

Abstract
Dynamic changes in alternative splicing during the life cycle of neurons supports development, plasticity, and is implicated in disease pathology. Cell-specific alternative splicing programs coordinate exon selection across networks of functionally connected genes. In this opinion piece we highlight recent publications that identify some of the molecular mechanisms – RNA and DNA binding proteins and epigenetic modifications – which direct cell specific exon selection during pre-mRNA splicing. Aberrant splicing patterns are signature features of a growing number of diseases of the nervous system. Recent publications demonstrate the value of delineating basic mechanisms that dictate exon choice to inform the development of new therapeutic strategies that correct or compensate for damaging deficits in alternative splicing.
Alternative Splicing of Neuronal Genes: New mechanisms and New Therapies
Alternative splicing – a universal mechanism
Alternative splicing is a form of RNA processing that is critical for virtually every stage in the life cycle of a neuron – starting from early neuronal differentiation, to axonal guidance and synapse formation, to supporting cell signaling and plasticity, and for programmed cell death [1–6]. The capacity of multi-exon genes to generate hundreds to thousands of splice variants is on full display in the nervous systems of animals (e.g. neurexin gene; see Fig. 1) [7]. There are technical challenges associated with identifying alternative mRNA splice isoforms across cell-types of multi-cellular organisms. However, transcriptome analyses of tissues and single cells are revealing the rich palettes of alternative splice isoforms across cell-types, development, adaptation, and in disease [1,4,6,8,9]. In the nervous system, alternative splicing of pre-mRNA is a central mechanism underlying many neuronal functions including adaptation in response to the ever-changing external environment. Cell-specific changes in the patterns of alternative splicing of pre-mRNA, and the ensuing modifications in protein activity, adjust cell function and network dynamics on timescales that are faster; more subtle; and through processes that are potentially less energetically demanding, as compared to those associated with changes in gene expression.
Figure 1. RNA and DNA binding proteins regulate alternative splicing of Neurexin by different mechanisms and in response to different cellular factors.
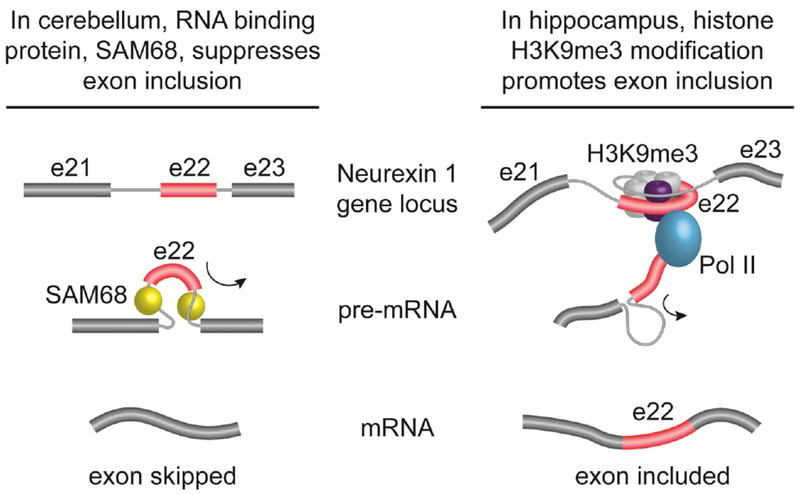
The Neurexin 1 gene (Nrxn1) generates multiple splice isoforms and contains many alternatively expressed exons. Nrxn1 exon 22 is alternatively expressed. Left: Exon 22 of Nrxn1 is repressed in both cerebellum and hippocampus by RNA binding protein SAM68 [56]. SAM68 binds cis elements in 5’ and 3’ introns flanking exon 22 (red). Right: In response to neuronal activity and memory consolidation, H3K9me3 modification is induced in hippocampus. H3K9me3 modified histone associate with DNA at Nrxn1 exon 22 locus. Elongation of Pol II is slowed, promoting exon 22 inclusion during pre-mRNA splicing. Activity-induced H3K9me3 underlies a change in the pattern of alternative splicing of Nrxn1 in hippocampal neurons [1].
