

SUMMARY
Maintaining proteostasis in eukaryotic protein folding involves cooperation of distinct chaperone systems. To understand how the essential ring-shaped chaperonin TRiC/CCT cooperates with the chaperone Prefoldin/GIMc (PFD) we here integrate cryoEM, crosslinking- mass-spectrometry and biochemical and cellular approaches to elucidate the structural and functional interplay between TRiC/CCT and PFD. We find these hetero-oligomeric chaperones associate in a defined architecture, through a conserved interface of electrostatic contacts that serves as a pivot point for a TRiC–PFD conformational cycle. PFD alternates between an open “latched” conformation and a closed “engaged” conformation that aligns the PFD–TRiC substrate binding chambers. PFD can act after TRiC bound its substrates to enhance the rate and yield of the folding reaction, suppressing non-productive reaction cycles. Disrupting the TRiC- PFD interaction in vivo is strongly deleterious, leading to accumulation of amyloid aggregates. The supra-chaperone assembly formed by PFD and TRiC is essential to prevent toxic conformations and ensure effective cellular proteostasis.
Keywords: Chaperonin, Chaperone, TRiC, CCT, Prefoldin, GIMc, Proteostasis, CryoEM, XL–MS
Graphical Abstract
INTRODUCTION
Ensuring efficient protein homeostasis (proteostasis) in eukaryotic cells depends on an elaborate network of molecular chaperones (Balchin et al., 2016). Among these, the ATP- dependent 1 MDa chaperonin complex TRiC (also called CCT) is essential for cell viability as it acts co- and post-translationally to assist folding of ~10 % of all newly translated proteins (Yam et al., 2008). TRiC also prevents aggregation and toxicity of proteins linked to amyloid neurodegenerative disease (Shen and Frydman, 2012; Sot et al., 2017). TRiC consists of two identical rings stacked back to back in a two-fold symmetrical architecture (Leitner et al., 2012). Each ring consists of eight paralog subunits in an evolutionarily conserved arrangement that creates a large internal chamber where unfolded polypeptides bind and fold (Meyer et al., 2003). All individual subunits possess an equatorial domain that binds ATP, an apical domain that binds unfolded substrate and an intermediate domain which transmits nucleotide-induced conformational changes (Lopez et al., 2015). At the apex of the complex, flexible apical loops serve as an ATP-regulated built-in lid that is open in the nucleotide-free state, but closes upon ATP-hydrolysis, confining the substrate within the central chamber for productive folding. The highly allosteric ATPase cycle involves both positive cooperativity within a ring and negative cooperativity between rings (Reissmann et al., 2007).
The diversification of TRiC subunits into eight paralogs underlies its unique ability to assist folding of select proteins. Each distinct apical domain of TRiC has unique recognition specificities, allowing substrate recognition through polyvalent contacts with distinct regions in the polypeptide (Balchin et al., 2018; Joachimiak et al., 2014; Spiess et al., 2006). TRiC subunits also exhibit dramatic differences in nucleotide affinity that segregate into a high and low ATP affinity hemispheres within each ring (Reissmann et al., 2012). Together with the distinct substrate binding topology, the staggered closure is proposed to order the folding trajectory of substrates upon release into the closed chamber (Balchin et al., 2018; Joachimiak et al., 2014; Lopez et al., 2015). This uniquely complex architecture of TRiC likely evolved to enable folding of aggregation-prone eukaryotic proteins with complex topologies (Yam et al., 2008).
While TRiC can itself promote substrate folding, in vivo it cooperates with other chaperones, such as Hsp70 (Frydman et al., 1994; Melville et al., 2003) and prefoldin/GimC (Siegers et al., 1999; Vainberg et al., 1998). Prefoldin (PFD) is a ~100 kDa ATP-independent chaperone initially identified genetically as GIMc (Geissler et al., 1998) and biochemically as PFD (Vainberg et al., 1998). Early studies showed PFD contributes to actin and tubulin folding (Geissler et al., 1998; Hansen et al., 1999; Vainberg et al., 1998), while subsequent studies indicate a broader cellular function, including cancer (Lopez et al., 2013; Wang et al., 2015; Xu and Her, 2013), neurodegeneration (Abe et al., 2013; Tashiro et al., 2013) and misfolded protein degradation (Abe et al., 2013; Comyn et al., 2016). Eukaryotic PFD is a hetero-hexamer with a jellyfish like architecture consisting of two ~23 kDa α and four ~17 kDa β subunits assembled in a poorly understood arrangement (Siegert et al., 2000). PFD/GIMc exhibits genetic interactions with TRiC (Costanzo et al., 2016; Geissler et al., 1998). PFD was proposed to maintain the solubility of unfolded actin or tubulin and directionally deliver them to TRiC for subsequent folding in a passive handoff (Hansen et al., 1999; Vainberg et al., 1998). A low resolution EM structure suggested that PFD binds to the apex of the TRiC chamber allowing substrate transfer (Martin-Benito et al., 2002). However, the mechanistic and functional underpinnings of the TRiC–PFD interaction remain poorly understood.
Here we define the structural and functional interplay between TRiC and PFD using a hybrid structural effort integrating cryoEM, XL–MS and modelling combined with biochemical and genetic analyses. PFD engages TRiC via a conserved electrostatic interface acting as a pivot point, and alternates between an open “latched” conformation and an “engaged” conformation aligning the substrate binding chambers of TRiC and PFD. Biochemical experiments demonstrate PFD acts on a TRiC-actin complex to enhance the yield and rate of the folding reaction. PFD likely suppresses non-productive reaction cycles by modifying the environment experienced by the TRiC bound substrate. Importantly, disrupting the TRiC–PFD interaction in vivo renders PFD lethal and leads to widespread amyloid aggregation, highlighting how inter- chaperone physical contacts establish the networks that maintain proteostasis.
RESULTS
A strategy for production of functional recombinant human TRiC
Functional studies of TRiC/CCT have been hindered by the lack of a robust recombinant expression system. To address this, we used baculovirus to co-express all eight human TRiC (hTRiC) subunits (Figure 1A). To aid structural studies, we also tagged hTRIC with GFP fiducial markers at flexible surface-exposed loops in yeast and human CCT1 and CCT6. In addition, affinity tags were inserted into surface exposed loops of yeast subunit CCT2 (a calmodulin binding peptide, CBP) and human CCT7 (a 6XHis tag) respectively (Figure 1A). In yeast, these insertions fully support growth (Figure S1A). Both yeast and human recombinant complexes were purified combining affinity and chromatographic purification steps (Ferreyra and Frydman, 2000). SDS–PAGE comparing these complexes to endogenous TRiC purified from bovine testes (bTRiC) confirmed the presence of the tagged subunit in addition to the characteristic pattern of TRiC subunits (Figure 1B). For each complex, native gel analysis (NPAGE) confirmed the presence of a single complex with the expected mobility (Figure 1C) and mass spectrometry (MS) analyses verified the presence of all eight subunits in similar abundances based on label free quantification.
Figure 1. A recombinant system for human TRiC.

