

Abstract
Infection with chikungunya virus (CHIKV) typically causes an acute illness characterized by fever, rash, and arthralgia. However, CHIKV infection can sometimes progress to chronic arthritis or even lethal disease. CHIKV continues to cause significant morbidity worldwide as its vector mosquitoes expand and spread. There are currently no approved vaccines or antiviral drugs available for the prevention or treatment of CHIKV. Although antibody therapy has shown promise in the prevention or treatment of CHIKV disease in preclinical models, challenges remain for implementing such therapies. Here, from the B cells of a survivor of natural CHIKV infection, we isolated ultrapotent neutralizing human monoclonal antibodies (mAbs) and encoded their sequences into mRNA molecules delivered by infusion. One human mAb, CHKV-24, was expressed to biologically significant levels in vivo following infusion of mRNAs in lipid nanoparticles in mice. We evaluated the protective capacity of CHKV-24 mAb IgG protein or mRNA in mouse models of CHIKV infection. Treatment with CHKV-24 mRNA protected mice from arthritis, musculoskeletal tissue infection, and lethality in a dose-dependent manner and reduced viremia to undetectable levels at 2 days post-inoculation. Infusion of macaques with CHKV-24 mRNA achieved a mean maximal human mAb concentration of 10.1 to 35.9 μg/mL, with a half-life of 23 days, a level well above that needed for protection in mice. Studies with CHKV-24 mRNA in macaques demonstrated a dose-response effect after the first dose of mRNA and maintained levels after second dose. These preclinical data with CHKV-24 mRNA suggest it might be useful to prevent human disease.
One Sentence Summary
Lipid nanoparticles carrying mRNA encoding a neutralizing human antibody against chikungunya virus protect against infection and arthritis.
Introduction
Monoclonal antibody (mAb) therapy has become one of the central tools in the pharmacological armamentarium for treatment of chronic conditions such as cancer and autoimmune diseases. MAbs also have been shown in many preclinical studies to have promise for treatment or short-term prevention of virus infection, especially emerging infections (1). Antibody treatment or prevention of infectious diseases has several theoretical advantages over vaccine development strategies, since antibodies have an extraordinary history of safety in humans (2, 3), a rapid development pathway of years (in contrast to decades for vaccines), and can be used in any age or virtually any high-risk or immunocompromised population (4). Passive immunization by administration of antibodies has the potential for a near immediate onset of action, compared with vaccines that require weeks to months to induce protective effects. However, to date, only palivizumab (Synagis; Medimmune), a humanized murine mAb for respiratory syncytial virus, has been licensed for use in humans. The barriers to common use of mAbs for management of infectious diseases stem mainly from the complexity and high cost of manufacture of recombinant antibody proteins.
Recently, gene transfer methods using adeno-associated virus (AAV) as a method for generating antiviral neutralizing antibodies in vivo (5, 6) or methods based on delivery of DNA (7, 8) or RNA (9) have been developed that enable injection of recipients with vectors encoding antibody sequences for rapid in vivo production of recombinant antibodies. DNA-encoded human antibodies have been shown to mediate beneficial effects in small animal models of infection (8, 10, 11), cancer immunotherapy (12, 13), and metabolic disease (14).These approaches obviate the need for the complex manufacturing processes inherent in production and quality control of large amounts of recombinant proteins. Instead, nucleic acids encoding antibodies can be manufactured quickly and likely can be produced commercially at much lower cost than the equivalent protein therapeutic. This approach could revolutionize the feasibility of widespread use of human mAb therapy and prophylaxis for infectious diseases. Administration of nucleic acids for foreign protein expression was described first in 1990 (15) and for purposes of active vaccination since 1993 (16), but the transfer of cDNA or mRNA encoding recombined antibodies as a means of passive immunization is more recent.
mRNA immunizations are promising but face some limitations, because the delivery of large amounts of RNA can trigger innate immune recognition by TLRs and/or RIG-I-like receptors that limit the level and duration of protein expression. Major improvements in sustainability of expression from exogenously delivered mRNA have been achieved by the use of modified nucleosides (17). We, and others, have shown that active vaccination with modified mRNA vaccine candidates for influenza, Zika, and cytomegalovirus induce robust and protective adaptive immune responses (18–20). Nucleoside-modified mRNA encapsulated into lipid nanoparticles (LNPs) also has been shown to be an effective tool for protein therapy (9, 21).
Here, we investigated if transfer of mRNAs encoding potently neutralizing human mAbs could achieve levels of expression in vivo that confer protection against infection and severe disease sequelae. For these studies, we isolated a new, potently inhibitory human mAb against chikungunya virus (CHIKV), a single-stranded RNA virus of the genus Alphavirus in the Togaviridae family. CHIKV is a mosquito-transmitted virus that causes systemic infection in humans characterized by acute onset of fever and severe polyarthralgia and is associated with chronic arthritis in many cases. Persistent debilitating arthralgia was reported in 5 to 61% of subjects in a series of 20 clinical studies (22). Mortality occurs more rarely, mostly in newborns, the immunocompromised, or older adults (23–25). Because of its high attack rate, CHIKV has caused large epidemics in Asia, Africa, Europe and the Pacific and Indian Oceans (26). A large outbreak in the Western Hemisphere began in 2013 in the Caribbean islands, and local transmission in the Americas rapidly led to over a million reported cases in a two-year span (27). U.S. travelers returning from the Caribbean to most U.S. states were affected, and local transmission was reported in the U.S. in Florida, Puerto Rico and the U.S. Virgin Islands (28).
We recently showed that some human mAbs to CHIKV prepared as IgG proteins have potent neutralizing activity (29–31) and confer protective effects against virus replication in mice (29, 32) and nonhuman primates (33). Here, we developed new ultrapotent human mAbs to CHIKV and used mRNA encoding the antibodies to treat and protect against CHIKV. The experiments revealed that delivery of optimized mRNA molecules encoding a potent human antibody resulted in expression at biologically significant levels in the serum of both mice (14.9 μg/mL) and nonhuman primates (10.1 to 35.9 μg/mL) and elicited protection against arthritis, musculoskeletal disease, and lethal challenge in mouse models.
Results
Donor selection.
We screened plasma samples from 44 subjects for the presence for neutralizing antibodies to CHIKV using virus replicon particles (VRPs) based on the Sri Lankan strain SL15649 (34). Most (40 of 44) donors had endpoint plasma neutralizing titers of >40, and ten subjects had titers >5,000. The highest titer observed was a remarkable value of 31,766. We used the PBMCs from this donor for mAb discovery experiments.
Generation of CHIKV-specific mAbs.
Following EBV transformation, we generated 59 lymphoblastoid cell lines with supernatants containing CHIKV-reactive antibodies that bound to CHIKV particles [181/25 vaccine strain (35)] and exhibited 66% or greater neutralizing activity. We fused the lines with the highest level of CHIKV reactivity and recovered 18 as hybridomas that secreted CHIKV-specific antibodies. Nucleotide sequence analysis of the antibody heavy chain variable genes for the 18 recovered cloned hybridoma lines revealed that each of the mAbs was encoded by a distinct V-D-J recombination. Eleven of the 18 recovered mAbs that bound to CHIKV 181/25 virion particles in ELISA also possessed neutralizing activity. The values for concentration of mAb that gave half-maximal inhibitory response (IC50) in the neutralization assay ranged from 4 to 2,266 ng/mL (Table 1). We chose the most potent inhibitory antibody (CHKV-24, an IgG1 with an IC50 of 4 ng/mL) for further study in the mRNA delivery experiments.
Table 1.
Neutralizing activity of CHKV-specific human mAbs
| MAb clone (CHKV-) |
IC50 FRNT* (ng/mL) against CHIKV |
|---|---|
| 24 | 4 |
| 35 | 11 |
| 27 | 17 |
| 8 | 24 |
| 12 | 25 |
| 48 | 37 |
| 29 | 86 |
| 32 | 81 |
| 53 | 319 |
| 31 | 684 |
| 50 | 2,266 |
| 1 | > |
| 4 | > |
| 9 | > |
| 13 | > |
| 19 | > |
| 22 | > |
| 23 | > |
Fifty percent maximal inhibitory concentration of antibody in a focus reduction neutralization test.
Protection of mice by delivery of CHKV24 mAb.
