

Abstract
Telomere alterations represent one of the major molecular changes in the development of human cancer. We have previously reported that telomere lengths in most serous tubal intraepithelial carcinomas (STIC) are shorter than they are in ovarian high-grade serous carcinomas (HGSC) or in normal-appearing fallopian tube epithelium from the same patients. However, it remains critical to determine if similar telomere alterations occur in TP53-mutated but histologically unremarkable “p53 signature” lesions, as well as incidental STICs without concurrent HGSC. In this study, we quantitatively measured telomere lengths by performing telomere-specific fluorescence in situ hybridization in conjunction with p53 immunolabeling in 15 p53 signatures and 30 incidental STICs without concurrent HGSC. We compared these new results with our previous data in paired STICs and concurrent HGSCs. We found that most p53 signatures (80%) and incidental STICs without HGSC (77%) exhibited significant telomere shortening compared with adjacent normal-appearing fallopian tube epithelium (p<0.01). Interestingly, however, p53 signatures and incidental STICs without HGSC displayed longer telomeres and less cell-to-cell telomere length heterogeneity than STICs associated with HGSC (p<0.001). These findings indicate that telomere shortening occurs in p53 signatures, the earliest pre-cancer lesion. Moreover, incidental STICs without concurrent HGSC are indeed similar to p53 signatures as they have less telomere shortening and less cell-to-cell telomere length heterogeneity than STICs associated with HGSC.
Keywords: ovarian cancer, fallopian tube, p53 signature, serous tubal intraepithelial carcinoma, telomere
Introduction
The high mortality associated with ovarian cancer is largely due to challenges in detecting its precursor before cancer develops. Extensive clinico-pathological, molecular, and epidemiologic studies have supported a new paradigm that fallopian tube lesions are the likely precursors of most high-grade serous carcinomas (HGSC) and contain a repertoire of pre-cancerous lesions at different stages of development enroute to HGSCs (1–9). The lesions contain pre-cancerous “p53 signature,” serous tubal intraepithelial carcinoma (STIC), and serous tubal intraepithelial lesion (STIL) (10, 11). The p53 signature is a minute lesion composed of histologically unremarkable, non-ciliated tubal epithelial cells which harbor TP53 missense mutations (reflected by diffuse and intense nuclear p53 immunoreactivity). In contrast, STICs exhibit nuclear atypia, presence of mitosis and/or apoptotic bodies, contain either missense or deleterious TP53 mutations, and have a high proliferation index as reflected by increased Ki-67 labeling. STILs can be biologically considered as dormant STICs, which are distinguished from STICs by a low proliferative capacity (10, 11). The reported incidence of tubal lesions varies; however, when rigorous sampling was performed in a large cohort of fallopian tubes from a high-risk population, the incidence of p53 signatures and STICs/STILs was as high as 27% and 12%, respectively (12). The five-year cause-specific survival rates for fallopian tube carcinoma in situ including STICs/STILs was reported to be high, 97.9%, compared with less than 50% for late-stage HGSCs of the fallopian tube and/or ovary (13).
Recent exome-wide studies have demonstrated that STICs and p53 signatures are clonally related to ovarian HGSCs when all are present in the same specimens, consistent with the paradigm that most ovarian HGSCs originate in the fallopian tube (9, 14–16). Studies aimed at analyzing the molecular pathogenesis of these precursor lesions and their relationship to ovarian HGSCs have compared ovarian cancers and fallopian tubal lesions from the same patients (7, 9, 15–17). However, there are at least two limitations concerning this approach. The overwhelming numbers of cancer cells present at diagnosis can outnumber their putative precursors by a factor of billions, arguing against the legitimacy of STIC as the precursor of ovarian HGSC. For instance, the massive carcinoma may have destroyed the site of the initial precursor lesions in the fallopian tubes, thus obscuring reconstruction of the genetic landscape during early tumor evolution (14, 15, 18, 19). Furthermore, cells disseminated from concurrent HGSCs, or from carcinomas arising from uterus or elsewhere, may have spread to the surface of fallopian tubes, and may deceptively mimic STIC lesions. Thus, those extant “STICs,” detected at the time of cancer diagnosis, may represent either independent STICs from the residual fallopian tube tissues or the carcinoma itself. As described by Alves et al., this imposes major conceptual difficulties for inferring tumor evolution from phylogenetic analysis in cases of concurrent STIC with advanced ovarian cancer (20).
To circumvent these limitations, we have analyzed early precursor fallopian tubal lesions, including p53 signatures and incidental STICs without concurrent HGSC from women whose tubes were removed due to benign gynecologic diseases. We performed telomere-specific fluorescence in situ hybridization (FISH) in conjunction with p53 immunolabeling to determine if telomere alterations occurred in these lesions prior to cancer development. Additionally, we compared the results with our previously published data from patients with concurrent STIC and HGSC (21). Telomeres are essential for genomic integrity; however, telomeres progressively shorten as a direct result of incomplete DNA replication in the lagging strand (22). As a consequence of abrogation of cell-cycle checkpoints, continued cell division may lead to short, destabilized telomeres. There is a limit to the number of doublings somatic cells can undergo before triggering successive rounds of chromosome breakage–fusion–bridge cycles, which drive chromosomal instability and clonal variation – both prerequisites for natural selection favoring tumor development (23). This study focuses on analyzing the telomere length landscape of p53 signatures and incidental STICs without concurrent HGSC. The findings shed new light on our understanding of ovarian cancer pathogenesis and provide essential information for designing better strategies for early detection and cancer prevention, two key requirements for reducing ovarian cancer mortality.
