.Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 2008; 4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du N, Cong H, Tian H, Zhang H, Zhang W, Song L, et al. Cell surface vimentin is an attachment receptor for enterovirus 71. J Virol 2014; 88:5816–5833. doi: 10.1128/JVI.03826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mak TN, Bruggemann H. Vimentin in bacterial infections. Cells 2016; 5:18.doi: 10.3390/cells5020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byun Y, Chen F, Chang R, Trivedi M, Green KJ, Cryns VL. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ 2001; 8:443–450. doi: 10.1038/sj.cdd.4400840. [DOI] [PubMed] [Google Scholar]
- 17.Moisan E, Girard D. Cell surface expression of intermediate filament proteins vimentin and lamin B1 in human neutrophil spontaneous apoptosis. J Leukoc Biol 2006; 79:489–498. doi: 10.1189/jlb.0405190. [DOI] [PubMed] [Google Scholar]
- 18.Dinsdale D, Lee JC, Dewson G, Cohen GM, Peter ME. Intermediate filaments control the intracellular distribution of caspases during apoptosis. Am J Pathol 2004; 164:395–407. doi: 10.1016/S0002-9440(10)63130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ise H, Goto M, Komura K, Akaike T. Engulfment and clearance of apoptotic cells based on a GlcNAc-binding lectin-like property of surface vimentin. Glycobiology 2012; 22:788–805. doi: 10.1093/glycob/cws052. [DOI] [PubMed] [Google Scholar]
- 20.Hsu PC, Liao YF, Lin CL, Lin WH, Liu GY, Hung HC. Vimentin is involved in peptidylarginine deiminase 2-induced apoptosis of activated Jurkat cells. Mol Cells 2014; 37:426–434. doi: 10.14348/molcells.2014.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang S, Liu Y, Wang J, Zhang Y, Rui Y, Zhang Y, et al. Cardioprotective effects of monocyte locomotion inhibitory factor on myocardial ischemic injury by targeting vimentin. Life Sci 2016; 167:85–91. doi: 10.1016/j.lfs.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Stevens C, Henderson P, Nimmo ER, Soares DC, Dogan B, Simpson KW, et al. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut 2013; 62:695–707. doi: 10.1136/gutjnl-2011-301775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang SH, Chi F, Peng L, Bo T, Zhang B, Liu LQ, et al. Vimentin, a novel NF-kappaB regulator, is required for meningitic Escherichia coli K1-induced pathogen invasion and PMN transmigration across the blood-brain barrier. PLoS One 2016; 11:e0162641.doi: 10.1371/journal.pone.0162641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol 2003; 5:59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]