(A) Purification strategies for yeast and human TRiC. (B) Recombinant TRiC and bovine TRiC have similar banding patterns by SDS–PAGE and (C) migrate similarly on Blue Native (BN)- PAGE. (D) XL/MS strategy. (E) Identified XLs mapped onto the TRiC subunit arrangement, top ring subunits (black), bottom ring subunits (grey) and number of observed XLs between adjacent subunits (red). (F) Histogram of shortest measured intra/interring Cα-Cα distances for identified XLs under the acceptable XL length (G) Measured inter vs intra ring Cα-Cα distances for hTRiC (black) and hTRiC-GFP (green), grey lines: acceptable Cα-Cα distance. (H) Histogram of the number of satisfied XLs for all possible subunit arrangements of TRiC, the correct arrangement has the most satisfied XLs (inset). (I) CryoEM reconstructions of CCT1-GFP and CCT6-GFP hTRiC, Extra density of GFP (green) shown as a difference map between the original (~7Å) and lowpass filtered maps (20Å). (J) Autoradiogram from NPAGE of [35S]actin folding assay: all TRiC variants bind and fold Actin upon ATP addition with efficiencies similar to bovine TRiC.
We examined if recombinant hTRiC and or hTRiC–GFP are assembled into the correct and evolutionarily conserved subunit arrangement using DSS crosslinking and MS analyses (XL– MS) (Leitner et al., 2012) (Figure 1D). For both recombinant hTRiC and GFP-hTRiC, the correct subunit arrangement satisfied the majority of inter-subunit peptide crosslinks (XLs) distributed throughout the structure (Figure 1E–G). Importantly, an unbiased combinatorial approach assessing the number of XL distance constraints for recombinant hTRiC statistically satisfied only the one correct arrangement among the 40,320 possible subunit combinations (Figure 1H).
Single particle cryoEM also validated the architecture of CCT1–GFP and CCT6–GFP hTRiC through (Figure 1I, S1C–E, Table S1). Maps for the open conformation were obtained to ~7Å resolution (Figure S1C), clearly resolving all sixteen subunits, with defined helical features at the equatorial domains and lower resolution for the flexible apical domains (Figure S1D). The extra densities of the fiduciary GFP tags observed in the 20 Å lowpass map located to opposite sides of the complex for CCT1–GFP, and adjacent across the rings for CCT6–GFP (Figure 1I, S1E) as predicted by the known TRiC arrangement (Leitner et al., 2012).
We next compared the ability of recombinant hTRiC and bTRiC to fold denatured [35S] – Actin, an obligate TRiC substrate. Chaperone association and ATP-dependent folding were monitored as described previously (Meyer et al., 2003), by migration of [35S]actin on NPAGE followed by autoradiography (Figure 1J). Actin aggregates in the absence of TRiC regardless of ATP addition (Figure 1J, lanes 1,2). In the presence of either bovine bTRiC or any of the hTRiCs tested, we observed efficient [35S]actin binding to TRiC (Figure 1J, lanes 3, 5, 7, 9). ATP addition led to folded [35S]actin production by all hTRiC complexes, with similar efficiency and kinetics as bTRiC (Figure 1J and S1F). Taken together, our results demonstrate that recombinant hTRiC is correctly assembled into the evolutionally conserved native subunit arrangement and bears native folding activity and structural characteristics.
Generation and characterization of recombinant PFD
To obtain natively assembled PFD, yeast yPFD was produced in bacteria while a baculovirus system produced human hPFD in insect cells (Figure 2A). Purification using affinity and conventional chromatography resulted in homogeneous PFD complexes (Figure 2B), that were active in binding non-native actin, as indicated by formation of a [35S]actin-PFD complex in NPAGE analysis (Figure 2C).
Figure 2. Recombinant systems for PFD and XL/MS arrangement.

(A) Purification strategies for human and yeast PFD. (B) SDS–PAGE of purified PFD complexes (Figure S2D). (C) Autoradiogram of NPAGE of yPFD and hPFD bound to denatured [35S]actin. (D) SDS–PAGE and Native-PAGE of hPFD −/+ DSS crosslinking. (E) Obtained XLs mapped onto homology models of hPFD and yPFD (solid black lines). (F) Histogram of number of XLs satisfied for every PFD subunit arrangement, the two arrangements satisfying the most constraints are mirror images in which each subunit has the same neighboring subunits. (G) Histograms of Cα-Cα distance of XLs mapped onto homology models of the two subunit arrangements that satisfied the most constraints. (H) Example immunoblot of serially diluted PFD (4–.06 μM) bound to TRiC (0.25 μM). (I) Quantification of immunoblots of TRiC-bound vs free PFD establish apparent dissociation constant, error bars represent SEM. (J) Role of ATP in TRiC–PFD interaction: Measured relative rotational diffusion (free Alexa647-PFD set to 1) of Alexa647-PFD mixed with TRiC ATP-AlF4 (closed), TRiC no nucleotide (open), and absence of TRiC.
XL–MS examined the subunit arrangement of both hPFD and yPFD (Figure 2D–G). DSS XLs were mapped onto homology models for either hPFD or yPFD derived from the archaeal PFD structure (PDB 1FXK) (e.g. Figure 2E). Unbiased combinatorial analyses for satisfied distance constraints in each of the 24 possible PFD subunit arrangements favored two mirror- image arrangements for both yPFD (not shown) and hPFD (Figure 2F). While one arrangement had significantly (t-test p< 0.003) smaller average Cα-CαXL distances than the mirror image for both yPFD and hPFD complexes, (Figure 2G and Figure S2C), we could not unequivocally distinguish between these mirror structures as subunits in both have the same nearest neighbors. We conclude that both PFD complexes are active in binding non-native substrates and share a unique and evolutionarily conserved subunit architecture.
To examine the interaction of purified recombinant PFD with TRiC, increasing concentrations of hPFD (0.06 to 4 μM) were incubated with 0.25 μM hTRiC and formation of the PFD–TRiC complex analyzed by NPAGE followed by anti-PFD immunoblot (Figure 2H). The ratio of TRiC-bound versus free PFD was quantified by fluorescent Western analysis (Figure 2I). Binding curves displayed a plateau at a 1:1 molar ratio of TRiC to PFD, yielding an apparent dissociation constant (Kd) of ~ 0.2 μM. Binding of a second PFD to TRiC was observed at higher PFD concentrations (Figure S2), this interaction appeared weaker, consistent with binding studies on archaeal homologues (Zako et al., 2006). We conclude that PFD stably binds to TRiC in a 1:1 stoichiometry. Of note, this assay did not detect stable PFD binding to TRiC locked in the closed state by incubation with ATP-AlF4. The effect of ATP-induced lid closure on the PFD–TRiC association was further examined by fluorescence polarisation using Alexa647-labeled hPFD (Figure 2J). Without ATP, TRiC caused a substantial decrease in the rotational diffusion of Alexa647-PFD, consistent with PFD–TRiC binding. In contrast, the closed TRiC-ATP-AlFx complex did not change the Alexa647–PFD anisotropy signal, consistent with a loss of interaction. Therefore, ATP-induced lid closure substantially decreases TRiC’s affinity for PFD.