Materials and Methods
Case selection
All cases were retrieved from the surgical pathology files in the Department of Pathology at the Johns Hopkins Hospital under institutional review board approval. Tissue acquisition followed the guidelines of the Institutional Review Board of the Johns Hopkins Hospital. Only deidentified information pertaining to diagnosis, treatment, and outcome were obtained. Neither personal health information nor medical records were collected or reviewed for this study. Informed consent could not be obtained, precluding access to HIPAA (Health Insurance Portability and Accountability Act of 1996, USA) protected information.
The inclusion criteria were salpingectomy specimens which had tubal precursor lesions including p53 signature, STIL, and STIC, with or without HGSC, obtained in the period from 2011 to 2017. When STIC lesions were present in separate fallopian tissue sections, we considered them as separate lesions. There were 15 p53 signatures and 30 incidental STICs including STILs (dormant STICs) without concurrent HGSCs from 16 patients included in this study. All samples were obtained as formalin-fixed and paraffin-embedded (FFPE) specimens. Cases were reviewed by three gynecologic pathologists (S.A., SF.L., and IM.S.) using previously described criteria (10, 11). Telomere length data from 15 patients who had concurrent stage III/IV ovarian/pelvic HGSC were extracted from data collected during our previous study (21), and were re-analyzed for the present study. Of these 15 patients, 22 STICs and 12 concurrent HGSCs were assessed. A total of 40 normal-appearing fallopian tube (NFT) epithelial tissues adjacent to each ovarian and fallopian lesion were assessed from 31 patients; 25 from the 16 patients in the present cohort and 15 from the 15 patients in the previous cohort (21).
Immunohistochemistry
p53 and Ki-67 immunostaining were performed for the classification of fallopian tube lesions according to previously established criteria (10, 11). Serial paraffin sections (4 μm) were prepared from each block, and whole sections were stained with hematoxylin and eosin (H&E) for histological examination. Immunostaining with an anti-p53 antibody (Clone DO-7, Cat# NCL-L-p53-DO7, 1:600 dilution, Leica Biosystems Inc, Buffalo Grove, IL, U.S.A) and an anti-Ki-67 antibody (Clone D2H10, Cat# 9027, 1:400, Cell Signaling Technology, Danvers, MA, U.S.A.) was manually performed. Briefly, unstained sections were deparaffinized and subjected to antigen retrieval by incubating slides in DAKO Target Retrieval Solution, Citrate pH 6 (Agilent, Santa Clara, CA, U.S.A.) at 90°C for 30 minutes. Tissue sections were then incubated with the primary antibody at 4°C overnight. Immunoreactivity was detected using DAKO EnVision™+ System-HRP-Labeled Polymer Anti-Mouse (Agilent). For all cases, we evaluated p53 staining patterns according to previously published guidelines (24), and estimated the approximate percentage of Ki-67 positive cells in all cancerous and precancerous lesions.
Telomere-specific Fluorescence in situ Hybridization combined with p53 Immunolabeling
Telomere lengths were assessed by telomere-specific fluorescence in situ hybridization (FISH) in combination with p53 immunofluorescence (IF)-labeling as previously described (25). In brief, after deparaffinization and hydration, slides were placed in citrate buffer (Vector Laboratories, Burlingame, CA, U.S.A.), and antigen retrieval was performed using a pre-heated steamer for 25 min. Cy3-labeled telomere-specific peptide nucleic acid (PNA) (Cat #F1002, Panagene, South Korea) in hybridization buffer (0.3 μg/ml PNA in 70% formamide, 10 mmol/L Tris, pH 7.5) was applied to the sample, denatured at 84°C for 5 minutes, and then incubated overnight at room temperature. A FITC-labeled PNA probe specific for human centromeric DNA repeats (CENP-B binding sequence) was included in the hybridization buffer as a positive control of hybridization efficiency. The slides were washed twice in wash buffer (70% formamide, 10 mmol/L Tris, pH 7.5), followed by PBST washes, and then incubated with an anti-p53 antibody (Clone DO-7, Cat# NCL-L-p53-DO7, Leica Biosystems Inc) at a dilution of 1:600 at 4°C overnight. After washing, a goat anti-mouse IgG conjugated with Alexa Fluor 647 (Cat# A32728, Thermo Fisher Scientific, Rockford, IL, U.S.A.) was applied at a dilution of 1:100, and incubation at room temperature was continued for 30 min. Finally, after washing, the slides were counterstained with 4’−6-diamidino-2-phenylindole (DAPI) and mounted with ProLong Gold Antifade reagent (Invitrogen, Carlsbad, CA, U.S.A.).
Image Analysis
To quantitatively measure telomere lengths, the slides were imaged using the TissueFAXS Plus (Tissue Gnostics, Vienna, Austria) automated microscopy workstation equipped with a Zeiss Z2 Axio Imager microscope (Carl Zeiss Microscopy, LCC, Thornwood, NY, U.S.A.). The digitized telomere FISH signals were quantified using the TissueQuest 6.0 software (Tissue Gnostics) module to analyze the fluorescent images with precise nuclear segmentation. The telomere fluorescence intensity was calculated as the ratio of total Cy3 intensity to the total DAPI intensity for each sampled nucleus to correct for possible ploidy differences or nuclear cutting artefacts. All nuclei with p53 immunolabeling in p53 signatures, at least 30 nuclei in STICs and HGSCs, and at least 30 nuclei in NFT epithelium without p53 immunolabeling were analyzed.