AS is the rule rather than the exception
Next-generation sequencing studies suggest that more than 90% of human multi-exon genes undergo alternative splicing and improved methods for single molecule sequencing are revealing the degree of coordinated splicing in individual RNAs as well as across genes, according to cell state [6,7,9]. Recent discoveries provide exciting insights into the neuronal-specific mechanisms and factors that regulate exon selection and have motivated research on identifying the regulators that control the action of splicing factors. In this opinion piece, we focus on recently published data that identify cellular mechanisms that regulate alternative splicing in neurons and, for a couple of example, how these findings informed promising new therapies.
Cellular factors that control alternative splicing
Central nervous systems are hotbeds of alternative splicing of pre-mRNAs and some splice sites are evolutionarily conserved, consistent with a central role in encoding properties essential for neuronal function. We now know many of the trans-acting nuclear splicing factor proteins that bind cis-elements in pre-mRNAs to either promote, or hinder recruitment of the spliceosome at intron/exon boundaries (see Fig. 1). Splicing factors bind to single or clusters of RNA motifs that are typically located in introns, to enhance or inhibit target exon inclusion. A subset of known RNA binding protein families includes polypyrimidine track binding proteins (PTB), RBFOX, NOVA, SR-rich (serine/arginine-rich), STAR, and heterogeneous nuclear ribonucleoproteins (hnRNPs). Genome-wide splicing factor binding maps at different developmental time points, in different tissues, and in disease form the initial framework of a code to eventually predict coordinated state- and cell-specific splicing patterns for networks of genes. However, as highlighted in a recent analysis of the binding specificities of >70 recombinant human RNA binding proteins, as assessed in vitro using an array of oligonucleotides, several factors including RNA secondary structure and neighboring nucleotide sequence, have the potential to influence RNA binding protein interactions with target RNAs [10].
Epigenetic modifications have also been shown to influence alternative splicing by mechanisms that are different from those governing the actions of RNA binding proteins. Transcription and pre-mRNA processing are coupled events; transcription rates influence the pattern of alternative splicing and alternative splicing influences transcription [11,12]. DNA-binding proteins, including histones, influence RNA Polymerase II (Pol II) kinetics and can alter exon choice during pre-mRNA splicing (see Fig. 1) [11,12]. DNA-binding proteins physically tether Pol II and spliceosome components, to slow Pol II elongation and impact splicing. Slow transcription elongation rates tend to favor alternative exon inclusion by promoting recruitment of splicing factors and spliceosome to intron-exon boundaries, whereas faster rates favor alternative exon skipping [13–20]. DNA binding proteins that have been implicated in alternative splicing include CTCF binding to Cd45 gene [16], HP1γ binding to Cd44 gene [21] and a recent publication, discussed below, shows that histone H3K9me3 modification controls cell-specific splicing of a number of genes, including Nrxn1 gene, during memory consolidation in mice [1] (see Fig. 1). Recent genome-wide analyses of epigenetic markers shows how their occupancy correlates with alternative splicing outcome (exon inclusion or repression) [22–24].