Functional cooperation of TRiC and PFD in actin folding
Actin folding in vivo and in vitro exhibits an obligate requirement for TRiC. PFD is proposed to function directionally to deliver substrates to TRiC, hence its name “pre-foldin”. Indeed, as reported (Vainberg et al., 1998), PFD efficiently binds actin and delivers it to TRiC for ATP-dependent folding (Figure S3A). However, PFD can also bind actin that was pre-bound to TRiC; notably this also occurs in an ATP-independent manner. This indicates that rather than acting directionally, PFD and TRiC bidirectionally transfer the bound substrate. Kinetic analyses monitored whether and how PFD affects the rate and yield of actin folding (Figure 3A). To this end, ~13 μM [35S]actin denatured in guanidinium chloride was rapidly diluted 100-fold (~0.13 μM final) into 0.25 μM hTRiC (Meyer et al., 2003; Thulasiraman et al., 2000). When indicated, 2.5 μM hPFD was included (Figure 3B, condition ② and ③). In the absence of chaperones, actin forms aggregates that are either removed by centrifugation or retained in the top of the native gel (Figure 3B, aggr). TRiC and PFD form binary complexes with denatured [35S]actin, that can be visualized by NPAGE (Figure 3B). Addition of 1 mM ATP initiated the TRiC-mediated [35S]actin folding reaction, leading to time-dependent production of folded [35S]actin with a characteristic NPAGE migration (Figure 3B, condition ①). Autoradiography followed by quantification of aggregated, TRiC-bound, PFD-bound and folded [35S]actin allowed us to determine the kinetics and yield of the actin folding reaction.
Figure 3. Prefoldin acts on the TRiC-substrate complex to increase folding processivity.

(A) Actin folding assays: ①TRiC alone, ②TRiC and PFD during initial Actin binding, and③PFD added after initial Actin binding to TRiC. Folding was initiated by raising temperature to 37 °C and ATP addition. (B) Representative autoradiograms of the 2 minute and 16 minute timepoints of folding, note appearance of aggregates in TRiC alone sample at 16 min. (C) Plots of quantified Actin folding for each reaction, dotted lines represent linear fits to first 3 time points (0–4 min), solid lines represent linear fits to later time points (4–64 min), the average amount of folded actin for the final 3 time points of TRiC alone is set as 1 in each replicate, error bars represent SEM. (D) Initial and steady state folding rates for each reaction. (E) Plot of amount of aggregated Actin with minimal migration into the native gels, note significant decrease in folding rates for TRiC alone reaction correlate with appearance of aggregated Actin (red arrow). (F) Actin bound to TRiC was purified from unbound Actin and folding assays were carried out in the absence or presence of PFD, strikingly Actin transferred from TRiC to PFD both in the absence and presence of ATP
In the absence of PFD, TRiC mediates actin folding (Figure 3, condition ①) with biphasic kinetics. Upon ATP addition, a burst of folding activity (~ 4 min) is followed by a much slower and inefficient folding phase that extended up to 60 min (Figure 3C), consistent with rapid but low yield folding from a stoichiometric TRiC actin complex (Balchin et al., 2018). Quantification indicated that ~35% of initially TRiC-bound actin reached the native state (Figure 3D, condition ①), in line with previously published reports (Gao et al., 1992; Meyer et al., 2003). Most folding-incompetent actin remained TRiC-bound throughout the time course of the reaction, despite ongoing ATP hydrolysis. However, after the 4 min inflexion point, we also observed the appearance of actin aggregates retained at the top of the NPAGE (Figure 3B,E condition ①). Since all reactions were clarified prior to folding initiation, these aggregates result from release of an aggregation-prone species from TRiC. Therefore, only a fraction of TRiC- bound actin is folding competent and reaches the native state within a few rounds of ATP hydrolysis, while a significant fraction of actin binds in a folding incompetent conformation that either remains TRiC bound or aggregates even through multiple rounds of ATP hydrolysis. We speculate that the two-fold excess of TRiC over actin suffices to efficiently capture the released substrate unable to fold, except for a subpopulation that is released in an aggregation-prone conformation.
To examine the effects of PFD on TRiC-mediated actin folding, we tested whether addition of PFD prior to substrate delivery (Figure 3A, condition ②) enhanced TRiC binding to actin. We did not observe more actin binding to TRiC, perhaps because TRiC was already present in molar excess over actin. Instead, a significant fraction of [35S]actin bound to PFD; reducing the actin-fraction initially bound to TRiC (Figure 3B, condition ② note that [35S]actin- TRiC now forms a ternary complex with PFD). Upon ATP addition, actin folding kinetics remained biphasic, but the later, slower phase was significantly more productive resulting in a higher yield of folded actin (Figure 3C, D, condition ②). Furthermore, inclusion of PFD abrogated the formation of actin aggregates at later time points (Figure 3E). Surprisingly, even when added prior to TRiC-substrate binding, PFD contribution to the TRiC-mediated folding reaction was primarily in the later phase. Indeed, linear fits generated for the actin folding kinetics indicated that the initial phase was unchanged by PFD, if not slightly slowed (Figure 3D). We thus considered whether PFD may act on the TRiC-actin complex after it is already formed. To test this, we generated a TRiC-actin complex as above, subjected it to high-speed centrifugation to remove any unbound actin aggregates, and added PFD to the preformed complex, together with 1 mM ATP (Figure 3A–E, condition ③). Strikingly, adding PFD to a preformed TRiC-actin complex had the same effect on actin folding kinetics and yield as when PFD was added at the start of the reaction, prior to TRiC-substrate binding. These findings strongly suggest that PFD plays a major role to facilitate folding of actin, even after it was bound to TRiC. Since PFD primarily enhanced the slow kinetic phase of actin folding, our data suggests that PFD action returns kinetically-trapped actin to a productive folding pathway, either by acting on TRiC-bound substrate or by acting on released species and returning them to TRiC.
The above results were inconsistent with PFD acting in a purely directional manner to deliver substrates to TRiC (Hansen et al., 1999). Particularly puzzling was that PFD can bind actin when added to a preformed TRiC actin complex. Actin that was initially bound to TRiC was transferred to PFD at early time points post-addition (Figure 3B, condition ③) even in the absence of ATP (Figure S3A and Figure 3F). One interpretation of these experiments is that there is transfer from TRiC-bound actin to PFD. Alternatively, the actin that binds PFD in these experiments could arise from a soluble unbound actin pool that was not cleared in the sedimentation step. To directly determine if TRiC can transfer its bound actin to PFD, we generated a pure [35S]actin-TRiC complex, that is separated from unbound [35S]actin by anion exchange chromatography as described (Frydman and Hartl, 1996; Reissmann et al., 2007). Prolonged incubation of the purified [35S]actin-TRiC complex at 37 °C in the absence of ATP, did not produce any significant levels of folded or aggregated actin (Figure 3F lane 1), consistent with removal of any unbound bound actin and a low actin off-rate in the absence of ATP. Without PFD addition, this purified TRiC-actin complex yielded ~30% folded actin in an ATP- dependent reaction (Figure 3F lane 2). ATP-incubation led to ~8% of the initially bound actin to aggregate; likely arising from actin released in a folding-incompetent, aggregation-prone conformation. The remaining actin stayed TRiC-bound even after a 60 min incubation with ATP, perhaps reflecting ATP-dependent release and rebinding to TRiC. Addition of PFD, to this complex resulted in the ATP-independent transfer of a substantial amount of [35S]actin from TRiC to PFD, as well as a shift in the TRiC-[35S]actin band to reflect the ternary complex with PFD (Figure 3F lane 3). Notably, following ATP addition, PFD enhanced the yield of actin folding and disfavored actin aggregation (Figure 3F lane 4), as observed in Figure 3C. Thus, even when we purify the TRiC-actin complex away from unbound or loosely bound actin, we still observe ATP independent actin transfer from TRiC to PFD. We conclude that PFD is not merely capturing actin that was released from TRiC due to ATP cycling. Instead, it appears that transfer is mediated by a ternary TRiC-actin-PFD complex, from which actin partitions between TRiC and PFD.