AG129 mice that lack receptors for interferon–α/β and –γ are highly vulnerable to infection with CHIKV (36) and thus provide a highly stringent model for testing antiviral compounds (37–39) or the protective efficacy of CHKV-24 mAb. Mice were treated by the intravenous route with a single administration of purified IgG for CHKV-24 mAb at doses of 10, 2 or 0.4 mg/kg. A dose-dependent concentration of human IgG in mouse serum was observed, as expected (Figure 1A). At 24 hours, mice were challenged by subcutaneous injection in the footpad and hock of the right leg with a total volume of 0.1 mL of the diluted virus (0.05 mL each site) with a lethal dose of CHIKV (102.5 TCID50). All mice survived after prior infusion with the dose of 10 mg/kg or 2 mg/kg of the CHKV-24 mAb (Figure 1B). Half (50%) of animals treated with 0.4 mg/kg of mAb survived (Figure 1B). All animals treated with a control antibody mAb against influenza A virus died, whereas all unchallenged (naïve) animals survived (Figure 1B). Comparison of the survival experiments and the level of serum human IgG levels achieved suggested that the CHKV-24 IgG could protect AG129 mice in a lethal challenge model at systemic levels of 10 μg/mL of antibody at the time of challenge.
Figure 1. Prophylactic efficacy of CHKV-24 IgG protein.
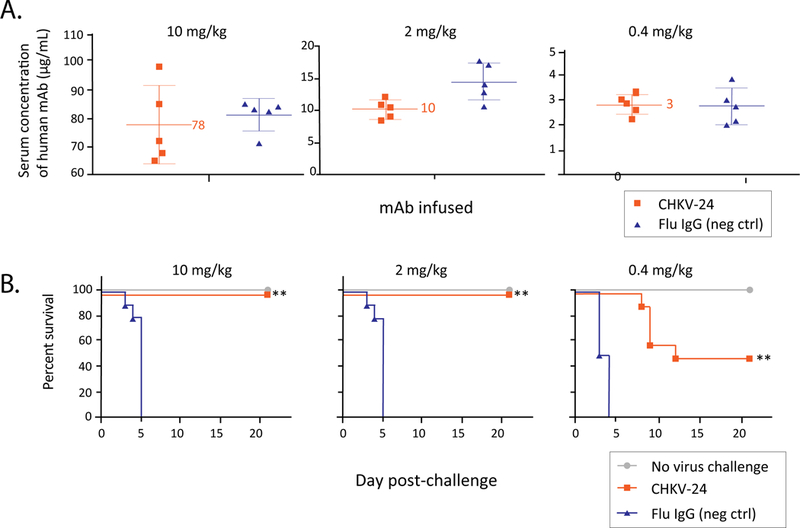
A. Concentration of human IgG in AG129 mouse serum after CHKV-24 IgG protein infusion. Total human IgG levels were measured 24 h after infusion of purified human mAb IgG protein for CHIKV-24 (red) or an irrelevant control mAb to influenza (flu; green). Animals receiving 10 mg/kg (200 μg), 2 mg/kg (40 μg) or 0.4 mg/kg (8 μg) of recombinant CHKV-24 IgG protein had mean systemic IgG concentrations of CHKV-24 of 78 μg/mL, 10 μg/mL or 3 μg/mL, respectively. The serum concentration of the influenza control antibody was similar. Five animals per group were tested. The mean values are indicated, and error bars show the standard deviation.
(B) Survival of AG129 mice treated with mAb CHKV-24 IgG protein and challenged with CHIKV. Mice were treated with a single intravenous injection of 10 mg/kg (left), 2 mg/kg (middle), or 0.4 mg/kg (right) of mAb CHKV-24 (red) or an irrelevant human IgG to influenza A virus (flu; green) 24 h prior to virus challenge with 102.5 TCID50 of CHIKV-LR06. The group shown in blue contained animals that were neither treated nor challenged. Challenged animals were anesthetized with isoflurane prior to subcutaneous injection in the footpad and hock of the right leg with a total volume of 0.1 mL of the diluted virus (0.05 mL each site). Kaplan-Meier survival plot is shown. Survival data were analyzed using the Wilcoxon log-rank survival analysis. ** indicates P<0.01, as compared with control. The number of animals in each group was 10. Animals receiving 2 or 10 mg/kg of CHKV-24 were completely protected (100% survival) from lethal challenge. Animals receiving 0.4 mg/kg of chikungunya IgG were partially protected (50% survival). All animals receiving the flu IgG at 10, 2 or 0.4 mg/kg succumbed (0% survival) to infection by day 5.
Protection of immunocompromised mice against lethal challenge by delivery of CHKV-24 mRNA.
Next, we determined if an mRNA encoding CHKV-24 also could confer a protective effect. The CHKV-24 antibody mRNA was formulated in a lipid nanoparticle (LNP) and stored at 4°C until use. AG129 mice were treated by the intravenous route with a single administration of CHKV-24-encoding mRNA at doses of 0.5, 0.1 or 0.02 mg/kg. The mRNA infusion resulted in expression of human antibody in vivo, with a dose-dependent concentration of human IgG detected in mouse serum 24 hours after infusion (Figure 2A). The mean peak serum concentration of the 0.5 mg/kg treated group was 14.9 μg/mL. Complete survival of mice (100%) was observed after treatment with the highest dose of 0.5 mg/kg of CHKV-24 mRNA (Figure 2B). 40% of the animals survived after treatment with 0.1 mg/kg mRNA, whereas survival was not observed at the lowest dose of 0.02 mg/kg mRNA (Figure 2B). Despite the lower level of protection at the two lower doses of mRNA, the survival curves were improved (P < 0.01) compared with placebo treatment, demonstrating a benefit of the CHKV-24 mRNA treatment even at the lower doses tested. We compared the level of serum human IgG levels achieved by mRNA infusion in a parallel group of non-challenged animals receiving 0.5 or 0.1 mg/kg of IgG (Figure 2A) with the results of the survival experiments (Figure 2B). The comparison suggested that the CHKV-24 mRNA treatment could completely protect AG129 mice in the lethal challenge model when a 10 μg/mL concentration of systemic CHKV-24 was achieved, while at least half of the animals were protected at CHKV-24 serum levels of about 3 μg/mL.
Figure 2. Prophylactic efficacy of CHKV-24 mRNA.
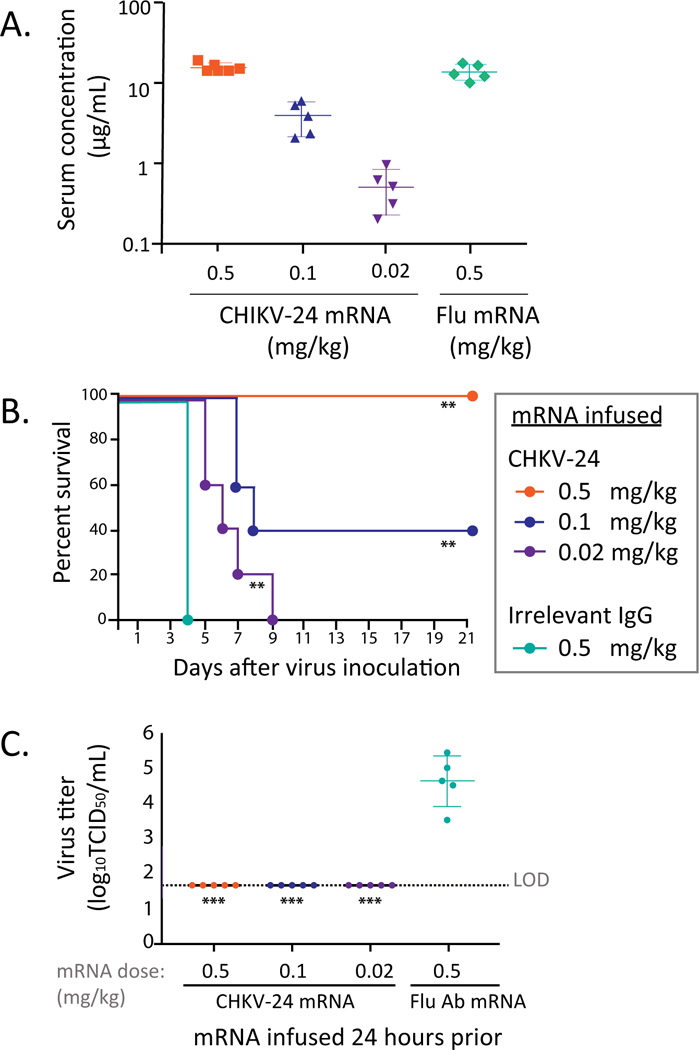
(A) Expression of human mAb IgG in serum following intravenous infusion of mRNA. Expression levels of human IgG in the serum of AG129 mice following infusion of mRNA encoding CHKV-24 or a control (influenza; flu) mAb mRNA. The CHKV-24 mRNA was administered to mice at a dose of 0.5 (red), 0.1 (green) or 0.02 (purple) mg/kg or the influenza mRNA at a dose of 0.5 mg/kg (orange), by intravenous tail vein injection. Animals were bled at 24 h after infusion to measure systemic levels of IgG. Each group had 5 animals. An additional group of 10 animals was infused with these mRNAs and doses at the same time and challenged with virus 24 h after infusion (results shown in panel B and C). The mean values are indicated, and error bars show the standard deviation.