Factors that modify splicing factor action
Many cellular factors regulate the action of RNA binding protein splicing factors to influence their cell-specific actions, these factors affect splicing factor expression levels, high order assembly of protein-RNA complexes, posttranslational modification, autoregulation, and alternative splicing [10,25–27]. A recent study by Black’s group illustrates the divergent actions of two splice isoforms of the splicing factor RBFOX1 in hippocampal neurons: one isoform localizes to, and acts in the nucleus to affect splicing of target pre-mRNAs; while the other localizes to, and acts in the cytoplasm to affect target mRNA stability. Cytoplasmic acting RNA binding proteins including RBFOX have been shown to bind 30UTRs of several mRNAs to influence mRNA stability [26]. Vamp1 mRNA is a target of RBFOX1 and encodes for VAMP1/synaptobrevin, a v-SNARE that is involved in synaptic vesicle priming and fusion at the neuromuscular junction and in a subpopulation of hippocampal inhibitory neurons (Gtexportal). Relative to VAMP2, VAMP1 is expressed at overall lower levels in brain, but it sits at the center of a protein phosphorylation network suggesting that phosphoregulation of VAMP1, and its associated proteins, is important for regulating neuronal function [57]. Cytoplasmic RBFOX1 stabilizes Vamp1 mRNA by preventing the binding of a microRNA (miR-9) to the 30UTR of Vamp1 mRNA. In the absence of RBFOX1 binding, miR-9 promotes Vamp1 mRNA degradation [26]. This cytoplasmic action of RBFOX1 is cell-specific and by stabilizing Vamp1 mRNA in inhibitory hippocampal neurons, RBFOX1 upregulates inhibitory output and influences the balance of excitation/ inhibitory signaling in hippocampal circuits [26]. This study emphasizes the dual actions of RBFOX1—as a splicing factor or as a regulator of RNA stability—depending on as yet unidentified factors that influence exon choice during alternative splicing of Rbfox1 pre-mRNA.
Long-term changes in nervous system function, including memory formation, are strongly correlated with a range of epigenetic modifications [22,28,29]. While most studies typically focus on documenting changes in gene expression levels, there is evidence of substantial alteration in exon choice during memory consolidation in mice [28]. The impact of memory-associated alterations in histone methylation marks was recently demonstrated for the neurexin 1 gene, Nrxn1. Histone modification accompanies memory formation, altering alternative splicing of Nrxn1 specifically in memory-activated neurons in the dentate gyrus of the hippocampus [1].
Activity-dependent Nrxn1 contains exon 22 (Nrxn1+22), an isoform which was found to protect memories from extinction [1]. Ding et al., demonstrated that the histone mark, H3K9me3, pauses RNA polymerase II, promoting exon 22 inclusion during pre-mRNA processing, and consequently leading to synapse restructuring (Fig. 1). The shift in the pattern of Nrxn1 splicing from Nrxn1 Δ22 to Nrxn1 + 22, and the subsequent impact on behavior, were shown to be long-lasting and dependent on the combined action of several proteins: A zinc-finger domain protein, p66α, recognizes a TGATAA motif in exon 22; p66α is phosphorylated by AMP-dependent protein-activated kinase following neuronal activity; HDAC2 binds activated p66α; and Suv39h1 a histone methyltransferase is recruited to exon 22 by p66α-HDAC2 assembly. This set of molecular interactions stabilizes the interaction of H3K9me3 with exon 22 of Nrxn1 [1].
While Ding and colleagues focused primarily on Nrxn1 in their study, they also reported H3K9me3-mediated co-transcriptional changes in alternative splicing of several genes implicated in memory formation including Nrxn2, Nrxn3, Gephyrin, and Scn1a [1]. Methyl-binding protein 2 (MeCP2) is also a well-known regulator of neuronal gene expression, and it too has been shown to influence alternative splicing of a large number of genes by RNA Pol II pausing [30]. Mutations in MeCP2 are the major causes of neurodevelopmental disorder Rett syndrome.
Cell-specific epigenetic markers reshape gene expression programs but, as highlighted for Nrxn1 gene, such modifications are also now being recognized as having profound impacts on alternative splicing of neuronal genes.
New therapies informed by alternative splicing
A growing number of diseases are linked to aberrant alternative splicing either caused by damaging mutations that disrupt splicing directly, by interfering with cis-acting elements and the action of trans-acting protein splicing factors; or indirectly by sequestering nuclear and cytoplasmic proteins that regulate pre-mRNA splicing [31]. Aberrant splicing is linked to a growing number of disease pathologies including Rett Syndrome [32], epilepsy [33], autism spectrum disorders [34,35], schizophrenia, bipolar disorder [8], spinal muscular atrophy (SMA)[36,37], frontotemporal dementia [38], parkinsonism [39,40], myotonic dystrophy [41], chronic pain [42], amyotrophic lateral sclerosis [31], and cancer [43].