These results suggest that actin binds to TRiC in distinct conformations, and only some are on-pathway for productive folding. PFD enhances the processivity of the TRiC folding reaction, and hence the overall actin folding rates. While our data do not conflict with PFD delivering substrates to TRiC they indicate PFD plays a major role after TRiC has already bound to substrates. Furthermore, the ability of actin to bi-directionally partition between TRiC and PFD without ATP indicates that the binding energy of actin for these chaperones is similar.
CryoEM structural analysis reveals a dynamic TRiC–PFD interaction
We next used cryoEM to obtain a structure of the TRiC–PFD complex (Figure 4, Table S1). CCT1–GFP hTRiC incubated with a two-fold molar excess of hPFD was imaged using single particle cryoEM. Images of vitrified TRiC–PFD revealed monodisperse particles (Figure 4A) and 2D class averages showed multiple orientations of TRiC with extra signals capping either one or both rings (Figure 4B, arrow). A single model refinement with all particle images produced a 6.5 Å cryoEM map (Figure S4D) showing unique asymmetric structural features in the open conformation of TRiC/CCT (Figure 4C, D) with detectable extra densities for PFD and the fiduciary GFP label on CCT1 at the lower contour level (Figure S4C). The TRiC map displayed well resolved structural characteristics, including clearly defined helices at the equatorial domain (Figure 4C, D). However, the extra-density capping the apex of TRiC was diffused and did not resolve any recognizable structural features of PFD (Figure 4C).
Figure 4. CryoEM map reconstructions of TRiC–PFD complex.

(A) Electron micrograph of ice-embedded TRiC–PFD complex. Representative particle views are indicated: top (red), tilted (orange) and side (blue). (B) Selected 2D reference free averages showing top, tilted and side views and diffused signal from PFD binding (Arrows). (C) Single model refinement of TRiC–PFD showing diffused densities shown in red around apical tips of TRiC. CCT1–8 shown in different color codes and red arrows indicate the location of protruding TRiC subunit (CCT1). (D) Two stacked CCT6 subunits are shown as a representative for the quality of the map and model. Apical domain (pink box) displays relatively lower resolution while equatorial domain (blue box) does higher resolution. (E) The structural dissection of the compositional and conformational heterogeneity in TRiC–PFD data set. (F) Segmented PFD from each PFD bound TRiC map showing six helical bundles. (G) Highlighted area (Orange color) on top-view of TRiC showing the direct contact of PFD to TRiC in each class.
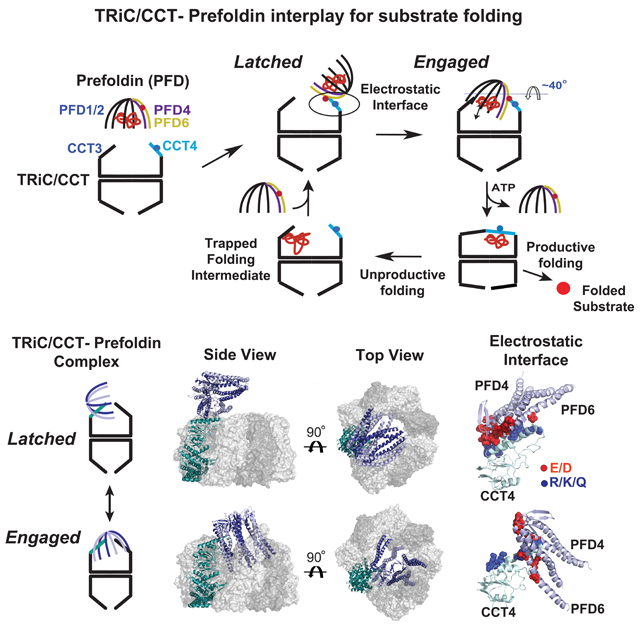
Since the conformational heterogeneity of the TRiC–PFD complex likely hindered our analysis (Figure 4B, C), we next adopted localized 3D reconstruction and focused classification approaches (Ilca et al., 2015; Roh et al., 2017) (Figure S4A). Strikingly, this approach revealed six major structural classes: one class corresponded to unbound TRiC rings and five classes corresponded to structures interpretable as conformationally distinct binding modes of PFD to TRiC (Figure 4E, S4B). In each map, the characteristic structural features of PFD were well resolved, including the coiled-coiled bundles of six PFD subunits (Figure S4D, S4E). This analysis indicates that PFD engages TRiC in a conformationally dynamic interaction that ranges from a single to multiple contact sites. All conformational states involve a key contact with the apical domain of CCT4 and the progressive engagement of additional TRiC subunits. We designate the two extreme states as the “latched” state (Class 2), where both substrate chambers in PFD and CCT appear independent to each other, and a tightly “engaged” state (Class 6) where the two chambers appear aligned in a unique chemical environment (Movie S1).
A molecular model of the TRiC–PFD complex
The correct subunit arrangement of eukaryotic PFD and its relative orientation to TRiC were resolved through a XL–MS strategy combined with cryoEM (Figure 5A and S5A, Table S2). We obtained 41 unique XLs between hTRiC and hPFD and 13 unique XLs for the yeast complex. Dominant XLs between PFD4/6 and CCT4/2/5/7, as well as PFD1/2 and CCT3/6/8 unequivocally oriented PFD onto the TRiC ring (Table S2). Mapping inter-complex XLs onto homology models of the mirror PFD arrangements bound to TRiC identified a unique PFD arrangement, identical in both the human and yeast complexes (Figure 5B for human and Figure S5A for yeast). The average XL length in the correct arrangement was 18.7 Å for human and 26 Å for the yeast complex, i.e. within the expected distance for DSS; in contrast, the distances for the incorrect mirror arrangement were 51.3 Å for human and 65 Å for yeast, (Figure 5B and Figure S5A). We conclude that a unique PFD subunit arrangement and binding orientation to TRiC are conserved in evolution.
Figure 5. TRiC and PFD interact in a subunit specific manner.

(A) DSS crosslinked TRiC–PFD analyzed by SDS-PAGE and Native-PAGE. (B) Histograms of Cα-Cα distances establish the correct arrangement of the PFD subunits: XLs mapped onto TRiC–PFD models with the two possible PFD subunit arrangements determined from PFD alone XLs show one conformation has a much smaller average XL distance (18.7 vs 51.3 Å) consistent with the ~ 24 Å DSS length. (C) CryoEM map of Class 6 showing overall architecture of TRiC- PFD complex. CCT1–8 and PFD1–6 subunits are shown in different color codes and red arrows indicate the location of protruding TRiC subunit (CCT1). (D) Ribbon diagram of PFD into segmented PFD density to show the map and model quality. (E) Segmented CCT4 subunit is shown as a representative for the quality of the map and model. (F) Surface electrostatic distribution and (G) Hydrophobicity distribution inside of TRiC chamber at the viewpoint from equatorial to apical domains.
Using the identified PFD architecture, we generated homology models for the PFD–TRiC cryoEM maps (Figure 5C, D, E, Class 6 shown here as a representative), which display the different PFD binding modes on TRiC (Figure 4, S5B). Overall, the architecture of TRiC/CCT and PFD are unchanged among the classes, with only minor local rearrangements on TRiC’s apical domains and PFD’s tentacles (Figure S5C, D). Of note, all structures share a subunit specific interaction between PFD4/6 and the CCT4 apical domain (Helix 268–293) (Figure S5B, E), which creates a unique asymmetric directionality on TRiC. Along the distinct classes, the PFD tentacles extend across specific apical helices of TRiC/CCT, becoming progressively inserted into the TRiC chamber. In the completely engaged Class 6, the substrate binding chambers of PFD and TRiC are aligned (Figure 5C). Of note, even in this engaged conformation, the TRiC chamber remains accessible to the environment through large fenestrae (e.g. see 40 Å opening indicated in Figure S5F) that render the substrate accessible to the exterior of the chamber.