(B) Protection against lethal CHIKV infection mediated by human mAb expressed from mRNA. CHKV-24 mRNA was administered to mice as a prophylaxis at 0.5 (blue), 0.1 (orange) or 0.02 (green) mg/kg by intravenous tail vein injection. An irrelevant IgG mRNA was used at 0.5 mg/kg as a control (red). Each group of animals was challenged 24 h after infusion with CHIKV strain LR06 and monitored for mortality. The number of animals in each group was 10. **(P<0.01) Indicates the survival differed significantly from that of the group treated with 0.5 mg/kg of the irrelevant IgG (Wilcoxon log-rank survival test).
(C) Titer of CHIKV in AG129 mice treated with mRNA encoding mAb CHKV-24 IgG or an mRNA encoding an irrelevant control mAb. Serum samples obtained two days after virus challenge were assayed on Vero cell monolayer cultures to determine virus titer (log10TCID50/mL). The limit of detection (LOD) was 1.7. The mean values are indicated, and error bars show the standard deviation. Comparisons were made by Kruskal Wallis test with Dunn’s post-test. *** indicates P<0.0003, as compared with control IgG. The number of animals in each group was 5.
Virus titer in serum two days after challenge was reduced below the level of detection in all mice treated with CHKV-24 mRNA, as compared with an average of 4.6 log10 50% tissue culture infectious doses (TCID50) in placebo-treated controls (Figure 2C). Although virus was not observed in the serum in the low-dose treatment group, virus likely replicated in other tissues, since mortality occurred. The reduction of viremia to the limit of detection corroborated a therapeutic effect against viral replication.
Protection of immunocompetent mice against arthritis and musculoskeletal disease by delivery of CHKV-24 mRNA.
While immunocompromised mice provide a stringent protection model, CHIKV infection is rarely fatal in humans but instead causes severe, acute and chronic polyarthralgia and polyarthritis. Accordingly, we evaluated whether post-exposure treatment with CHKV-24 mRNA LNP could protect in the immunocompetent mouse model of CHIKV-induced arthritis and musculoskeletal disease, where subcutaneous infection results in a biphasic swelling of the infected foot peaking at 3 and 7 dpi (34, 40). When CHKV-24 mRNA was administered 4 h post-infection, wild-type (WT) C57BL/6 mice did not develop foot swelling compared to the mice that received an mRNA LNP encoding an irrelevant IgG control (Figure 3A). At 2 dpi, serum from the majority of mice receiving CHKV-24 mRNA had titers at the limit of detection, whereas high levels of viremia were observed in the control treated mice (Figure 3B). At 7 dpi, CHKV-24 mRNA-treated mice had an 80-fold reduction in viral RNA in the ipsilateral ankle, with no spread to the contralateral ankle compared to the control mRNA treated mice (Figure 3C). Histological analysis of the ipsilateral foot at 7 dpi showed large cellular infiltration into the joint space of the control mRNA treated mice, whereas this finding was absent in the CHKV-24 mRNA-treated group (Figure 3D). Slides from two of five mice administered CHKV-24 mRNA showed minimal cellular infiltration in the midfoot (Figure 3D; right panel), although the remainder had detectable cellular infiltration in the soft tissue (Figure 3D; middle panel). However, the extent of immune cells and edema in the midfoot was reduced markedly compared to the control mRNA-treated mice (Figure 3D; left panel). These results show that CHKV-24 mRNA therapy also confers protection in an immunocompetent mouse model of CHIKV arthritis.
Figure 3. Therapeutic administration of CHKV-24 mRNA reduces clinical disease and viral titer in WT mice.
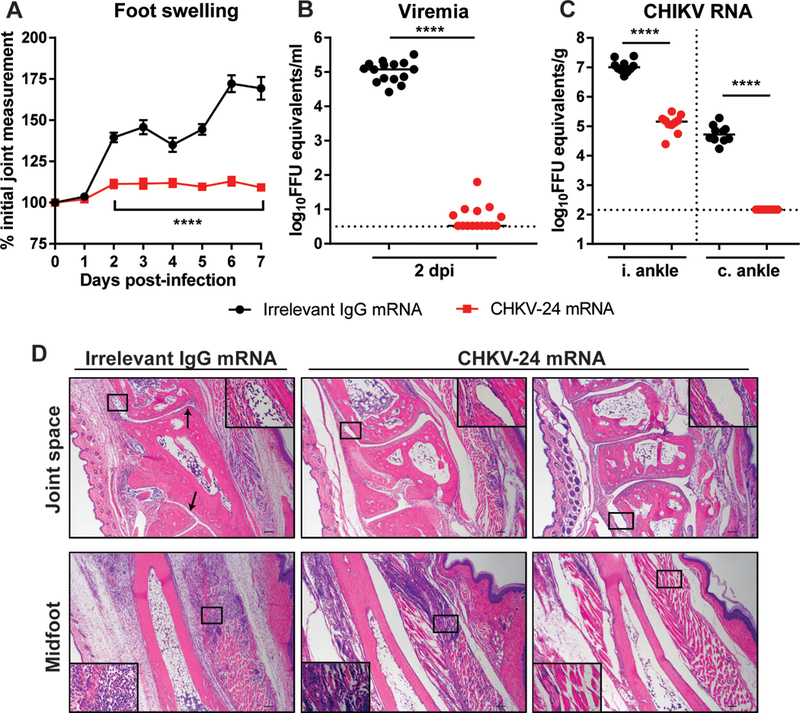
C57BL/6 mice received human IgG mRNA (10 mg/kg) by intravenous injection 4 h after inoculation with CHIKV-LR06. (A) Foot swelling was monitored by digital calipers (n = 15/group, two experiments, two-way ANOVA with Sidak’s post-test). Line indicates significance between the groups at each time point. Error bars indicate standard error of the mean. (B) Serum was collected at 2 dpi or (C) ipsilateral (i.) and contralateral (c.) ankles were harvested on 7 dpi, and viral RNA was quantified by qRT-PCR (serum: n = 15/group, two experiments; ankles: n = 10/group, two experiments, Mann Whitney test for each tissue). Bars indicate median values. Dotted lines indicate the limit of detection. (D) Ipsilateral feet were collected at 7 dpi, fixed in PFA, decalcified, paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Images show low-magnification (scale bar 100 μm) with a high magnification inset (scale bar 10 μm). Top and bottom panels are representative images of the joint space and midfoot, respectively (n = 5/group, two experiments). Arrows indicate cellular infiltrate in joint space.
CHKV-24 expression from modified RNA in cynomolgus macaques.
We next tested whether infusion of CHKV-24 mRNA LNPs could induce expression of human IgG in the serum of monkeys that corresponds to the protective concentrations observed in mice. A group of 4 macaques was infused by the intravenous route with mRNA encoding CHKV-24 at 0.5 mg/kg. This study was repeated with 6 macaques per group. There were no test article-related clinical signs, changes in body weight, or changes in food consumption during the study. Human IgG1 expression peaked at 24 hours after the start of infusion for animals dosed at 0.5 mg/kg, with mean human IgG levels of 10.1 to 35.9 μg/mL) (Figure 4 and Table 2). The half-life of the mRNA-expressed CHKV-24 was 23 days in macaques (Table 2).
Figure 4. Pharmacodynamics of CHK-24 mRNA in cynomolgus monkeys.