In recent years, DNA engineering technologies, such as CRISPR-Cas9, have been employed to correct pathogenic mutations in neurons that interfere with alternative splicing and to induce compensatory shifts in alternative splicing, away from a nonfunctional, toward a functional splice isoform [44–46].
Most CRISPR-Cas9 applications that target alternative splicing mechanisms have so far involved editing genomic DNA. In particular, cytidine deaminase mediated mutagenesis has been applied to edit highly conserved cis-elements at intron-exon junctions necessary for exon recognition and splicing [44,46]. This tool was applied recently to restore the reading frame of the Duchenne muscular dystrophy (DMD) gene and found to rescue its function in induced pluripotent stem cells (iPSCs) derived from patients carrying the DMD pathogenic mutation [44].
A related strategy, targeting RNA, was recently applied in patient iPSCs to shift the pattern of alternative splicing in the Tau encoding MAPT gene, to compensate for pathogenic mutations in MAPT that cause frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [45]. Two forms of MAPT are expressed in neurons in humans, 4R and 3R, that contain or lack exon 10 which encodes one of 4 microtubule binding domains in tau. Importantly, higher levels of Tau-4R in human brain is linked to FTDP-17 [38,47]. Damaging mutations in MAPT can occur in the intron downstream of exon 10, disrupting an intronic splice silencer and elevating 4R Tau 17 [38,47]. The CRISPR-Cas9 tool dCasRx targets RNA and, when linked to a splice factor, can modify the pattern of alternative splicing [45]. Konermann et al. 2018 successfully employed dCasRx fused to hnRNP splicing factors in iPSCs, to shift splicing from Tau-4R toward Tau-3R in cortical neurons. This was achieved by targeting dCasRx-hnRNPs to exon 10 splice acceptors, and two putative exonic splice enhancer sites in MAPT gene, thereby promoting exon skipping. It will be exciting to see if this approach has therapeutic benefits.
Finally, one of the most studied examples of alternative splicing linked to a major neurological disorder involves the Survival of Motor Neuron (SMN) protein, an RNA binding protein that is necessary for small ribonucleoproteins (snRNPs) assembly and for RNA splicing. Pathogenic mutations in SMN1 gene cause spinal muscular atrophy (SMA), a severe hereditary neuromuscular disease linked to high levels of infant mortality [48,49]. All affected individuals carry damaging mutations (often large deletion) of SMN1, but copy number of an adjacent, partially functional paralog gene, SMN2, scales inversely with phenotypic severity [49,50]. SMN2 fails to compensate for the loss of SMN1 because of protein instability relative to SMN1. SMN1 and SMN2 only differ in few nucleotides, but this difference leads to exon 7 skipping in SMN2. Critically, the non-truncated SMN2 exon 7-containing protein is fully functional and sufficient to support neuronal survival in both SMN1-null mice and in human iPSCs derived from SMA patients [36,51–53].
These studies motivated therapeutic strategies to promote exon 7 inclusion in SMN2 thereby compensating for SMN1 loss of function in SMA. In December 2016, the US Food and Drug Administration (FDA) approved Nusinersen (Spinraza™, also known as ISIS–SMNRx or ISIS 396443) for use in treating SMA. Nusinersen, an antisense oligonucleotide (ASO) designed to promote SMN2 exon 7 inclusion, was developed by Ionis Pharmaceuticals and taken into clinical trial in partnership with Biogen. Nusinersen is a 2′- O-methoxyethyl phosphorothioate-modified ASO specifically designed to alter splicing of SMN2 and thus increase the amount of functional SMN protein that is deficient in SMA patients. Clinical trials have reported that Nusinersen promotes full-length SMN protein leading to improved motor function in SMA infants compared to untreated children [54,55].