The aligned chambers of TRiC and PFD in the engaged state dramatically change the environment surrounding the bound substrate (Figure 5F, G). First, PFD binding to TRiC creates a tunnel-like environment with an asymmetrical distribution of positive and negative charges in the chamber (Figure 5F). The polar PFD tentacles combine with subunits CCT 2/4/5 to create a negatively charged region and with subunits CCT 1/3/6/8 to create a positively charged surface (Figure 5F). Of note, this asymmetric charge distribution can affect the solvent properties of the folding environment and may interact with charged substrate regions (Chakraborty et al., 2010). Intriguingly, upon ATP-dependent lid closure, TRiC fully encapsulates the substrate in a chamber that maintains this same charge asymmetry (Leitner et al., 2012). Second, an evolutionarily conserved large hydrophobic pocket within the PFD chamber profoundly remodels the hydrophobic surfaces available to the TRiC-bound substrate (Figure 5G). These structural analyses provide a rationale for our observed TRiC–PFD functional interplay to achieve actin folding (Figure 3). Thus, the PFD chamber properties contribute to modulation of trapped folding intermediates on– and off– the TRiC chamber, to return them to a productive folding pathway.
The TRiC–PFD interaction interface functions as an electrostatic pivot point
All TRiC–PFD structural states share a contact interface between PFD4/6 and the apical domain of CCT4 (Figure 4G, S5B). In the more engaged states (Class 3–6), we observed an additional contact between CCT3 and PFD1 (Figure 6A, Class 6 shown as a representative), suggesting a temporal sequence of increased contributions from each TRiC subunit to PFD engagement. To examine whether individual apical domains of TRiC mediate stable PFD binding, we expressed and purified the apical domains of each yTRiC subunit (herein yApi1–8) and tested their binding to PFD by gel-shift assay on NPAGE. Only the apical domain of CCT4 (yApi4) stably associated with human or yeast PFD (Figure 6B, S6A). Next, the contribution of Api4 to TRiC–PFD binding was assessed by competition assays adding purified Api4 to a TRiC–PFD binding reaction. Api4 efficiently prevented, in a concentration dependent manner, the formation of both yeast and human TRiC–PFD complex (Figure 6C, S6B); higher concentrations produced a corresponding shift in PFD mobility due to binding to Api4. We conclude that the apical domain of CCT4 has an evolutionarily conserved role mediating the TRiC–PFD interaction. Of note, denatured [35S]actin bound to PFD in the presence or absence of Api4, indicating Api4 binding does not preclude PFD-substrate association (Figure 6D). Thus, PFD can simultaneously bind substrate and the apical domain of CCT4, substantiating the presence of distinct binding sites for these PFD ligands.
Figure 6. TRiC–PFD interaction site mediated by an electrostatic pivot point on CCT4.

(A) CryoEM map of Class 6 highlighting the PFD and TRiC interaction. Segmented map with fitted model showing the direct contact of PFD to apical helix of CCT4 (Cyan) and 5 (Gold). (B) Binding of purified CCT apical domains to PFD analyzed by NPAGE and immunoblot for PFD. (C) Competitive inhibition of TRiC–PFD binding by apical domain of CCT4 (Api4), analyzed by NPAGE and immunoblot. TRiC binding to PFD is inhibited at near equimolar concentrations of Api4. (D) 35S-Actin binding to PFD alone, PFD with Api4, and Api4 alone analyzed by NPAGE followed by fluorescence and autoradiography. (E) Api4–PFD interface highlighting positive charges in Api4 (blue) and negative charges in PFD (red). (F) TRiC–PFD binding is not competed away by Api4–EEE mutant (Yeast: R268E, K276E and K277E, Human: R280E, K288E, Q289E), but is competed away by WT Api4 (G); indicating loss of Api4-EEE binding to PFD. (H) Model of hTRiC and hPFD interface between Pfd4 and Pfd6 and TRiC: electrostatics modeled in blue to red color gradient indicating positive to negative charge. (I) Surface conservation of TRiC and PFD interface obtained from alignment of 100 orthologous sequences mapped onto the hTRiC and hPFD structures. Color gradient of red to green indicates decreasing conservation.
Comparing the structural models of all PFD–TRiC states reveals that PFD pivots on the long helix 8 (H269–293) of CCT4 apical domain (Figure S5D, 6A). All states involve contacts between positively charged residues (R/K/Q) in this CCT4 helix with negatively charged residues (E/D) in the PFD4/6 interface (Figure 6E, shown for Classes 2 and 6). To test whether TRiC–PFD binding is driven by this electrostatic interaction, we introduced charge-reversal mutations in Helix 8 (Yeast: R268E, K276E and K277E, Human: R280E, K288E, Q289E) of Api4 (herein Api4–EEE) (Figure 6F). Strikingly, Api4–EEE could neither bind PFD (Figure 6F) nor compete with TRiC for PFD binding (Figure 6G) even at the highest concentrations tested. This demonstrates that this charged CCT4 Helix is the key determinant of the TRiC–PFD interaction. These complementary electrostatic patches are conserved across eukaryotic PFD and TRiC (Figure 6H, I). We conclude that PFD is recruited to TRiC through a conserved electrostatic interaction between extended negatively and positively charged patches in PFD and CCT4 respectively (Figure S6C). These extended interfaces allow PFD to pivot while bound to TRiC, in order to align their substrate binding chambers (Figure 6H).
Direct TRiC–PFD contacts are essential for proteostasis in vivo
To examine the biological relevance of the PFD–TRiC interaction site the Api4–EEE mutations were integrated into the chromosomal copy of Saccharomyces cerevisiae CCT4. (Figure 7). The CCT4–EEE cells were slow-growing but viable (Figure 7A). NPAGE analysis comparing the conformation of yTRiC–WT and yTRiC–EEE with or without ATP–AlFx showed both TRiC variants migrated as a single complex with characteristic mobility and could form the ATP-induced closed lid complex (Figure 7B). We next examined whether the CCT4–EEE mutation affects TRiC binding to PFD (Figure 7C). Lysates from either TRiC–WT or TRiC–EEE cells were incubated with PFD immobilized on CNBr beads and washed with increasing KCl concentrations; followed by anti TRiC immunoblot. TRiC–WT, exhibited robust and salt- sensitive binding to PFD; consistent with an electrostatic interaction (Figure 7C). In contrast, TRiC–EEE did not bind PFD even at the lowest salt concentration. These experiments establish that the Helix 8 in CCT4 is not only sufficient but also necessary for binding of the full TRiC complex to PFD.
Figure 7. TRiC–PFD electrostatic interaction is essential for successful folding through PFD.