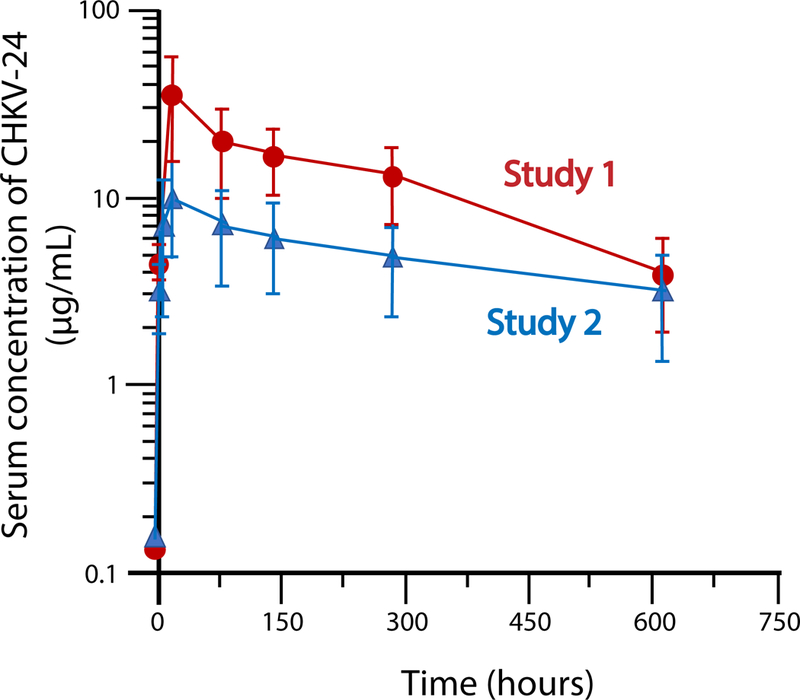
Data shows the total human IgG1 concentrations from two NHP studies in which animals were treated with 0.5 mg/kg of CHKV24 mRNA by intravenous infusion. CHKV-24 mRNA was delivered over 60 min, in a volume of 5 mL/kg, and dose concentration of 0.02 mg/mL. Four or six animals were tested in each group for study 1 or 2, respectively. The mean values are indicated, and error bars show the standard deviation. Maximum concentration of 35.9 μg/mL and 10.1 μg/mL was observed at 24 h after infusion for Study 1 (red curve) and Study 2 (blue curve), respectively.
Table 2.
Human IgG pharmacokinetic parameters of CHKV-24 in macaques following delivery of antibody-encoding mRNA*
| Tmax (hr) | Cmax (μg/mL) | AUC0–720hr (hr*μg/mL) |
t1/2 (hr) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | CV% | Mean | SD | CV% | Mean | SD | CV% | Mean | SD | CV% |
| 24 | 0 | 0 | 10.1 | 5.36 | 53 | 3,720 | 1,950 | 52.4 | 561 | 65.8 | 11.7 |
The CHKV-24 NHP serum samples from study 2 were analyzed using a Human Therapeutic IgG1 ELISA Kit, with serum dilutions ranging from 1:100 to 1:1,000. Parameters were calculated using Phoenix pharmacokinetic software (Certara, USA). A standard curve of absorbance at 450 nm versus log (concentration) was fit with a 4-parameter logistic equation for IgG1 quantification.
We next tested whether or not the function of the antibodies in macaque serum expressed following mRNA infusion was comparable to that of the recombinant mAb CHKV-24 IgG protein. The 24-hour timepoint serum samples from the studies shown in Figure 4 were tested for anti-CHIKV activity. Antibody function was assessed by a 50% plaque reduction neutralization test (PRNT50) and ELISA; a standard curve for concentration versus activity in each assay was generated using dilution curves of purified recombinant CHKV-24 at defined concentrations (Figure 5). These analyses suggested that the mRNA-expressed antibody was fully functional.
Figure 5. Functional concentrations of mRNA-expressed CHKV-24 IgG in NHP serum.
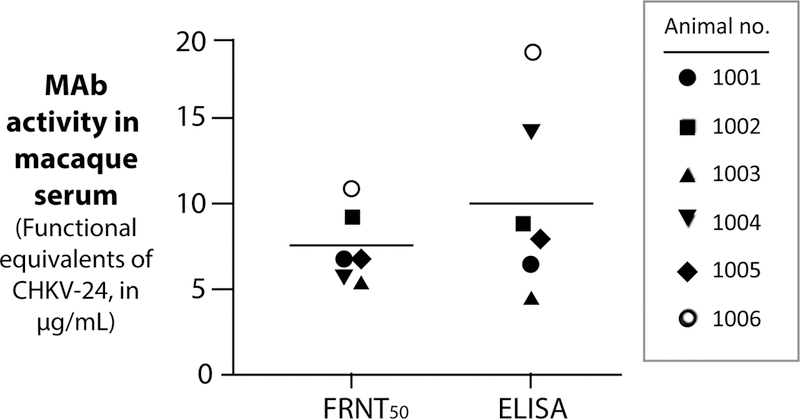
Macaques were infused with 0.5 mg/kg of mRNA encoding CHKV-24, and 24 h later serum samples were obtained and tested for the presence of CHIKV-specific binding or neutralizing antibodies. Antibody function was assessed by a focus reduction neutralization test (FRNT50; left) and by ELISA (right); a standard curve for concentration versus activity in each assay was generated using dilution curves of purified CHKV-24 at defined concentrations. A group of 6 animals was tested, and the in vitro experiments were performed twice. The mean values are indicated, and error bars show the standard deviation.
CHKV-24 expression from modified RNA in cynomolgus macaques after multiple doses.
Following a single-dose study in NHPs using the CHKV-24 mRNA, we tested expression of CHKV-24 IgG after multiple mRNA doses in a NHP study under GLP conditions. Macaques were administered two intravenous doses (PBS control or 0.3, 1.0 or 3.0 mg/kg of CHKV-24 mRNA) one week apart on days 0 and 7, followed by a necropsy on main study animals on day 8 or following a 12-week treatment free recovery period (day 98). The study design contained the following endpoints: clinical observations, body weights, food consumption, hematology, coagulation, clinical chemistry, cytokine analysis, C3a and Bb complement analysis, toxicokinetics analysis, human IgG protein expression, gross necropsy, organ weights, and histopathology. The only observed findings were 1) a dose-dependent increase in splenic weight without microscopic findings was observed in animals 24 hours post second dose (day 8) which was not observed in the recovery animals (day 98) and 2) 3 out of 5 male macaques in the 3.0 mg/kg dose group exhibited an increase in CCL2 levels at 2 hours after the second dose (day 8), which returned to baseline by 6 hours. An increase in CCL2 levels was not observed in females. Liver function tests were normal in all samples tested. Multiple serum samples were collected throughout the duration of the study to measure expression after multiple doses.
CHK24 IgG was detected in macaque plasma samples from all animals after mRNA administration. Increasing CHKV-24 IgG concentrations were observed with increasing doses of mRNA. At 24 hours after dosing, maximum CHKV-24 IgG concentrations of 16.2 μg/mL (after dose 1) or 28.8 μg/mL (after dose 2) were observed for animals administered the high dose of 3.0 mg/kg mRNA (Figure 6). Sex-based differences were not detected in CHKV-24 IgG plasma levels. In animals in the group treated with the highest dose (3.0 mg/kg), CHKV-24 IgG plasma levels were detected 90 days after the second dose, with an average serum concentration of 2.9 μg/mL (Figure 6).
Figure 6. Concentrations of mRNA-expressed CHKV-24 IgG in NHP serum after repeat mRNA dosing.
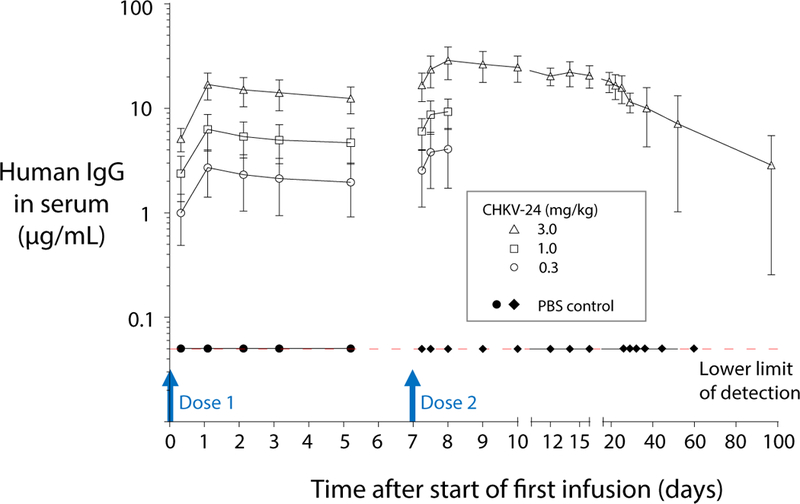
In a non-clinical GLP repeat dose study using groups of four animals per treatment, macaques were infused with mRNA encoding CHKV-24 on day 0 and day 7, and serum samples were collected for human IgG1 quantification at 6, 24, 48, 72 or 120 hours after the start of infusion of dose 1 and 6, 12, 24, 48, 72, 120, 168, 216, 288, 360, 432, 528, 720, 1,080 or 2,160 hours after the start of infusion of dose 2. A dose response was observed after each administration of mRNA encoding CHKV-24. Antibody concentrations after day 8 were calculated only for the highest dose level (3 mg/kg). At 24 hours after dosing with 3.0 mg/kg mRNA, the maximum CHKV-24 IgG serum concentration was 16.2 μg/mL or 28.8 μg/mL for dose 1 or dose 2, respectively. The mean values are indicated, and error bars show the standard deviation.