The publications discussed above are powerful examples of the critical importance of basic research informing the design of potentially highly specific, novel therapies for the treatment of severe neurological diseases. Aberrant splicing is linked to a growing number of disease pathologies. As discussed, for technologies to correct and to control alternative splicing defects we need to continue effort to understand the mechanisms that regulate alternative splicing of neural genes. Knowing the cell-specific signals and proteins that control RNA processing will be key to understanding the mechanisms that generate unique cell-specific patterns of mRNA isoforms and control expression levels of splice isoforms to influence cell function.
Highlights.
Alternative splicing is dynamic and supports all stages of neuronal function
Studies reveal molecular mechanisms that direct cell-specific exon selection
Epigenetic markers regulate exon choice to support development and plasticity
New therapeutic approaches can correct or compensate for splicing defects
Acknowledgements:
Funded by NIH grant NS055251.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
Contributor Information
Diane Lipscombe, Robert J and Nancy D Carney Institute for Brain Science, Department of Neuroscience, Brown University. Providence, RI 02912, USA. diane_lipscombe@brown.edu.
Eduardo Javier Lopez Soto, Robert J and Nancy D Carney Institute for Brain Science, Department of Neuroscience, Brown University. Providence, RI 02912, USA. ejlopezsoto@gmail.com, eduardo_lopez_soto@brown.edu.
References
- 1.°°.Ding X, Liu S, Tian M, Zhang W, Zhu T, Li D, Wu J, Deng H, Jia Y, Xie W, et al. : Activity-induced histone modifications govern Neurexin-1 mRNA splicing and memory preservation. Nat Neurosci 2017, 20:690–699. [DOI] [PubMed] [Google Scholar]; Demonstrates the cell-specific importance of histone H3K9me3 modification and DNA binding partners in Neurexin 1 gene alternative splicing during memory consolidation. Illustates the different mechanisms by which RNA and DNA binding proteins, and epigenetic marks control exon choice, and illustrates how these mechanisms direct cell-specific alternative splicing of Nrxn1 in memory preservation.
- 2.Fiszbein A, Giono LE, Quaglino A, Berardino BG, Sigaut L, von Bilderling C, Schor IE, Steinberg JH, Rossi M, Pietrasanta LI, et al. : Alternative Splicing of G9a Regulates Neuronal Differentiation. Cell Rep 2016, 14:2797–2808. [DOI] [PubMed] [Google Scholar]
- 3.Jacko M, Weyn-Vanhentenryck SM, Smerdon JW, Yan R, Feng H, Williams DJ, Pai J, Xu K, Wichterle H, Zhang C: Rbfox Splicing Factors Promote Neuronal Maturation and Axon Initial Segment Assembly. Neuron 2018, 97:853–868 e856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.°.Traunmuller L, Gomez AM, Nguyen TM, Scheiffele P: Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 2016, 352:982–986. [DOI] [PubMed] [Google Scholar]
- 5.Vogt MA, Ehsaei Z, Knuckles P, Higginbottom A, Helmbrecht MS, Kunath T, Eggan K, Williams LA, Shaw PJ, Wurst W, et al. : TDP-43 induces p53-mediated cell death of cortical progenitors and immature neurons. Sci Rep 2018, 8:8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.°.Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffie R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, et al. : Precise temporal regulation of alternative splicing during neural development. Nat Commun 2018, 9:2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Treutlein B, Gokce O, Quake SR, Sudhof TC: Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci U S A 2014, 111:E1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.°°.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. : Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive analysis showing the level of disruption of alternative splicing in human brain associated with autism specturm disorder, schizophrenia and bipolar disorder.