Statistical Analysis
Telomere fluorescence intensities of each fallopian tubal lesion and ovarian cancer (p53 signatures, incidental STICs without concurrent HGSC, STICs associated with HGSC, or HGSCs) were compared with the adjacent NFT epithelium using Wilcoxon test (Mann-Whitney test). Each lesion, compared to its matched NFT, was classified as either displaying significantly shorter, longer, or no significant change of telomere lengths. The relative telomere length (%) to the adjacent NFT epithelium (mean telomere signal intensity per nucleus of the lesion/mean telomere signal intensity per nucleus of the adjacent NFT epithelium × 100%) was calculated and compared among p53 signatures, incidental STICs without HGSC, and STICs associated with HGSC using Wilcoxon test (Mann-Whitney test). Next, we analyzed cell-to-cell heterogeneity in telomere lengths between nuclei using the coefficient of variation, which is a standardized measure of dispersion of the distribution. The coefficients of variation of telomere lengths were compared among NFT epithelium, p53 signatures, incidental STICs without HGSC, STICs associated with HGSC, and HGSCs using Wilcoxon test (Mann-Whitney test). The data were analyzed using JMP software (version 13.0; SAS Institute, Cary, NC, U.S.A.); p<0.01 was considered significant.
Quantitative analysis data are shown in Tables 1 and 2 for p53 signatures and incidental STICs without HGSCs, and in supplemental Tables S1 and S2 for STICs associated with HGSC and HGSCs from our previous report (21). The data include 1) mean telomere signal intensities per nucleus in individual lesions and their adjacent NFT epithelium, 2) relative telomere lengths compared to NFT epithelium (and concurrent STIC for HGSCs) (%), 3) significance (p) calculated by Wilcoxon test (Mann-Whitney test) for the comparison of telomere signal intensities between lesions and NFT epithelium (or concurrent STIC for HGSCs), 4) coefficients of variation (%) of the telomere signal intensities in each cell of the lesion, and 5) p53 immunostaining patterns and ki-67 labeling index in each group.
Table 1.
Telomere length changes in p53 signature lesions
| Lesion No. |
Mean telomere signal intensity | Relative telomere length to adjacent NFT (A/B × 100%) |
p-value* compared with adjacent NFT | CV | Immunohistochemistry | ||
|---|---|---|---|---|---|---|---|
| p53 signature (A) |
Adjacent NFT (B) |
p53 staining pattern | Ki-67 labeling index | ||||
| 1 | 33.39 | 35.93 | 93% | <0.0001 | 31% | Diffuse | 0% |
| 2 | 27.39 | 35.93 | 76% | <0.0001 | 43% | Diffuse | 0% |
| 3 | 17.06 | 33.96 | 50% | <0.0001 | 41% | Diffuse | 1% |
| 4 | 27.95 | 54.67 | 51% | <0.0001 | 31% | Diffuse | 7% |
| 5 | 6.88 | 11.14 | 62% | <0.0001 | 87% | Diffuse | 2% |
| 6 | 10.11 | 11.14 | 91% | 0.8404 | 45% | Diffuse | 0% |
| 7 | 14.93 | 18.88 | 79% | 0.0774 | 40% | Diffuse | 1% |
| 8 | 10.65 | 18.88 | 56% | <0.0001 | 54% | Diffuse | 3% |
| 9 | 29.57 | 37.96 | 78% | <0.0001 | 30% | Diffuse | 1% |
| 10 | 17.21 | 21.74 | 79% | 0.0001 | 69% | Diffuse | 8% |
| 11 | 16.31 | 21.74 | 75% | <0.0001 | 47% | Diffuse | 7% |
| 12 | 17.26 | 21.74 | 79% | <0.0001 | 50% | Diffuse | 7% |
| 13 | 18.27 | 21.74 | 84% | 0.0144 | 46% | Diffuse | 9% |
| 14 | 41.59 | 55.56 | 75% | <0.0001 | 35% | Diffuse | 9% |
| 15 | 17.59 | 25.05 | 70% | <0.0001 | 39% | Diffuse | 9% |
Analyzed by Wilcoxon test (Mann-Whitney test).
NFT, normal-appearing fallopian tube; CV, coefficient of variation.
Table 2.
Telomere length changes in incidental STIC lesions without concurrent HGSC
| Lesion No. |
Mean telomere signal intensity | Relative telomere length to adjacent NFT (A/B × 100%) |
p-value* compared with adjacent NFT | CV | Immunohistochemistry | ||
|---|---|---|---|---|---|---|---|
| Incidental STIC without HGSC (A) |
Adjacent NFT (B) |
p53 staining pattern | Ki-67 labeling index | ||||
| 1 | 38.86 | 37.96 | 102% | 0.6795 | 39% | Diffuse | 9% |
| 2 | 19.78 | 21.74 | 91% | 0.0007 | 42% | Diffuse | 6% |