Discussion
Here, we show that an mRNA-encoded Ab with virus neutralizing activity has potency at equivalent levels as observed with the corresponding purified IgG form of the mAb. We showed that infusion of mRNA encoding a potent virus neutralizing antibody can induce concentrations of human IgG in the serum of treated mice that protect immunocompromised and immunocompetent mice against lethal challenge and arthritis, respectively. The same mRNA infusions achieved protective concentrations of CHIK-24 in macaques with peak concentrations achieved at 24 hours after infusion of 10.1 to 35.9 μg/mL. The differences in peak expression level across the two NHP studies are attributed to assay and study variability and the outbred population of animals. Furthermore, the serum half-life of this antibody in macaques was found to be 23 days following a single infusion. These studies provide a rational basis for use of similar RNA LNP formulations in humans and point the way toward defining the human dose of mRNA needed to accomplish biologically meaningful expression of human IgGs in vivo.
Treatment with CHKV-24 mRNA or mAb significantly protected mice from lethality in a dose-dependent manner. Viremia was reduced to the limit of detection on 2 dpi, further supporting the efficacy of CHKV-24 in this mouse model. Protection was mediated by systemic levels of 10 μg/mL of CHKV-24 mAb, which has an in vitro neutralization IC50 value of 4 ng/mL. The higher concentration needed for effect in vivo may be explained in part by the stringency of the testing in immunocompromised AG129 mice, which lack interferon–α/β and –γ responses as well as the expected antigen excess, which effectively shifts the neutralization to a requirement for greater antibody concentrations (41). Determining the ratio of effective in vitro and in vivo concentrations for antiviral antibodies is complex, and often requires combined experimental-mathematical approaches that include precise estimates of virion-antibody interaction stoichiometry, tissue distribution and half-life (42). Post-exposure treatment in WT mice reduced viremia, diminished infection in the ipsilateral foot, prevented spread to the contralateral foot, and protected against foot swelling. Administration of CHKV-24 antibody mRNA, when given as a single or two 60-minute intravenous infusions, was well tolerated in monkeys at all dose levels tested. Human IgG1 antibodies were detectable through day 83 when dosed once with CHKV-24 mRNA at 0.5 mg/kg and through day 100 after two doses at 3 mg/kg of CHKV-24 mRNA.
These studies suggest that passive immunization or treatment of humans by administration of LNP formulations containing mRNAs encoding for an anti-CHIKV antibody may be feasible. The ability to deliver sufficient protective levels of antibodies in humans using such LNP RNA formulations can only be determined in human clinical studies. Based on our results, the CHKV-24 mRNA has been selected as a development candidate for testing in humans. The high levels of mAb expression achieved here with CHKV-24 mRNA in mice and NHPs, and the complete protection of mice against lethal disease or arthritis, suggest that additional studies are warranted to determine the promise of this approach for prevention or treatment of CHIKV disease. Prophylaxis with this antibody treatment could be considered for travelers to affected areas, and clinical testing of therapy of infected patients could be evaluated to see if reductions of virus load prevent the development of acute and/or chronic arthritis. If successful, such studies could suggest a platform for rapid development and deployment of mRNA-encoded mAbs for many other emerging infectious diseases. Although there are no licensed mRNA-encoded therapeutic antibodies yet, and the final cost of such a product has not yet been determined, the cost of production is likely to be far less than that of the corresponding protein IgG molecule made in cultured cells. This antibody delivery modality also could incorporate the entire range of recent antibody engineering innovations, including Fc alterations for extended half-life (43, 44), optimized effector functions (45), or Fc mutations eliminating Fc receptor interactions (46, 47), to prevent the potential for antibody-mediated enhancement, which may occur in some natural flavivirus infections.
Materials and Methods
Study design.
We sought to develop an mRNA-encoded human monoclonal antibody for CHKV. The study was designed to isolate human B cells secreting CHIK-neutralizing antibodies from an immune donor and then obtain the antibody variable genes encoding neutralizing antibodies. The antibody genes were synthesized as RNA and formulated in lipid nanoparticles, then expressed following intravenous infusion of mice or nonhuman primates to test for level of expression and ability to protect against virus challenge.
Isolation of human mAbs.
The studies were approved by the Ethics Review Committee of the Medical Faculty, University of Colombo, Sri Lanka (serving as the NIH-approved Institutional Review Board (IRB) for Genetech Research Institute) and the Institutional Review Board of Vanderbilt University Medical Center. Sri Lankan blood samples obtained were discarded buffy coats from routine blood donations at the National Blood Center in Colombo, Sri Lanka. All samples were de-identified prior to removal from the National Blood Center. Peripheral blood mononuclear cells (PBMCs) and plasma samples were separated at Genetech Research Institute by density gradient centrifugation and then cryopreserved and stored on liquid nitrogen until transfer to Vanderbilt using a liquid nitrogen dry shipper.
We did not prescreen donors prior to PBMC and plasma collection for the presence of CHIKV-specific antibodies, although it was known that CHIKV infection was common in Colombo during the years 2006–2008 (prior to collection). PBMCs for the selected donor were thawed rapidly, and the B cells in the sample were transformed with EBV in the presence of CpG to generate B cell lymphoblastoid cell lines. Supernatants from these cell lines were screened for the presence of human CHIKV-specific binding antibodies by ELISA using the live attenuated CHIKV vaccine virus strain 181/25 (35) as the antigen. Transformed B cells with a high ELISA binding signal were collected and fused by electrofusion to the myeloma cell line HMMA2.5, distributed into culture plates and expanded in culture, and selected by growth in hypoxanthine-aminopterin-thymidine medium containing ouabain. The supernatants of cell lines plated from the fusion reactions were tested after two weeks for the presence of human antibodies binding to CHIKV strain 181/25. Hybridoma cells for CHIKV-reactive lines were cloned by single-cell sorting using a FACSAria III sorting flow cytometer, in a Baker laminar flow hood for aerosol containment. The clonal lines were adapted to serum-free medium. Supernatants from cloned hybridomas growing in serum-free medium were collected, purified, and concentrated from clarified medium by protein G chromatography.
Cell lines for virus assays.
BHK-21 cells (ATCC CCL-10) were maintained in α-minimal essential medium (αMEM; Gibco) supplemented to contain 10% fetal bovine serum (FBS) and 10% tryptose phosphate (Sigma). Vero 81 cells (ATCC CCL-81) were maintained in αMEM supplemented to contain 5% FBS. Medium for all cells was supplemented to contain 0.29 mg/mL L-glutamine (Gibco), 100 U/mL penicillin (Gibco), 100 μg/mL streptomycin (Gibco), and 500 ng/mL amphotericin B. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
Viruses.
Virus suspensions of CHIKV attenuated vaccine strain 181/25 (35, 48) were grown on Vero cell monolayer cultures, and supernatant was harvested 36 h post-inoculation and clarified by centrifugation at 2,000 x rpm for 10 min at 4°C. The CHIKV East/Central/South African [ECSA] genotype strain used for neutralization screening in this study was SL15649 (Genbank accession number GU189061). For in vivo studies, the Reunion Island CHKV isolate LR2006-OPYI was obtained from Robert Tesh (UTMB, WRCEVA). Stocks for these viruses were prepared in C6/36 Aedes albopictus cells.
Focus reduction neutralization assay.
MAbs were diluted serially and incubated with 102 focus-forming units (FFU) of CHIKV for 1 h at 37°C in duplicate wells. MAb-virus mixtures were added to Vero cell monolayer cultures for 90 min at 37°C followed by an overlay with a 1% methylcellulose in Minimum Essential Medium (MEM, Invitrogen) supplemented with penicillin and streptomycin, 10 mM HEPES, and 2% HI-FBS. Cells were fixed 18 h later after the addition of 1% paraformaldehyde (PFA) in PBS. Infected cells were incubated with 500 ng/mL of murine mAb CHK-11 (49). After washing and incubation with HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich), foci of infection were developed using TrueBlue substrate (KPL) and counted using a BioSpot plate reader (Cellular Technology, Inc.). Inoculated wells containing mAb were compared to wells inoculated in the absence of mAb. The half maximal effective concentration (EC50 value) was calculated using non-linear regression analysis constraining the bottom to 0 and top to 100.