(A) Surface charge of WT and EEE yTRiC highlighting Api4 region neutralized by EEE mutations. (B) NPAGE analysis of WT and EEE TRiC in the absence and presence of ATP- AlFx. (C) Electrostatic interactions facilitate PFD–TRiC binding: WT or EEE yTRiC binding to yPFD coupled to CNBr-beads examined at 100, 200 or 400 mM KCl. (D) In vivo flux through a PFD–TRiC pathway. In the absence of PFD, nascent chains can directly associate with TRiC, while in the EEE-TRiC nascent chains will accumulate on PFD. (E) pfd5 suppresses EEE-TRiC phenotype. Yeast strains (WT, Δpfd5, EEE, EEE-Δpfd5 carrying either a PFD5 expressing vector or the empty vector control were serially diluted and grown at 20 °C, 30 °C or 37 °C. (F) Proteostat staining (red) of yeast strains to detect aggregation, nuclei are stained with DAPI (blue). (G) Aggregate analysis of indicated yeast strains: (T) total lysates; supernatant (S); insoluble pellet fraction (P). (H) Available protein folding pathways for TRiC substrates in WT, Δpfd5 and TRiC-EEE yeast strains. (I) Pivot motion of PFD on TRiC displayed through top and side views of the different particle classes from CryoEM.
Unlike TRiC, PFD is not essential for cell viability. However, deletion of individual PFD subunits leads to cold sensitivity (Geissler et al., 1998) (Figure 7D, E). Strikingly, cells bearing TRiC–EEE, unable to bind PFD, had an extremely severe phenotype, showing strong reductions in growth rate not only at low temperature but even at 30 °C (Figure 7E). We reasoned that if the inability to form a direct TRiC–PFD contact results in increased misfolding, loss of PFD might rescue the severe TRiC–EEE phenotype. Indeed, deletetion of core PFD subunit PFD5 (Δpfd 5), required for PFD assembly, mitigated the TRiC-EEE phenotype, restoring growth to near WT levels (Figure 7E). To verify that PFD is indeed toxic in TRiC-EEE, we introduced a PFD5 expressing plasmid into TRiC-EEE Δpfd5 double mutant cells. Remarkably, expression of PFD5 restored the slow growth phenotype observed in the TRiC-EEE cells. Next, we tested whether the slow growth phenotype observed above arose from the disruption of normal proteostasis. The presence of toxic misfolded species was assessed by staining cells with Proteostat, a fluorescent dye that selectively associates with protein aggregates (Figure 7F). In control wild type (WT) cells, little Proteostat staining is observed at 30 °C. Deletion of PFD5 caused a mild increase in Proteostat staining at 30°C. In contrast, in TRiC-EEE cells we observed a strong increase in Proteostat fluorescence. Most cells contained one perinuclear and at least one peripheral cytoplasmic inclusion that were positive for Proteostat staining, consistent with the previously described JUNQ and IPOD (Kaganovich et al., 2008). Of note, Proteostat staining in TRiC-EEE cells was reduced by additional deletion of PFD5 (Figure 7F, TRiC-EEE Δpfd5). Biochemical analyses of aggregation in these cells (Willmund et al., 2013) confirmed and extended the conclusions of the Proteostat staining, the TRiC-EEE strain had substantially more aggregated proteins compared to the other strains (Figure 7G, Figure S7). Resolution of multiple proteins aggregated in these cells (Figure 7G, red dots), suggest a pleiotropic effect on protein solubility. Of note, both assays confirmed that the widespread aggregation in the TRiC-EEE strain was alleviated by the additional deletion of PFD.
We next characterized the protein aggregates in TRiC-EEE cells compared to WT. Immunoblot analysis found virtually no change in TRiC levels or solubility across all four strains (Figure S7B). TRiC-substrate α-tubulin was strongly depleted in TRiC-EEE cells, which was alleviated by concomitant deletion of Δpfd5. By contrast, β-tubulin and actin, also abundant TRiC substrates, were highly enriched in the aggregates of TRiC-EEE (Table S3). Thus, α- tubulin is targeted to degradation in TRiC-EEE cells, while β-tubulin and actin aggregate. MS analyses indicated that a wide range of cellular proteins aggregate in TRiC-EEE cells, including known TRiC substrates, such as β -tubulin, actin, cell cycle regulators, mitochondria-targeted proteins and ribosome biogenesis factors. While PFD was not found in the aggregates, other chaperones, including the Hsp70 SSB, Hsp104 and Hsp90 aggregated in TRiC-EEE cells. These experiments indicate that disrupting the TRiC–PFD interaction in vivo leads to widespread proteostasis impairment, causing both aggregation and degradation of cellular proteins. We conclude that a direct interaction between TRiC and PFD is essential to maintain proteostasis.
DISCUSSION
Here we establish the structural and functional basis for TRiC–PFD cooperation in protein folding. A conserved electrostatic interaction allows PFD to bind to the open TRiC state and pivot to a conformation that aligns their two substrate-binding chambers and alters the chemical properties of the substrate environment. Presumably this returns TRiC-bound trapped intermediates to a productive folding pathway. A direct physical contact between TRiC and PFD is essential for effective proteostasis. Disrupting their interaction renders PFD toxic and leads to accumulation of misfolded amyloid aggregates. Taken together, our work illuminates a poorly understood but critical node of eukaryotic proteostasis.
Kinetic studies of TRiC mediated actin folding led to surprising insights into the molecular role of PFD. TRiC-bound actin folds with biphasic kinetics. Most folding occurs in the initial phase, consistent with most productive actin folding not requiring many cycles of release into solution (Balchin et al., 2018; Frydman and Hartl, 1996). Despite ongoing ATP hydrolysis by TRiC, the remaining actin folded slowly and inefficiently, either remaining TRiC-bound or aggregating. Recent work found that actin adopts a specific TRiC-bound topology that appears required for productive folding (Balchin et al., 2018). We speculate this productive conformation is responsible for the fast folding phase, while actin molecules bound in non-productive topologies require remodeling by PFD. Consistent with this idea, PFD addition to TRiC, either before or after addition to substrate, only marginally affected the first kinetic phase of actin folding, but greatly enhanced the rate and yield of the second phase, and abrogated actin aggregation. These results indicate that PFD acts on actin bound to TRiC in a non-productive conformation to return it to a productive folding pathway. Whether such remodeling of actin folding intermediates occurs in the shared TRiC–PFD chamber or whether PFD receives actin from TRiC following ATP-release for rebinding to the chaperonin in a different conformation are important questions for future investigation. There is no requisite directionality in the substrate interaction between TRiC and PFD. Since PFD enhances actin folding kinetics and suppresses aggregation after TRiC binding, it could be argued that PFD functions as a processivity factor rather than, or in addition to, in substrate delivery. Future studies should clarify whether PFD also acts upstream of TRiC in a co-translational folding pathway and whether in vivo, TRiC and PFD function as a complex to bind substrate.
CryoEM classification showed that TRiC-bound PFD adopts a series of conformations. While cryoEM cannot tell the sequence of events among these conformations, it is logical to consider a conformational cycle where the “latched” (Class 2) state represents an initial binding event between TRiC and PFD through contacts with CCT4. Class 2 is likely a transient stage, with minor population on our cryoEM analyses (~5%), while the “engaged” (Class 6) state, where the two substrate-binding chambers are aligned, shows the largest population (20%). Class 3–5 (10% each) would represent intermediate conformations between Class 2 and 6. Using contact area analyses (Krissinel and Henrick, 2007) and minimum RMSD between the models, we obtained a comprehensive model (Figure 7H, Movie S2) for a mechanical transition of PFD binding to TRiC. All conformations maintain electrostatic contacts between Helix 269–293 of CCT4 inserted to bridge the space between PFD4 and 6; the short helix 310–320 of CCT4 also makes direct contact with PFD6 (Figure S5E), which provide an anchor point along which PFD pivots through the different classes. Additional interactions between the apical helices of CCT3 and PFD1 (Figure 6A) likely facilitate the hinge-like motion (Movie S2).