| 3 | 15.70 | 20.32 | 77% | <0.0001 | 34% | Diffuse | 6% |
| 4 | 19.16 | 38.29 | 50% | <0.0001 | 39% | Diffuse | 7% |
| 5 | 27.98 | 33.34 | 84% | <0.0001 | 47% | Diffuse | 3% |
| 6 | 46.65 | 60.69 | 77% | <0.0001 | 35% | Completely Negative | 6% |
| 7 | 54.94 | 60.69 | 91% | <0.0001 | 29% | Completely Negative | 1% |
| 8 | 22.14 | 34.88 | 63% | <0.0001 | 58% | Diffuse | 3% |
| 9 | 36.00 | 59.72 | 60% | <0.0001 | 59% | Diffuse | 8% |
| 10 | 59.30 | 59.72 | 99% | <0.0001 | 62% | Diffuse | 2% |
| 11 | 17.81 | 24.79 | 72% | <0.0001 | 68% | Diffuse | 2% |
| 12 | 46.84 | 55.14 | 85% | 0.0008 | 43% | Diffuse | 1% |
| 13 | 25.41 | 45.71 | 56% | <0.0001 | 45% | Diffuse | 3% |
| 14 | 14.57 | 14.69 | 99% | 0.8729 | 38% | Diffuse | 31% |
| 15 | 12.06 | 18.88 | 64% | <0.0001 | 46% | Diffuse | 24% |
| 16 | 13.53 | 18.88 | 72% | <0.0001 | 49% | Diffuse | 23% |
| 17 | 23.65 | 21.74 | 109% | <0.0001 | 42% | Diffuse | 21% |
| 18 | 15.86 | 20.32 | 78% | <0.0001 | 38% | Diffuse | 25% |
| 19 | 49.40 | 64.51 | 77% | <0.0001 | 38% | Diffuse | 47% |
| 20 | 37.16 | 41.30 | 90% | <0.0001 | 40% | Diffuse | 18% |
| 21 | 43.34 | 54.38 | 80% | <0.0001 | 41% | Diffuse | 52% |
| 22 | 49.51 | 54.38 | 91% | <0.0001 | 38% | Diffuse | 30% |
| 23 | 36.41 | 55.56 | 66% | <0.0001 | 40% | Diffuse | 54% |
| 24 | 35.20 | 36.35 | 97% | 0.0057 | 56% | Diffuse | 15% |
| 25 | 47.38 | 36.35 | 130% | <0.0001 | 44% | Diffuse | 29% |
| 26 | 63.48 | 49.81 | 127% | 0.0001 | 48% | Diffuse | 62% |
| 27 | 25.02 | 30.60 | 82% | <0.0001 | 68% | Diffuse | 79% |
| 28 | 32.86 | 30.60 | 107% | 0.6341 | 67% | Diffuse | 97% |
| 29 | 17.79 | 21.97 | 81% | <0.0001 | 43% | Diffuse | 63% |
| 30 | 21.28 | 21.97 | 97% | 0.6535 | 47% | Diffuse | 75% |
Analyzed by Wilcoxon test (Mann-Whitney test).
STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma; NFT, normal-appearing fallopian tube; CV, coefficient of variation.
Results
In total, telomere lengths from 79 fallopian and ovarian epithelial lesions and 40 adjacent areas showing normal-appearing fallopian tube epithelium were analyzed. These lesions included 15 p53 signatures, 30 incidental STICs without concurrent HGSC, 22 STICs associated with HGSC, and 12 HGSCs.
Using p53 immunolabeling to accurately identify the lesional cells and telomere-specific FISH to detect the telomeres, we compared the intensity of the telomere FISH signals between nuclei in the target lesion and those in the adjacent NFT epithelium both qualitatively (by pathologists) and quantitatively (by digital image analysis). H&E staining, p53 and Ki-67 immunostaining, p53 immunofluorescence labeling, and telomere-specific FISH images of a representative cases of a p53 signature and an incidental STIC are shown in Figure 1. The intensity of telomere FISH signals is associated with telomere length. Both p53 signature and incidental STIC lesions exhibited significantly shorter telomeres compared to the adjacent NFT epithelial cells (Figure 1).
Figure 1. Representative images of a p53 signature and an incidental STIC without concurrent HGSC.
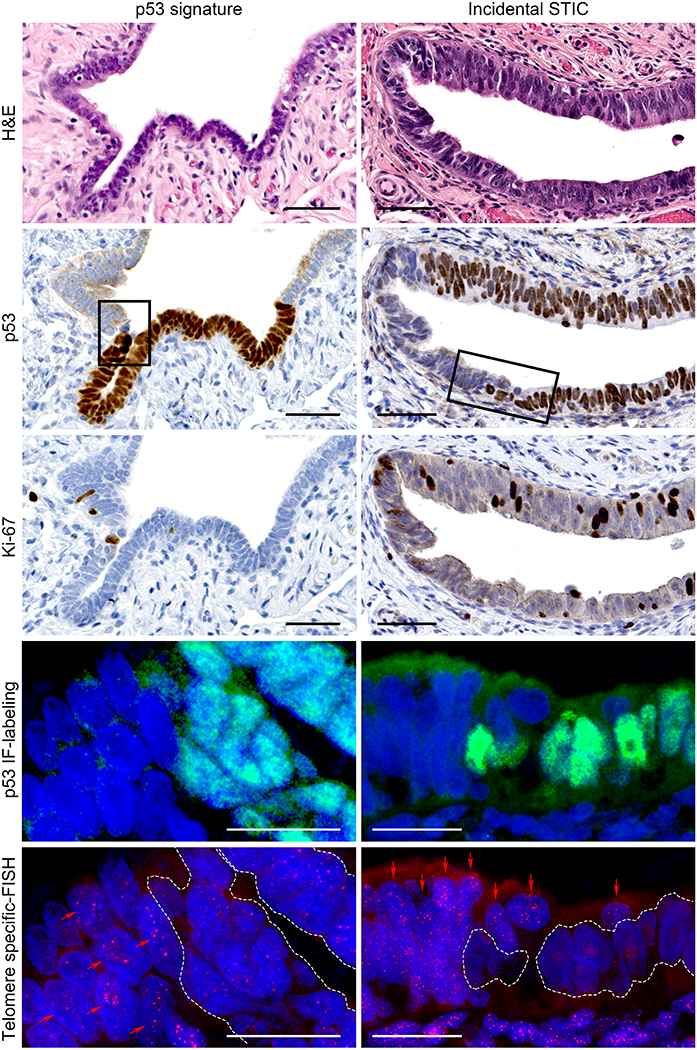
In H&E staining, the p53 signature is a cytologically benign secretory cell-rich segment. The lesion shows diffuse p53 staining pattern and a low Ki-67 labeling index by immunohistochemistry. STIC exhibits nuclear stratification, enlargement, and prominent nucleoli. It shows diffuse p53 expression and a high Ki-67 labeling index. p53 IF-labeling and telomere-specific FISH images represent higher magnification of the area marked by rectangle in p53 staining. The p53 IF-labeling shows p53-positive and -negative regions of the sections. In telomere-specific FISH, the telomere signals are weaker in the p53-positive cells within the p53 signature and the STIC lesion (white dotted lines) compared with the p53-negative, adjacent NFT epithelium (red arrows). Nuclei are stained with DAPI (blue), p53 is labeled with green nuclear fluorescence, and telomeres are hybridized with a Cy3-labeled telomere-specific FISH probe (small red dots in nuclei).