Virus capture ELISA for hybridoma screening.
Antibody binding to virus particles was performed by coating ELISA assay plates (Nunc 242757) with purified murine mAb CHK-152 (49), prepared at 1 μg/mL in 0.1 M Na2CO3 and 0.1 M NaHCO3 pH 9.7 binding buffer and incubating at 4°C overnight. After incubating plates for 1 h at RT with blocking buffer (5% powdered milk and 2% goat serum in PBS with Tween 20 [PBS-T]), plates were washed five times with PBS-T and incubated with 25 μL of culture supernatant from BHK21 cell monolayers infected with CHIKV vaccine strain 181/25. After incubation at room temperature for 1 h, plates were washed ten times with PBS-T, and 10 μL of B cell culture supernatant was added into 25 μL/well of blocking buffer. Plates were incubated at room temperature for 1 h prior to washing five times with PBS-T. A secondary antibody conjugated to alkaline phosphatase (goat anti-human Fc; Meridian Life Science, W99008A) was applied at a 1:5,000 dilution in 25 μL/well of blocking buffer, and plates were incubated at room temperature for 1 hour. Following five washes with PBS-T, phosphatase substrate solution (1 mg/mL phosphatase substrate in 1 M Tris aminomethane [Sigma, S0942]) was added at 25 μL/well, and plates were incubated at room temperature for 2 h before determining the optical density at 405 nm using a Biotek plate reader.
mRNA synthesis.
mRNA was synthesized in vitro by T7 RNA polymerase-mediated transcription from a linearized DNA template, which incorporates the 5′ and 3′ untranslated regions (UTRs) and a poly-A tail, as previously described (50, 51). The final mRNA uses a cap 1 structure to increase mRNA translation efficiency. After purification, the mRNA was diluted in citrate buffer to the desired concentration.
Lipid nanoparticle (LNP) formulation.
LNP formulations were prepared by ethanol drop nanoprecipitation, as previously described (52). Briefly, lipids were dissolved in ethanol at molar ratios of 50:10:38.5:1.5 (ionizable lipid: DSPC: cholesterol: PEG-lipid). The lipid mixture was combined with a 6.25 mM sodium acetate buffer (pH 5) containing mRNA at a ratio of 3:1 (aqueous:ethanol) using a microfluidic mixer (Precision Nanosystems). Formulations were dialyzed against phosphate-buffered saline (pH 7.4) in dialysis cassettes for at least 18 h. Formulations were concentrated using Amicon ultra centrifugal filters (EMD Millipore), passed through a 0.22-μm filter, and stored at 4°C until use. All formulations were tested for particle size, RNA encapsulation, and endotoxin and were found to be between 80 nm – 100 nm in size, with greater than 90% encapsulation, and <10 EU/mL endotoxin.
Protection studies in mice.
This lethal challenge study was conducted in accordance with the approval of the Institutional Animal Care and Use Committee of Utah State University (Protocol #2339). The work was performed in the AAALAC-accredited Laboratory Animal Research Center of Utah State University (PHS Assurance no. A3801–01) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Revision; 2010).
Male and female AG129 mice, bred in an in-house colony at Utah State University, were assigned randomly to experimental groups and individually marked with ear tags. The CHIKV-LR-2006 stock was prepared by passaging the virus twice in C6/36 Aedes albopictus cells. The CHIKV stock had a titer of 109.5 TCID50/mL. The CHIKV-specific mAb, CHKV-24 was collected from hybridoma supernatants and purified by protein G chromatography, and the antibody suspension was supplied in a ready-to-treat liquid form. Virus titers in sera were assayed using an infectious cell culture assay where a specific volume of serum was added to the first tube of a series of dilution tubes. Serial dilutions were made and added to Vero cell culture monolayers. Three days later cytopathic effect (CPE) was used to identify the end-point of infection. Four replicates were used to calculate the TCID50 per mL of serum.
The concentration of human IgG in AG129 mouse serum after CHKV-24 IgG protein infusion was determined by an IgG ELISA that detected human IgG (but not murine IgG). Thus, the total human IgG concentration was the concentration of the passively administered mAb. Total human IgG levels were measured 24 h after infusion of purified human mAb IgG1 protein for CHIKV-24 or an irrelevant control human IgG mAb to influenza virus hemagglutinin. Animals were administered 10 mg/kg (200 μg), 2 mg/kg (40 μg) or 0.4 mg/kg (8 μg) of recombinant CHKV-24 IgG protein or the same doses of the flu control antibody. Five animals were tested per group.
For protection studies, cages of mice were assigned randomly to groups of 5 animals. Groups of mice were treated with 0.5, 0.1 or 0.2 mg/kg of CHKV-24 IgG via a single intravenous tail vein injection 24 h prior to virus challenge. Alternatively, similar groups of animals were given mRNA encoding human antibodies by an intravenous route, at 10, 1, or 0.4 mg/kg mRNA. Mice then were anesthetized with isoflurane prior to subcutaneous injection in the footpad and hock of the right leg with 102.5 TCID50 of CHKV a total volume of 0.1 mL (0.05 mL each site). Survival was monitored twice daily through the critical period of disease to 7 days post-infection. Serum was collected by cheek vein bleed on day 2 post-infection to measure viremia.
Experiments with WT mice were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health after approval by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance Number: A3381–01). All injections with virus were performed under anesthesia with ketamine hydrochloride (80 mg/kg) and xylazine (15 mg/kg).
Four-week-old WT C57BL/6J mice (000664; Jackson Laboratories) were inoculated subcutaneously in the left footpad with 103 FFU of CHIKV-LR in Hanks’ Balanced Salt Solution (HBSS) supplemented 1% heat-inactivated (HI)-FBS. mRNA encoding human mAbs was administered by an intravenous route 4 h post-infection at 10 mg/kg mRNA. Ipsilateral foot swelling was monitored via measurements (width x height) using digital calipers. Serum was collected on 2 dpi. To measure tissue viral titers, mice were sacrificed, perfused extensively with 20 mL of PBS, and tissues were collected on 7 dpi. Serum and tissues were titered for CHIKV RNA by qRT-PCR using RNA isolated from viral stocks as a standard curve to determine FFU equivalents, as previously described (30). For histology, animals were sacrificed and perfused with 4% PFA on 7 dpi. Ipsilateral feet were collected, and hair was removed using Nair (Church & Dwight). Tissue was fixed for 24 h in 4% PFA, rinsed with PBS and water, and then decalcified for 14 days in 14% EDTA free acid (Sigma) at pH 7.2. Decalcified tissue was rinsed, dehydrated, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H & E). Images were acquired on a Nikon Eclipse E400 microscope.
Expression of human antibody protein in nonhuman primates after infusion of mRNA.
Nonhuman primate studies were conducted at Charles River Laboratories (Sherbrooke, Quebec, Canada). Animal experiments and husbandry followed NIH guidelines (NIH Publications No. 8023, eighth edition) and the USA National Research Council and the Canadian Council on Animal Care (CCAC) guidelines. No treatment randomization or blinding methods were used for any of the animal studies. Sample sizes were determined by the resource equation method. The repeat-dose NHP study was conducted under GLP conditions.
Macaques used for study were 2 to 3 years old males and weighed between 2.3 and 2.8 kg at the initiation of dosing. Tuberculin tests were carried out on arrival at the test facility and were negative. Animals were housed socially (up to 3 animals of same sex and same dosing group together) in stainless steel cages equipped with a stainless-steel mesh floor and an automatic watering valve, with the exception of times when they were separated for designated study procedures/activities. Animals were housed in a temperature- and humidity-controlled environment (21–26 °C and 30–70%, respectively), with an automatic 12-hour dark/light cycle. Primary enclosures were as specified in the USDA Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and as described in the Guide for the Care and Use of Laboratory Animals (53). PMI Nutrition International Certified Primate Chow No. 5048 (25% protein) was provided twice daily, except during designated procedures. The chow was provided in amounts appropriate for the size and age of the animals. Municipal tap water after treatment by reverse osmosis and ultraviolet irradiation was made freely available to each animal via an automatic watering system (except during designated procedures).
Infusion of mRNA in macaques.