PFD and TRiC themselves also showed notable conformational changes upon binding, likely to accommodate PFD into the TRiC chamber (Figure S5C). In particular, the engaged state (Class 6) induces a slight expansion of the TRiC chamber and alters dramatically the environment of the bound substrate. The TRiC–PFD interaction sites are compatible with formation of a ternary complex with the substrate protein, consistent with our observations (e.g. Figure 3B, 6D). A substrate within the composite chamber of the engaged TRiC–PFD state will be surrounded by an asymmetric distribution of charged and hydrophobic residues (Figure 5D, E) that likely remodel the conformation of the bound substrate and therefore its folding trajectory. This explains the enhanced processivity of TRiC-mediated folding in the presence of PFD. These questions will need to be addressed by structural approaches in the presence of substrate.
Interestingly, even in the engaged TRiC–PFD conformation there are large openings (e.g. 40 Å in Figure 5C, S5E) communicating the central chamber with the outside environment. The structural flexibility of CCT2 in the open state, previously observed for yTRiC (Zang et al., 2016), and observed here for hTRiC, is unaffected by PFD binding (Movie S1). CCT2 conformational dynamics may further expand access to the central chamber as it provides an even larger side opening into the TRiC–PFD chamber. This could provide a gate to accommodate domain-wise TRiC-mediated folding (Russmann et al., 2012) in multidomain or cotranslationally binding substrates. Of note, we have been unable to image PFD complexes with closed TRiC–ATP, consistent with biochemical data suggesting PFD is released upon TRiC lid closure.
Yeast cells are estimated to have ~20,000 molecules of TRiC and 2–4,000 molecules of PFD (i.e. ca. 0.8 μM TRiC vs 0.08–0.16 μM PFD), raising the question of their respective roles within the eukaryotic chaperone networks. In vivo, disrupting the TRiC–PFD interaction is strongly deleterious and is accompanied by a dramatic accumulation of aggregates containing essential TRiC substrates as well as chaperones which could either be TRiC substrates or sequestered in the aggregates (Figure 7 and S7, Table S3). Such widespread aggregation and chaperone sequestration likely contribute to a pleiotropic deleterious phenotype. Misfolded proteins could arise from the inability of PFD to deliver substrates to TRiC or from inability of TRiC to fold bound substrates without PFD assistance. Of note, the absence of PFD alleviates these severe phenotypes, presumably because its function is compensated by other chaperone systems that ensure the processivity of TRiC folding. Our study highlights how direct communication between distinct chaperone systems orchestrates the pathways essential for efficient proteostasis.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
For materials and further information, please contact the lead contact, Judith Frydman (jfrydman@stanford.edu).
DETAILED EXPERIMENTAL PROCEDURES
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and growth conditions
CRISPR strains:
Primers were designed for guide sequence (see Table S4) and used to amplify the pCAS9 vector(Ryan et al., 2016). The yeast Api4 EEE plasmid (see Cloning) was used for amplifying homology sequence containing the EEE mutation for recombination, guide recognition sequence was mutated using overlapping PCR. The EEE CRISPR was carried out on WT BY4742 yeast and Δpfd5 strain by transforming pCAS9-Api4guide and PCR product into strains. Transformants were plated onto G418 and allowed to grow for 48 hrs at 37 °C. Individual colonies were selected and grown overnight in YPD. Overnight cultures were spread on YPD plates, and mutations in individual colonies were confirmed by PCR and sequencing.
Other strains:
PFD5 add back strains were generated by transforming clone YGPM10b11 from the yeast genomic tiling collection from Dharmacon into indicated strains and selecting on –Leu plates. GFP/CBP CCT yeast strains were generated by taking haploid strains containing the pCuCCTx(Ura3) vector (Tam et al., 2006) and deleted for the corresponding cctx gene and transforming with the pAB-CCTx (Leu) vector containing the desired insertion (for instance CCT1–GFP). Counter selection against the WT (Ura3) plasmid was performed using (5’–FOA) plates. Obtained colonies were tested for growth on –Leu plates to verify presence of pAB vector, and – Ura plates to verify loss of pCu vector.
Growth conditions:
For experiments involving the EEE strain, all strains in experiment were grown at 37 °C overnight, and then diluted to an OD600 under 0.1 and grown to an OD600 of 0.3 to 0.9 at reported temperatures (minimum of 3 hrs of growth). For experiments involving the PFD5 vector, cells were grown in –Leu media. For experiments not involving the EEE strain, strains were grown at 30 C, and treated as above. For drop tests involving the PFD5 vector, cells were plated on –LEU plates. For all other drop tests cells were plated on YPD. Plates were incubated at indicated temperatures for drop tests.
METHOD DETAILS
Cloning
CCT1–8 apical domains:
Each apical domain was cloned into the super folder GFP between Beta sheets 10 and 11 of the beta-barrel structure with a glycine serine linker added to provide some flexibility. Constructs were cloned into pST39(Tan, 2001) using XbaI and BamHI restriction sites for Api domains 1,2,3,4,5,7 and 8, and XbaI and HindIII for Api domain 6. For the yeast apical domains, pRS vectors containing genomic CCTX were used as template. Mutations were made by overlapping PCR with primers containing the desired mutations.
Fastbac Dual hTRiC construct cloning:
Individual TRiC subunits were cloned into the pFastbac dual vector (ThermoFisher Scientific) using SalI/NotI for CCT1,2,6 and 7 and XhoI/KpnI for CCT3,4,5 and 8. The dual vectors consisted of CCT1,8, CCT2,4, CCT3,6 and CCT5,7 with CCT2, CCT5, CCT6 and CCT8 being cloned initially followed by CCT4, CCT7, CCT3 and CCT1 respectively. For GFP tagged vectors, BspEI sites were cloned into flexible loops using overlapping PCR. GFP was inserted into BspEI sites using standard techniques.
Fastbac Dual hPFD construct cloning:
Individual Pfd subunits were cloned into the pFastbac dual vector (ThermoFisher Scientific) using SalI/NotI for Pfd1,4 and 5, and XhoI/KpnI for Pfd2,3, and 6. The dual vectors consisted of Pfd1,2, Pfd5,3 and Pfd4,6 with Pfd1, Pfd4, and Pfd5 being cloned initially followed by Pfd2, Pfd3 and Pfd6 respectively.
Yeast GFP and CBP vectors:
BspEI sites were cloned into flexible loops of CCT subunits using site directed mutagenesis in pAB vectors. Either GFP or CBP was cloned into BspEI sites using standard cloning techniques.