Scale bars: black, 50 μm; white, 20 μm.
H&E, hematoxylin and eosin; IF, immunofluorescence; FISH, fluorescence in situ hybridization; STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma; NFT, normal-appearing fallopian tube.
Telomere lengths were compared using the quantified telomere data (Tables 1 and 2, and Supplemental Tables S1 and S2). The distribution of telomere lengths was analyzed by Wilcoxon test (Mann-Whitney test) (Table 3 and Figure 2). The majority of p53 signatures and STIC with or without HGSC exhibited significant telomere shortening compared with the adjacent NFTs. Notably, 12 (80%) of 15 p53 signatures showed significantly shorter telomeres compared to adjacent NFT epithelium. In a comparison of mean telomere signal intensities between the p53 signature and the patient-matched NFT epithelium, the ratios ranged between 50% and 93% (Table 1). None of the p53 signatures showed significantly longer telomeres than adjacent NFT epithelium. Among incidental STICs without concurrent HGSC, 23 (77%) of 30 lesions exhibited significantly shorter telomeres, 4 (13%) exhibited no significant change, and 3 (10%) exhibited longer telomeres compared to their adjacent NFT epithelium (Table 2). From our previous report (21), similar to incidental STICs, most of the STICs associated with HGSC (82%) exhibited significant telomere shortening (Supplemental Table S1). In 12 HGSCs, there were 7 (58%) lesions with significantly shorter, 2 (17%) with no significant change, and 3 (25%) with longer telomeres than the adjacent NFT epithelium. Of these 12 HGSCs, 6 (50%) exhibited significantly longer telomeres than their concurrent STIC lesions in the same tissue sections (Supplemental Table S2).
Table 3.
The summary of lesions with significantly shorter or longer telomeres, and those without significant change in telomere lengths.
| Ovarian and fallopian tubal lesions | Total No. | Shorter* | No change* | Longer* |
|---|---|---|---|---|
| p53 signatures (vs. NFT) | 15 | 12 (80%) | 3 (20%) | 0 (0%) |
| STICs (vs. NFT) | 52 | 41 (79%) | 6 (11%) | 5 (10%) |
| Incidental STICs without concurrent HGSC (vs. NFT) | 30 | 23 (77%) | 4 (13%) | 3 (10%) |
| STICs associated with HGSC (vs. NFT) | 22 | 18 (82%) | 2 (9%) | 2 (9%) |
| HGSCs (vs. NFT) | 12 | 7(58%) | 2 (17%) | 3 (25%) |
| HGSC (vs. concurrent STIC) | 12 | 0 (0%) | 6 (50%) | 6 (50%) |
Analyzed by Wilcoxon test (Mann-Whitney test) (p <0.01)
NFT, normal-appearing fallopian tube; STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma.
Figure 2. Telomere length variation in ovarian and fallopian tubal lesions.
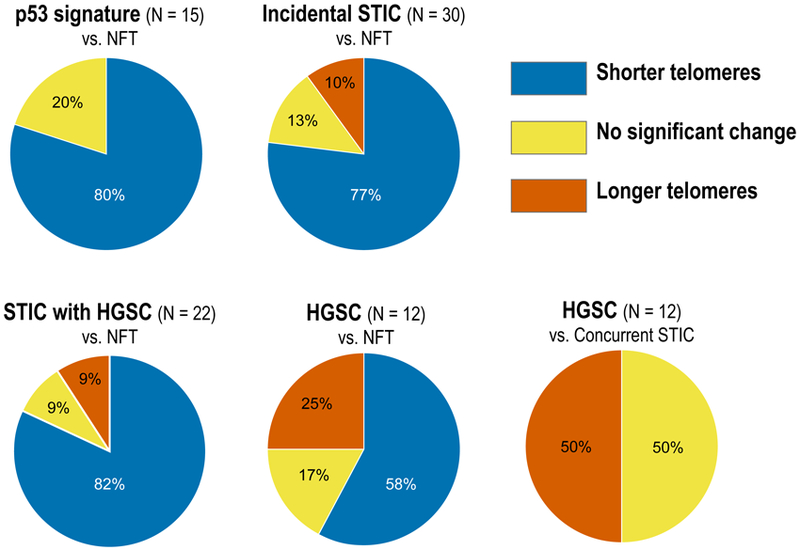
The charts show percent distribution of telomere lengths as significantly shorter, longer, or unchanged.
STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma; NFT, normal-appearing fallopian tube.
Next, we calculated the relative telomere lengths in individual lesions in relation to their adjacent NFT epithelium and expressed the data as a relative ratio (percent). We compared the relative telomere length among p53 signatures, incidental STICs without HGSC, and STICs associated with HGSC (Figure 3A). We observed that p53 signatures and incidental STICs without HGSC exhibited significantly longer telomeres than STICs associated with HGSC (p = 0.0009 and p < 0.0001, respectively). In other words, STICs associated HGSC exhibited significantly shorter relative telomere lengths compared to p53 signatures and incidental STICs.
Figure 3. Relative telomere length and variation.