We performed a single-dose study of lipid nanoparticle formulated mRNA encoding the CHKV-24 mAb in cynomolgus monkeys at Charles River Laboratories. We determined the pharmacokinetics of CHIKV antibody after a 60-min intravenous infusion of 0.5 mg/kg mRNA. The following parameters and end points were evaluated in this study: clinical signs, body weights, food evaluation, and human IgG expression in serum. The infusion was delivered at a dose rate of 5 mL/kg/h using a temporary indwelling catheter inserted in an appropriate peripheral vein once at the start of the study.
For the repeat-dose study, mRNA was administered to animals by 60-min intravenous infusion (5 mL/kg/h) via a tail vein, delivering doses on days 0 and 7). The dose volume for each animal was based on the most recent body weight measurement. The animals were restrained temporarily for dose administration and were not sedated. Each infused dose was administered using a temporary indwelling catheter inserted in a tail vein and an injection set connected to an infusion pump. The first day of dosing was designated as day 0. The injection areas were marked as frequently as required to allow appropriate visualization of administration sites. Hair was clipped or shaved to improve visualization of the injection sites.
Collection of serum samples from NHPs.
For the single-dose NHP study, blood samples (0.3 mL) were collected in serum separator tubes on day 1 (at pre-dose and 6, 24, 96, 168, 336, or 720 h after the start of infusion) and on day 82. For the repeat-dose study, samples were collected at 6, 24, 48, 72 or 120 hours after start of infusion (dose 1) and 6, 12, 24, 48, 72, 120, 168, 216, 288, 360, 432, 528, 720, 1,080 or 2,160 hours after start of infusion (dose 2). The blood samples were maintained at ambient temperature for a target of 30 min following collection, then processed to serum within 90 min of collection. The samples were centrifuged for 10 min in a refrigerated centrifuge (set to maintain 4°C) at 1,200 × g. The resulting serum was separated, aliquoted, and frozen immediately over dry ice before storage at −80°C.
Quantification of human IgG in NHP serum.
The CHKV-24 NHP samples were analyzed using a Human Therapeutic IgG1 ELISA Kit (Cayman Chemical, #500910). The kit instructions were followed exactly with serum dilutions ranging from 1:100 to 1:1,000. A standard curve of absorbance at 450 nm versus log (concentration) was fit with a 4-parameter logistic equation for IgG1 quantification.
Estimation of expressed human antibody half-life in NHPs.
Human IgG pharmacokinetic parameters were estimated using Phoenix software (Certara, USA) using a non-compartmental approach (NCA), consistent with the intravenous route of administration. Parameters were estimated using nominal sampling times relative to the start of each dose administration. Concentration values reported as Below Quantifiable Limit were assigned a value of zero. The area under the concentration versus time curve (AUC) was calculated using the linear trapezoidal method with linear interpolation. AUC values were reported to 3 significant digits, and t1/2 values were reported to one decimal place. The terminal elimination phase for each subject was estimated using at least three observed concentration values. The slope of the elimination phase was determined using log linear regression on the unweighted concentration data.
Statistical analysis.
Survival data were analyzed using the Wilcoxon log-rank survival analysis. Comparisons were made by Kruskal Wallis test with Dunn’s post-test on R, a language and environment for statistical computing.
Supplementary Material
Acknowledgments
We thank Erica Parrish and Ryan Irving at Vanderbilt University Medical Center for laboratory management support, Justin Julander of Utah State University for assistance with evaluation of antibodies, and Qing Tan at Washington University School of Medicine for help with animal studies.
Funding: The work was supported by Defense Advanced Research Projects Agency (DARPA) grant W911NF-13–1-0417, NIH grant R01 AI114816, and by Moderna Therapeutics. The views, opinions and/or findings expressed are those of the author and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government.
Footnotes
Competing interests: S.M.E., E.H-N., M.A.T. and S.H. are employees of Moderna Therapeutics. G.C. was an employee of Moderna Therapeutics when the studies were conducted. G.C. owns stock in Moderna. M.S.D. is a member of the Scientific Advisory Board of Moderna. J.E.C. has served as a consultant for Takeda Vaccines, Sanofi Pasteur, Pfizer, and Novavax, is on the Scientific Advisory Boards of CompuVax, GigaGen, and Meissa Vaccines, and is Founder of IDBiologics, Inc. Vanderbilt University Medical Center and Moderna have patent applications submitted pertaining to the CHKV-24 antibody and mRNA formulations of CHKV-24.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Material. The CHKV antibodies in this study are available by MTA with Vanderbilt University Medical Center. The research grade LNP-formulated mRNA encoding CHK24 IgG is available by MTA with Moderna Therapeutics.
References and Notes
- 1.Marston HD, Paules CI, Fauci AS, Monoclonal antibodies for emerging infectious diseases - Borrowing from history. N. Engl. J. Med. 378, 1469–1472 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Matucci A, Nencini F, Pratesi S, Maggi E, Vultaggio A, An overview on safety of monoclonal antibodies. Curr. Opin. Allergy Clin. Immunol. 16, 576–581 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Niebecker R, Kloft C, Safety of therapeutic monoclonal antibodies. Curr. Drug. Saf. 5, 275–286 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Sparrow E, Friede M, Sheikh M, Torvaldsen S, Therapeutic antibodies for infectious diseases. Bull. World Health Organ. 95, 235–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balazs AB, Ouyang Y, Hong CM, Chen J, Nguyen SM, Rao DS, An DS, Baltimore D, Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat. Med. 20, 296–300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson PR, Schnepp BC, Zhang J, Connell MJ, Greene SM, Yuste E, Desrosiers RC, Clark KR, Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat. Med. 15, 901–906 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muthumani K, Flingai S, Wise M, Tingey C, Ugen KE, Weiner DB, Optimized and enhanced DNA plasmid vector based in vivo construction of a neutralizing anti-HIV-1 envelope glycoprotein Fab. Hum. Vaccin. Immunother. 9, 2253–2262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott STC, Kallewaard NL, Benjamin E, Wachter-Rosati L, McAuliffe JM, Patel A, Smith TRF, Schultheis K, Park DH, Flingai S, Wise MC, Mendoza J, Ramos S, Broderick KE, Yan J, Humeau LM, Sardesai NY, Muthumani K, Zhu Q, Weiner DB, DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. N..PJ. Vaccines 2, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardi N, Secreto AJ, Shan X, Debonera F, Glover J, Yi Y, Muramatsu H, Ni H, Mui BL, Tam YK, Shaheen F, Collman RG, Kariko K, Danet-Desnoyers GA, Madden TD, Hope MJ, Weissman D, Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muthumani K, Block P, Flingai S, Muruganantham N, Chaaithanya IK, Tingey C, Wise M, Reuschel EL, Chung C, Muthumani A, Sarangan G, Srikanth P, Khan AS, Vijayachari P, Sardesai NY, Kim JJ, Ugen KE, Weiner DB, Rapid and long-term immunity elicited by DNA-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J. Infect. Dis. 214, 369–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel A, Park DH, Davis CW, Smith TRF, Leung A, Tierney K, Bryan A, Davidson E, Yu X, Racine T, Reed C, Gorman ME, Wise MC, Elliott STC, Esquivel R, Yan J, Chen J, Muthumani K, Doranz BJ, Saphire EO, Crowe JE, Broderick KE, Kobinger GP, He S, Qiu X, Kobasa D, Humeau L, Sardesai NY, Ahmed R, Weiner DB, In vivo delivery of synthetic human DNA-encoded monoclonal antibodies protect against ebolavirus infection in a mouse model. Cell Rep. 25, 1982–1993 e1984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muthumani K, Marnin L, Kudchodkar SB, Perales-Puchalt A, Choi H, Agarwal S, Scott VL, Reuschel EL, Zaidi FI, Duperret EK, Wise MC, Kraynyak KA, Ugen KE, Sardesai NY, Kim Joseph J., Weiner DB, Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immuno.l Immunother. 66, 1577–1588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duperret EK, Trautz A, Stoltz R, Patel A, Wise MC, Perales-Puchalt A, Smith T, Broderick KE, Masteller E, Kim JJ, Humeau L, Muthumani K, Weiner DB, Synthetic DNA-Encoded Monoclonal Antibody Delivery of Anti-CTLA-4 Antibodies Induces Tumor Shrinkage In Vivo. Cancer Res 78, 6363–6370 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khoshnejad M, Patel A, Wojtak K, Kudchodkar SB, Humeau L, Lyssenko NN, Rader DJ, Muthumani K, Weiner DB, Development of novel DNA-encoded PCSK9 monoclonal antibodies as lipid-lowering therapeutics. Mol. Ther., (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL, Direct gene transfer into mouse muscle in vivo. Science 247, 1465–1468 (1990). [DOI] [PubMed] [Google Scholar]