- 9.°.Tilgner H, Jahanbani F, Blauwkamp T, Moshrefi A, Jaeger E, Chen F, Harel I, Bustamante CD, Rasmussen M, Snyder MP: Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat Biotechnol 2015, 33:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. : Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell 2018, 70:854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G: Multiple links between transcription and splicing. RNA 2004, 10:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.°°.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T: Epigenetics in alternative pre-mRNA splicing. Cell 2011, 144:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review on how epigenetic markers influence alternative splicing.
- 13.de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR: A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003, 12:525–532. [DOI] [PubMed] [Google Scholar]
- 14.Kadener S, Fededa JP, Rosbash M, Kornblihtt AR: Regulation of alternative splicing by a transcriptional enhancer through RNA pol II elongation. Proc Natl Acad Sci U S A 2002, 99:8185–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T: Regulation of alternative splicing by histone modifications. Science 2010, 327:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S: CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sims RJ 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D: Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell 2007, 28:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, Ljungman M: Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res 2014, 24:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allo M, Buggiano V, Fededa JP, Petrillo E, Schor I, de la Mata M, Agirre E, Plass M, Eyras E, Elela SA, et al. : Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol 2009, 16:717–724. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez I, Munita R, Agirre E, Dittmer TA, Gysling K, Misteli T, Luco RF: A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat Struct Mol Biol 2015, 22:370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saint-Andre V, Batsche E, Rachez C, Muchardt C: Histone H3 lysine 9 trimethylation and HP1gamma favor inclusion of alternative exons. Nat Struct Mol Biol 2011, 18:337–344. [DOI] [PubMed] [Google Scholar]
- 22.Hu Q, Kim EJ, Feng J, Grant GR, Heller EA: Histone posttranslational modifications predict specific alternative exon subtypes in mammalian brain. PLoS Comput Biol 2017, 13:e1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agirre E, Bellora N, Allo M, Pages A, Bertucci P, Kornblihtt AR, Eyras E: A chromatin code for alternative splicing involving a putative association between CTCF and HP1alpha proteins. BMC Biol 2015, 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Zhao W, Olson SD, Prabhakara KS, Zhou X: Alternative splicing links histone modifications to stem cell fate decision. Genome Biol 2018, 19:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uzor S, Zorzou P, Bowler E, Porazinski S, Wilson I, Ladomery M: Autoregulation of the human splice factor kinase CLK1 through exon skipping and intron retention. Gene 2018, 670:46–54. [DOI] [PubMed] [Google Scholar]
- 26.Vuong CK, Wei W, Lee JA, Lin CH, Damianov A, de la Torre-Ubieta L, Halabi R, Otis KO, Martin KC, O’Dell TJ, et al. : Rbfox1 Regulates Synaptic Transmission through the Inhibitory Neuron-Specific vSNARE Vamp1. Neuron 2018, 98:127–141 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ying Y, Wang XJ, Vuong CK, Lin CH, Damianov A, Black DL: Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell 2017, 170:312–323 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, et al. : DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci 2016, 19:102–110. [DOI] [PubMed] [Google Scholar]
- 29.Kwapis JL, Alaghband Y, Kramar EA, Lopez AJ, Vogel Ciernia A, White AO, Shu G, Rhee D, Michael CM, Montellier E, et al. : Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat Commun 2018, 9:3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng TL, Chen J, Wan H, Tang B, Tian W, Liao L, Qiu Z: Regulation of mRNA splicing by MeCP2 via epigenetic modifications in the brain. Sci Rep 2017, 7:42790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, et al. : TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 2018, 21:228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY: Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999, 23:185–188. [DOI] [PubMed] [Google Scholar]
- 33.Lal D, Trucks H, Moller RS, Hjalgrim H, Koeleman BP, de Kovel CG, Visscher F, Weber YG, Lerche H, Becker F, et al. : Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia 2013, 54:265–271. [DOI] [PubMed] [Google Scholar]
- 34.Gonatopoulos-Pournatzis T, Wu M, Braunschweig U, Roth J, Han H, Best AJ, Raj B, Aregger M, O’Hanlon D, Ellis JD, et al. : Genome-wide CRISPR-Cas9 Interrogation of Splicing Networks Reveals a Mechanism for Recognition of Autism-Misregulated Neuronal Microexons. Mol Cell 2018, 72:510–524 e512. [DOI] [PubMed] [Google Scholar]
- 35.Quesnel-Vallieres M, Dargaei Z, Irimia M, Gonatopoulos-Pournatzis T, Ip JY, Wu M, Sterne-Weiler T, Nakagawa S, Woodin MA, Blencowe BJ, et al. : Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol Cell 2016, 64:1023–1034. [DOI] [PubMed] [Google Scholar]
- 36.°°.Singh NK, Singh NN, Androphy EJ, Singh RN: Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 2006, 26:1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]; First antisense oligonucleotide designed to alter splicing of SMN2 and thus increase the amount of functional SMN protein.