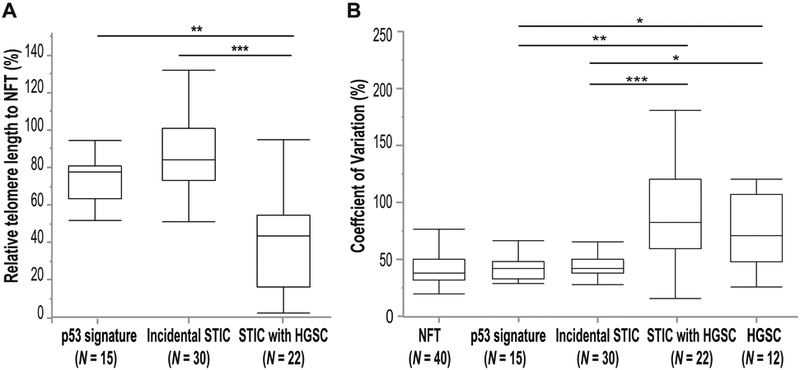
(A) Comparison of telomere lengths among p53 signatures, incidental STIC without concurrent HGSC, and STICs associated with HGSC relative to adjacent NFT epithelium. (B) Comparison of the coefficient of variation in telomere lengths among NFTs, p53 signatures, incidental STICs without HGSC, STICs associated with HGSC, and HGSCs.
*p <0.01; **p <0.001; ***p <0.0001 (Analyzed by Wilcoxon test [Mann-Whitney test]).
STIC, serous tubal intraepithelial carcinoma; HGSC, high-grade serous carcinoma; NFT, normal-appearing fallopian tube.
Next, we compared the coefficients of variation among NFT epithelium, p53 signatures, incidental STICs without HGSC, STICs associated with HGSC, and HGSCs (Figure 3B). Both p53 signatures and incidental STICs without HGSC had significantly lower coefficients of variation than STICs associated with HGSC (p = 0.0003 and p < 0.0001, respectively) or HGSCs (p = 0.0043 and p = 0.0010, respectively), indicating less heterogeneity in telomere length in the absence of HGSC among cells within the same lesion. In contrast, we did not observe any significant differences in coefficients of variation among NFT epithelium, p53 signatures, and incidental STICs without HGSC, or between STICs associated with HGSC and HGSCs.
Discussion
Telomeres are nucleoprotein complexes located at chromosome ends that consist of repetitive DNA (TTAGGGn) bound by shelterin, a protective protein complex (26). Telomeres function to protect the chromosome ends from being recognized as sites of DNA damage, thus preventing activation of DNA damage response pathways (27, 28). The current study explores the landscape of telomere length alterations in precancerous fallopian tube lesions of ovarian cancer. We found that telomere shortening occurs in histologically unremarkable p53 signatures, as well as in incidental STICs without concurrent HGSC. Moreover, STICs associated with HGSCs, in general, had the shortest telomeres among the lesions studied: p53 signatures, incidental STICs without HGSC, and HGSCs. Interestingly, cell-to-cell telomere length distributions within a lesion were more homogeneous in p53 signatures and incidental STICs without HGSC than in STICs associated with HGSC and HGSCs. Our data provide new insights into the molecular events of tumor initiation of ovarian cancer in the fallopian tubes, from the perspective of a dynamic change in telomere length. We note several biological and pathological implications of the findings.
Not all STICs are equivalent — incidental STICs without HGSC are distinct from STICs associated with HGSC with respect to relative telomere lengths and intra-lesional telomere length heterogeneity. STICs associated with HGSC are conventionally thought to be the immediate precursor lesions from which the carcinomas develop. As such, these cancer-associated STICs presumably are more evolutionarily advanced than incidental STICs without HGSC, most of which may not progress to carcinomas and represent an evolutionary “dead-end.” Indeed, morphologically, the degrees of disturbed nuclear polarity in incidental STICs without HGSC tended to be less prominent than those in STICs associated with HGSC, although further studies are needed to confirm this preliminary finding. The higher intra-lesional cell-to-cell heterogeneity of telomere length suggests an underlying genomic instability, creating diverse subclones with variable telomere length. The presence of different subclones allows microenvironment to select one or few of them to become carcinoma.
In general, telomeres shorten after each cell division unless telomerase which functions to maintain the telomere length is activated. Therefore, that telomere length can serve as a molecular clock and reflects a cellular replicative history. Our findings suggest that the presence of short telomeres in STICs associated with HGSC is a result of a longer chronological trajectory involving extensive clonal expansion. Among these cells, a clone(s) may arise and further expand, ultimately transitioning to a carcinoma due to telomerase activation, or other mechanisms that maintain telomere length compatible with incessant cell division. On the other hand, incidental STICs without HGSC may represent an earlier lesion (from its inception) that has yet to reach a critical threshold of telomere length.
The tubal precursors represent a unique group of lesions because they, like HGSCs, are all characterized by TP53 mutations even at the early tumor initiation stages such as p53 signatures and incidental STICs without concurrent HGSC (9, 29, 30). When the p53 pathway is intact, severe telomere attrition triggers the DNA damage response, promoting cellular senescence or programmed cell death; this phenomenon is a potent tumor suppressor mechanism conferred by the p53 pathway (31, 32). However, a defective p53 pathway, as occurs in all tubal precursor cells, allows some STICs to continue to proliferate with increasingly shortened telomeres, leading to genomic instability. Molecular mechanisms that are responsible for maintaining telomere length, such as telomerase activation, help cells escape crisis and attain cellular immortality (33, 34). p53 is a negative regulator of the hTERT promoter (that drives telomerase expression), and loss of p53 may contribute to hTERT reactivation, resulting in maintenance of telomere length (35, 36). Therefore, telomere dysfunction and p53 inactivation together may cooperate in tumor progression. The p53 signature lesions are morphologically unremarkable; the epithelium may continue brief clonal expansion but another tumor suppressor function quickly emerges to limit proliferation. Despite a relatively small temporal window of clonal expansion, the p53 signature epithelial cells, although morphologically indistinguishable from the adjacent TP53 wild-type epithelium, strikingly exhibit already shortened telomeres. However, the majority of these cells do not further progress to STICs, and may even alter their histological appearance because they, unlike STICs, do not harbor many molecular genetic changes including somatic mutations and copy number changes (30).