- 16.Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, Gromkowski SH, Deck RR, DeWitt CM, Friedman A, et al. , Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 259, 1745–1749 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Kariko K, Buckstein M, Ni H, Weissman D, Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS, Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125 e1110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindgren G, Ols S, Liang F, Thompson EA, Lin A, Hellgren F, Bahl K, John S, Yuzhakov O, Hassett KJ, Brito LA, Salter H, Ciaramella G, Lore K, Induction of robust B cell responses after influenza mRNA vaccination is accompanied by circulating hemagglutinin-specific ICOS+ PD-1+ CXCR3+ T follicular helper cells. Front. Immunol. 8, 1539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang F, Lindgren G, Lin A, Thompson EA, Ols S, Rohss J, John S, Hassett K, Yuzhakov O, Bahl K, Brito LA, Salter H, Ciaramella G, Lore K, Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol. Ther. 25, 2635–2647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An D, Schneller JL, Frassetto A, Liang S, Zhu X, Park JS, Theisen M, Hong SJ, Zhou J, Rajendran R, Levy B, Howell R, Besin G, Presnyak V, Sabnis S, Murphy-Benenato KE, Kumarasinghe ES, Salerno T, Mihai C, Lukacs CM, Chandler RJ, Guey LT, Venditti CP, Martini PGV, Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 21, 3548–3558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Aalst M, Nelen CM, Goorhuis A, Stijnis C, Grobusch MP, Long-term sequelae of chikungunya virus disease: A systematic review. Travel Med. Infect. Dis. 15, 8–22 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Economopoulou A, Dominguez M, Helynck B, Sissoko D, Wichmann O, Quenel P, Germonneau P, Quatresous I, Atypical Chikungunya virus infections: clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Reunion. Epidemiol. Infect. 137, 534–541 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Evans-Gilbert T, Chikungunya and neonatal immunity: Fatal vertically transmitted Chikungunya infection. Am. J. Trop. Med. Hyg. 96, 913–915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kee AC, Yang S, Tambyah P, Atypical chikungunya virus infections in immunocompromised patients. Emerg. Infect. Dis. 16, 1038–1040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weaver SC, Arrival of chikungunya virus in the new world: prospects for spread and impact on public health. PLoS Negl. Trop. Di.s 8, e2921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yactayo S, Staples JE, Millot V, Cibrelus L, Ramon-Pardo P, Epidemiology of Chikungunya in the Americas. J. Infect. Dis. 214, S441–S445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Centerrs for Disease Control and Prevention, Chikungunya virus in the United States, URL : https://www.cdc.gov/chikungunya/geo/united-states.html; accessed June 25, 2018.
- 29.Smith SA, Silva LA, Fox JM, Flyak AI, Kose N, Sapparapu G, Khomandiak S, Ashbrook AW, Kahle KM, Fong RH, Swayne S, Doranz BJ, McGee CE, Heise MT, Pal P, Brien JD, Austin SK, Diamond MS, Dermody TS, Crowe JE Jr., Isolation and characterization of broad and ultrapotent human monoclonal antibodies with therapeutic activity against Chikungunya virus. Cell Host Microbe 18, 86–95 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fox JM, Long F, Edeling MA, Lin H, van Duijl-Richter MKS, Fong RH, Kahle KM, Smit JM, Jin J, Simmons G, Doranz BJ, Crowe JE Jr., Fremont DH, Rossmann MG, Diamond MS, Broadly neutralizing alphavirus antibodies bind an epitope on E2 and inhibit entry and egress. Cell 163, 1095–1107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawman DW, Fox JM, Ashbrook AW, May NA, Schroeder KMS, Torres RM, Crowe JE Jr., Dermody TS, Diamond MS, Morrison TE, Pathogenic Chikungunya virus evades B cell responses to establish persistence. Cell Rep. 16, 1326–1338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miner JJ, Cook LE, Hong JP, Smith AM, Richner JM, Shimak RM, Young AR, Monte K, Poddar S, Crowe JE Jr., Lenschow DJ, Diamond MS, Therapy with CTLA4-Ig and an antiviral monoclonal antibody controls chikungunya virus arthritis. Sci. Transl. Med. 9(375) pii: eaah3438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broeckel R, Fox JM, Haese N, Kreklywich CN, Sukulpovi-Petty S, Legasse A, Smith PP, Denton M, Corvey C, Krishnan S, Colgin LMA, Ducore RM, Lewis AD, Axthelm MK, Mandron M, Cortez P, Rothblatt J, Rao E, Focken I, Carter K, Sapparapau G, Crowe JE Jr., Diamond MS, Streblow DN, Therapeutic administration of a recombinant human monoclonal antibody reduces the severity of chikungunya virus disease in rhesus macaques. PLoS Negl. Trop. Dis. 11, e0005637 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morrison TE, Oko L, Montgomery SA, Whitmore AC, Lotstein AR, Gunn BM, Elmore SA, Heise MT, A mouse model of chikungunya virus-induced musculoskeletal inflammatory disease: evidence of arthritis, tenosynovitis, myositis, and persistence. Am. J. Pathol. 178, 32–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levitt NH, Ramsburg HH, Hasty SE, Repik PM, Cole FE Jr., Lupton HW, Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 4, 157–162 (1986). [DOI] [PubMed] [Google Scholar]
- 36.Couderc T, Chretien F, Schilte C, Disson O, Brigitte M, Guivel-Benhassine F, Touret Y, Barau G, Cayet N, Schuffenecker I, Despres P, Arenzana-Seisdedos F, Michault A, Albert ML, Lecuit M, A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 4, e29 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaur P, Chu JJ, Chikungunya virus: An update on antiviral development and challenges. Drug Discov. Today, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Partidos CD, Weger J, Brewoo J, Seymour R, Borland EM, Ledermann JP, Powers AM, Weaver SC, Stinchcomb DT, Osorio JE, Probing the attenuation and protective efficacy of a candidate chikungunya virus vaccine in mice with compromised interferon (IFN) signaling. Vaccine 29, 3067–3073 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang E, Kim DY, Weaver SC, Frolov I, Chimeric Chikungunya viruses are nonpathogenic in highly sensitive mouse models but efficiently induce a protective immune response. J. Virol. 85, 9249–9252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner J, Anraku I, Le TT, Larcher T, Major L, Roques P, Schroder WA, Higgs S, Suhrbier A, Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84, 8021–8032 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pierson TC, Diamond MS, A game of numbers: the stoichiometry of antibody-mediated neutralization of flavivirus infection. Prog. Mol. Biol. Transl. Sci. 129, 141–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandenberg OF, Magnus C, Rusert P, Gunthard HF, Regoes RR, Trkola A, Predicting HIV-1 transmission and antibody neutralization efficacy in vivo from stoichiometric parameters. PLoS Pathog. 13, e1006313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S, Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 169, 5171–5180 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR, Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Mathieu M, Brezski RJ, IgG Fc engineering to modulate antibody effector functions. Protein Cell 9, 63–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chappel MS, Isenman DE, Everett M, Xu YY, Dorrington KJ, Klein MH, Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc. Natl. Acad. Sci. U.S.A. 88, 9036–9040 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tao MH, Morrison SL, Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 143, 2595–2601 (1989). [PubMed] [Google Scholar]
- 48.Mainou BA, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS, Reovirus cell entry requires functional microtubules. MBio 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pal P, Dowd KA, Brien JD, Edeling MA, Gorlatov S, Johnson S, Lee I, Akahata W, Nabel GJ, Richter MK, Smit JM, Fremont DH, Pierson TC, Heise MT, Diamond MS, Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 9, e1003312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS, Modified mRNA vaccines protect against Zika virus infection. Cell 169, 176 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Richner JM, Jagger BW, Shan C, Fontes CR, Dowd KA, Cao B, Himansu S, Caine EA, Nunes BTD, Medeiros DBA, Muruato AE, Foreman BM, Luo H, Wang T, Barrett AD, Weaver SC, Vasconcelos PFC, Rossi SL, Ciaramella G, Mysorekar IU, Pierson TC, Shi PY, Diamond MS, Vaccine mediated protection against zika virus-induced congenital disease. Cell 170, 273–283 e212 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, Lynn A, Bulychev A, McFadyen I, Chan J, Almarsson O, Stanton MG, Benenato KE, A novel amino lipid series for mRNA D\delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 26, 1509–1519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Council NR, Guide for the Care and Use of Laboratory Animals. (National Academies Press, Washington, DC, 2011). [8th edition] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.