- 37.Deshaies JE, Shkreta L, Moszczynski AJ, Sidibe H, Semmler S, Fouillen A, Bennett ER, Bekenstein U, Destroismaisons L, Toutant J, et al. : TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 2018, 141:1320–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blauwendraat C, Wilke C, Simon-Sanchez J, Jansen IE, Reifschneider A, Capell A, Haass C, Castillo-Lizardo M, Biskup S, Maetzler W, et al. : The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med 2018, 20:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quadri M, Mandemakers W, Grochowska MM, Masius R, Geut H, Fabrizio E, Breedveld GJ, Kuipers D, Minneboo M, Vergouw LJM, et al. : LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol 2018, 17:597–608. [DOI] [PubMed] [Google Scholar]
- 40.Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, Collins RL, Domingo A, Currall B, Stortchevoi A, et al. : Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172:897–909 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu N, Antoury L, Baran TM, Mitra S, Bennett CF, Rigo F, Foster TH, Wheeler TM: Non-invasive monitoring of alternative splicing outcomes to identify candidate therapies for myotonic dystrophy type 1. Nat Commun 2018, 9:5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manners MT, Ertel A, Tian Y, Ajit SK: Genome-wide redistribution of MeCP2 in dorsal root ganglia after peripheral nerve injury. Epigenetics Chromatin 2016, 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, Gleave M, Dong X, Qi J: Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci U S A 2018, 115:E4584–E4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.°.Yuan J, Ma Y, Huang T, Chen Y, Peng Y, Li B, Li J, Zhang Y, Song B, Sun X, et al. : Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol Cell 2018, 72:380–394 e387. [DOI] [PubMed] [Google Scholar]
- 45.°°.Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD: Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173:665–676 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]; Application of CRISPR-CAS9 linked to splice effectors to correct for the expression of damaging splice isoforms of Tau protein.
- 46.Gapinske M, Luu A, Winter J, Woods WS, Kostan KA, Shiva N, Song JS, Perez-Pinera P: CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol 2018, 19:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD: Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A 1999, 96:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wirth B, Herz M, Wetter A, Moskau S, Hahnen E, Rudnik-Schoneborn S, Wienker T, Zerres K: Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999, 64:1340–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. : Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80:155–165. [DOI] [PubMed] [Google Scholar]
- 50.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J: Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997, 16:265–269. [DOI] [PubMed] [Google Scholar]
- 51.Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR: Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010, 24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, Hua Y, Rigo F, Matson J, Hung G, et al. : Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 2011, 3:72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.d’Ydewalle C, Ramos DM, Pyles NJ, Ng SY, Gorz M, Pilato CM, Ling K, Kong L, Ward AJ, Rubin LL, et al. : The Antisense Transcript SMN-AS1 Regulates SMN Expression and Is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, Yamashita M, Rigo F, Hung G, Schneider E, et al. : Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016, 388:3017–3026. [DOI] [PubMed] [Google Scholar]
- 55.Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM: Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P: SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147:1601–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Ville´n J, Haas W, Sowa ME, Gygi SP: A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]