These findings are consistent with those reported in various precancerous lesions of oral cavity, lung, breast, esophagus, stomach, colon, pancreas, bile ducts, prostate, urinary bladder, and uterine cervix (37–45). Although analysis of precursor lesions prior to development of ovarian HGSCs has not been previously performed, we note that there are other studies, including our own (21), in which telomere lengths in ovarian precancerous lesions have been reported (46). In the Chene study, DNA was extracted from laser capture microdissected tissue sections of ovarian cancers, STICs, and tubo-ovarian dysplasia lesions. Quantitative real-time PCR was performed for analysis of telomere lengths. Similar to our present data, all the precancerous fallopian tube lesions analyzed exhibited shorter telomere lengths than cancer-free patient controls, and ovarian carcinomas exhibited longer telomeres than STICs and fallopian dysplasia lesions. In contrast to this previous study, in which the spatial context was lost, here we visually analyzed telomere length signals in individual cells, which enabled us to compare telomere lengths directly among target cells of the precancerous lesions and adjacent normal-appearing cells, and to assess intra-lesional cell-to-cell telomere length heterogeneity.
A potential limitation of our study is the employment of data from our previous study in which we analyzed telomere signals in STICs and their concurrent HGSCs. However, we used the same staining technique (i.e., telomere-specific FISH) and similar image analysis tools in both studies. Further, as the readout, we assessed the relative telomere length (i.e., the ratio of signals of the target lesions/those of the adjacent NFT epithelium from the same tissues). These measures minimize the effects of confounding factors (e.g., age and hormone status) on the telomere lengths and storage time of tissue blocks used in this study.
From our findings, we conclude that telomere shortening occurs in precancerous lesions before they can be detected by a morphological abnormality. Importantly, incidental STICs without HGSC are molecularly different from STICs associated with HGSC; STICs without HGSC are characterized by longer telomeres and less cell-to-cell telomere length heterogeneity compared to STICs associated with HGSC. Thus, more telomere shortening and increased cell-to-cell telomere length heterogeneity is likely associated with increased risk of developing ovarian HGSCs. Future efforts through multi-institutional consortium studies will be needed to verify these conclusions and determine their clinical importance.
Supplementary Material
Telomere length changes in STICs associated with HGSC from our previous report (21).
Telomere length changes in HGSCs from our previous report (21).
Conflict of interest and Sources of Funding:
This work was supported by the Department of Defense CDMRP W81XWH-11-2-0230; NIH/NCI P50CA228991; the Honorable Tina Brozman Foundation, Ovarian Cancer Research Alliance, Teal award, Roseman Foundation, Gray foundation and the Richard W. TeLinde Endowment from the Johns Hopkins University. All the authors claim no conflict of interest and nothing to be disclosed.
References:
- 1.Kindelberger DW, Lee Y, Miron A, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am J Surg Pathol. 2007; 31: 161–169. [DOI] [PubMed] [Google Scholar]
- 2.Crum CP, Drapkin R, Miron A, et al. The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol. 2007; 19: 3–9. [DOI] [PubMed] [Google Scholar]
- 3.Salvador S, Rempel A, Soslow RA, et al. Chromosomal instability in fallopian tube precursor lesions of serous carcinoma and frequent monoclonality of synchronous ovarian and fallopian tube mucosal serous carcinoma. Gynecol Oncol. 2008; 110: 408–417. [DOI] [PubMed] [Google Scholar]
- 4.Kurman RJ, Shih Ie M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010; 34: 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhn E, Kurman RJ, Shih IM. Ovarian Cancer Is an Imported Disease: Fact or Fiction? Curr Obstet Gynecol Rep. 2012; 1: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhn E, Kurman RJ, Sehdev AS, et al. Ki-67 labeling index as an adjunct in the diagnosis of serous tubal intraepithelial carcinoma. Int J Gynecol Pathol. 2012; 31: 416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuhn E, Kurman RJ, Vang R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J Pathol. 2012; 226: 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurman RJ, Shih Ie M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol. 2016; 186: 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Labidi-Galy SI, Papp E, Hallberg D, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. 2017; 8: 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visvanathan K, Vang R, Shaw P, et al. Diagnosis of serous tubal intraepithelial carcinoma based on morphologic and immunohistochemical features: a reproducibility study. Am J Surg Pathol. 2011; 35: 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vang R, Visvanathan K, Gross A, et al. Validation of an algorithm for the diagnosis of serous tubal intraepithelial carcinoma. Int J Gynecol Pathol. 2012; 31: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Visvanathan K, Shaw P, May BJ, et al. Fallopian Tube Lesions in Women at High Risk for Ovarian Cancer: A Multicenter Study. Cancer Prev Res (Phila). 2018; 11: 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trabert B, Coburn SB, Mariani A, et al. Reported Incidence and Survival of Fallopian Tube Carcinomas: A Population-Based Analysis From the North American Association of Central Cancer Registries. J Natl Cancer Inst. 2018; 110: 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDaniel AS, Stall JN, Hovelson DH, et al. Next-Generation Sequencing of Tubal Intraepithelial Carcinomas. JAMA Oncol. 2015; 1: 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert MA, Pan S, Hernandez KM, et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016; 6: 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducie J, Dao F, Considine M, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun. 2017; 8: 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhn E, Wang TL, Doberstein K, et al. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod Pathol. 2016; 29: 1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabban JT, Vohra P, Zaloudek CJ. Nongynecologic metastases to fallopian tube mucosa: a potential mimic of tubal high-grade serous carcinoma and benign tubal mucinous metaplasia or nonmucinous hyperplasia. Am J Surg Pathol. 2015; 39: 35–51. [DOI] [PubMed] [Google Scholar]
- 19.Singh R, Cho KR. Serous Tubal Intraepithelial Carcinoma or Not? Metastases to Fallopian Tube Mucosa Can Masquerade as In Situ Lesions. Arch Pathol Lab Med. 2017; 141: 1313–1315. [DOI] [PubMed] [Google Scholar]
- 20.Alves JM, Prieto T, Posada D. Multiregional Tumor Trees Are Not Phylogenies. Trends Cancer. 2017; 3: 546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn E, Meeker A, Wang TL, et al. Shortened telomeres in serous tubal intraepithelial carcinoma: an early event in ovarian high-grade serous carcinogenesis. Am J Surg Pathol. 2010; 34: 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Lange T How telomeres solve the end-protection problem. Science. 2009; 326: 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosme-Blanco W, Chang S. Dual roles of telomere dysfunction in initiation and suppression of tumorigenesis. Exp Cell Res. 2008; 314: 1973–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yemelyanova A, Vang R, Kshirsagar M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011; 24: 1248–1253. [DOI] [PubMed] [Google Scholar]
- 25.Meeker AK, Gage WR, Hicks JL, et al. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002; 160: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015; 350: 1193–1198. [DOI] [PubMed] [Google Scholar]
- 27.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010; 11: 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012; 336: 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vang R, Levine DA, Soslow RA, et al. Molecular Alterations of TP53 are a Defining Feature of Ovarian High-Grade Serous Carcinoma: A Rereview of Cases Lacking TP53 Mutations in The Cancer Genome Atlas Ovarian Study. Int J Gynecol Pathol. 2016; 35: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren-Chin Wu PW, Lin Shiou-Fu, Zhang Ming, et al. Genomic Landscape and Evolutionary Trajectories of Ovarian Cancer Early Precursor Lesions. J Pathol. 2018; DOI: 10.1002/path.5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cesare AJ, Hayashi MT, Crabbe L, et al. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell. 2013; 51: 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chin L, Artandi SE, Shen Q, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999; 97: 527–538. [DOI] [PubMed] [Google Scholar]
- 33.Jafri MA, Ansari SA, Alqahtani MH, et al. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016; 8: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266: 2011–2015. [DOI] [PubMed] [Google Scholar]
- 35.Kanaya T, Kyo S, Hamada K, et al. Adenoviral expression of p53 represses telomerase activity through down-regulation of human telomerase reverse transcriptase transcription. Clin Cancer Res. 2000; 6: 1239–1247. [PubMed] [Google Scholar]
- 36.Xu D, Wang Q, Gruber A, et al. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene. 2000; 19: 5123–5133. [DOI] [PubMed] [Google Scholar]
- 37.Meeker AK, Hicks JL, Iacobuzio-Donahue CA, et al. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin Cancer Res. 2004; 10: 3317–3326. [DOI] [PubMed] [Google Scholar]
- 38.Lantuejoul S, Raynaud C, Salameire D, et al. Telomere maintenance and DNA damage responses during lung carcinogenesis. Clin Cancer Res. 2010; 16: 2979–2988. [DOI] [PubMed] [Google Scholar]
- 39.Meeker AK, Hicks JL, Gabrielson E, et al. Telomere shortening occurs in subsets of normal breast epithelium as well as in situ and invasive carcinoma. Am J Pathol. 2004; 164: 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tahara T, Shibata T, Kawamura T, et al. Telomere length shortening in gastric mucosa is a field effect associated with increased risk of gastric cancer. Virchows Arch. 2016; 469: 19–24. [DOI] [PubMed] [Google Scholar]
- 41.Tahara T, Shibata T, Kawamura T, et al. Telomere length in non-neoplastic gastric mucosa and its relationship to H. pylori infection, degree of gastritis, and NSAID use. Clin Exp Med. 2016; 16: 65–71. [DOI] [PubMed] [Google Scholar]
- 42.Hong SM, Heaphy CM, Shi C, et al. Telomeres are shortened in acinar-to-ductal metaplasia lesions associated with pancreatic intraepithelial neoplasia but not in isolated acinar-to-ductal metaplasias. Mod Pathol. 2011; 24: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuda Y, Ishiwata T, Izumiyama-Shimomura N, et al. Gradual telomere shortening and increasing chromosomal instability among PanIN grades and normal ductal epithelia with and without cancer in the pancreas. PLoS One. 2015; 10: e0117575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansel DE, Meeker AK, Hicks J, et al. Telomere length variation in biliary tract metaplasia, dysplasia, and carcinoma. Mod Pathol. 2006; 19: 772–779. [DOI] [PubMed] [Google Scholar]
- 45.Meeker AK, Hicks JL, Platz EA, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002; 62: 6405–6409. [PubMed] [Google Scholar]
- 46.Chene G, Tchirkov A, Pierre-Eymard E, et al. Early telomere shortening and genomic instability in tubo-ovarian preneoplastic lesions. Clin Cancer Res. 2013; 19: 2873–2882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Telomere length changes in STICs associated with HGSC from our previous report (21).
Telomere length changes in HGSCs from our previous report (21).