Protein Expression and Purification
hTRiC:
We used the Bac to Bac baculovirus expression system for recombinant protein production (Invitrogren). P3 virus for human TRiC subunits was prepared as described in the protocol. 1 l of High-Five insect cells (Invitrogen) at a concentration of ~3X106 cells/ml were co- infected with 5 mls of each of the viruses. and cells were harvested 2 to 3 days after infection by centrifugation. Cells were resuspended in 100 mls lysis buffer (100 mM NaCl, 100 mM HEPES pH 7.4, complete protease inhibitors EDTA free (Roche), 1 mM DTT, PMSF, Benzonase 5 U/ml (Sigma-Aldrich)) and lysed using an emusiflex (Avestin). Lysate was clarified by ultracentrifugation (50,000XG, 1 h). Cleared lysate was passed over a FFQ column equilibrated with MQA buffer (50 mM NaCl, 20 mM HEPES pH 7.4, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10% glycerol), and then eluted with 40% MQB buffer (1 M NaCl, 20 mM HEPES pH 7.4, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 10% glycerol). TRiC eluted from FFQ column was passed over a Heparin column equilibrated with 20% MQB buffer and washed until UV returned to baseline. TRiC was eluted with a 200 ml gradient from 20% MQB to 100% MQB, and fractions containing TRiC identified by SDS-PAGE. TRiC containing fractions were concentrated down using an Amicon Ultra 100 kDa MWCO centrifugal filter (Millipore) to a volume of ~ 2 mls. The concentrated TRiC was further purified by size exclusion chromatography over a Superdex200 26/60 (GE Healthcare Life Sciences) equilibrated with column buffer (100 mM NaCl, 50 mM HEPES pH 7.4). Eluted TRiC was identified by SDS- PAGE and purified by Talon resin (Clontech). TRiC eluted from the talon resin was purified by anion exchange chromatography using a MonoQ 10/100 column equilibrated with MQA and eluted by a 150 ml gradient to 100% MQB. Fractions containing TRiC were identified by SDS- PAGE, concentrated to ~ 1 ml, aliquoted and snap frozen. Typical yields were greater than 10 mg/L and TRiC assayed by mass spec is ~ 95% pure.
yTRiC:
Approximately 6 L of yeast at an OD ~2 expressing CCT2 with a CBP tag was harvested by centrifugation. Cells were resuspended in equal volume lysis buffer (200 mM NaCl, 100 mM HEPES pH 7.4, 5 mM CaCl2, 20% glycerol, complete protease inhibitors (Roche), benzonase 5U/ml, PMSF). Cells were lysed using a MM301 mixer mill grinder MM301 (Retsch). Lysate was cleared by centrifugation 20,000Xg for 30 min. Approximately 9 mls of calmodulin resin was equilibrated in wash buffer A (200 mM NaCl, 50 mM HEPES pH 7.4, 5 mM CaCl2, 20% glycerol) and added to the cleared lysate. Binding was carried out at 4 C on a mutator for 2.5 h. Resin/lysate was loaded onto a gravity column and washed with 20 mls buffer A, 40 mls buffer B (500 mM NaCl, 50 mM HEPES pH 7.4, 5 mM CaCl2, 20% glycerol), and 20 mls buffer A. TRiC was eluted from the calmodulin resin with elution buffer (100 mM NaCl, 50 mM HEPES pH 7.4, 10 mM EDTA, 20% glycerol). Eluted protein was further purified by binding to two 5 ml HiTrap Heparin columns (GE Healthcare) equilibrated with MQA buffer. Heparin columns were washed with 20% MQB, and then eluted with a 60 ml gradient to 100% MQB. ATP to 1 mM was added to eluted TRiC, and TRiC was concentrated down to ~ 600 μl using an Amicon Ultra concentrator 10,000 MWCO. Concentrated protein was run over a Superose 6 gel filtration column equilibrated with MQA. Fractions containing TRiC were concentrated and snap frozen in liquid nitrogen.
hPFD:
1 L of High five insect cells (Invitrogen) at a concentration of ~3X10^6 cells/ml are infected with 8 mls of each of the viruses. and cells are harvested 2 to 3 days after infection by centrifugation. Cells are resuspended in 100 mls lysis buffer (100 mM NaCl, 100 mM HEPES pH 7.4, complete protease inhibitors EDTA free (Roche), PMSF, Benzonase 5 U/ml (Sigma- Aldrich), ~ 10 mM imidazole), and lysed using an emusiflex (Avestin). Lysate is clarified by ultracentrifugation (50,000*G, 1 hr). Lysate is cleared at 50,000 G for 30 min. Cleared lysate was run over a gravity column containing 10 mls of Nickel resin equilibrated with buffer A (50 mM HEPES pH 7.4, 100 mM NaCl). Resin was washed with buffer A, buffer A with an additional 400 mM NaCl, and buffer A again. PFD was eluted with buffer A containing 300 mM imidazole. Eluted protein was further purified on an MonoQ 10/100 anion exchange equilibrated with buffer A + 1 mM DTT. PFD was eluted from the MonoQ by a gradient to 100% buffer B (1 M NaCl, 50 mM HEPES pH 7.4, 1 mM DTT). Eluted PFD was concentrated down to ~ 1 ml using Amicon Ultra 30 KD MWCO and run over an SDX200 26/600 gel filtration column (GE Healthcare) equilibrated with buffer A + 1 mM DTT. Fractions containing PFD were concentrated using an Amicon Ultra 30 KD MWCO and 50% glycerol was added to a final concentration of 10%.
Alexa647-hPFD:
Alexa647 maleimide (Life Technologies) was used to label purified hPFD. 117 μM hPFD was mixed with freshly dissolved Alexa647 dye at 1.17 mM (10 molar excess), dye was reacted for 1 hr at room temperature, and quenched by addition of DTT. Labeled complex was purified by size exclusion chromotagraphy using an SDX200 10/300 (GE life sciences). Final concentrations were determined using extinction coeffiicient, labeling efficiency was 0.59 dyes/complex.
yPFD:
A polycistronic vector for co-expression of PFD subunits was transformed into BL21 Rosetta2 pLysS cells and grown to ~ 0.3 OD and induced O/N at 23 °C. Cells were pelleted, washed with PBS containing PMSF, and resuspended in lysis buffer (100 mM NaCl, 100 mM HEPES pH 7.4, complete protease inhibitors EDTA free (Roche), PMSF, Benzonase 5 U/ml (Sigma-Aldrich), ~ 10 mM imidazole). Cells were lysed using an emulsiflex and lysate cleared at 20,000 G for 30 min. ScPFD was purified from lysate as described for HsPFD.
Apical domains:
Apical domains were purified as described in (Joachimiak et al., 2014). Briefly, each Apical domain construct was transformed into BL21 Rosetta2 pLysS cells grown to ~ 0.3 OD and induced O/N at 23 °C. Cells were pelleted, washed with PBS containing PMSF, and resuspended in lysis buffer. Cells were lysed using an emusilflex, and lysate cleared at 20,000XG for 30 min. Cleared lysate was passed over Talon affinity resin (Clonetech), washed with column buffer + Triton X 100, column buffer + 500 mM NaCl, and column buffer + 1 mM ATP. Protein was eluted with column buffer + 300 mM imidazole and concentrated down to ~ 2 mls. Concentrated protein was run over an SDX200 26/60 size exclusion chromatography column equilibrated with column buffer + 1 mM DTT, the peak fraction was collected, concentrated and 50% glycerol was added to 10%. Protein was than aliquoted and snap frozen for future use.
Crosslinking-Mass Spectrometry
For hPFD with hTRiC samples, TRiC was brought to 1 μM, and Pfd to 2 μM, human PFD alone was prepared at 10 μM. For yPFD with yTRiC samples, TRiC was brought to 1 μM, and Pfd to 10 μM, and yPFD alone was prepared at 10 μM. All samples were prepared in 50 mM HEPES pH 7.4, 50 mM NaCl and 1 mM DTT. Fresh DSS crosslinker dissolved in DMSO was added to a final concentration of 1 mM, and samples were crosslinked for 2 h at room temperature for the human complexes and 37 °C for 30 min for the yeast complexes. Crosslinking was quenched by incubation for 30 minutes after addition of 1 M Tris pH 7.4 to a final concentration of 100 mM for human complexes, and addition of 1 M Ammonium Bicarbonate to 50 mM for yeast complexes.
Crosslinked samples were processed as described previously (Leitner et al., 2014). In brief, processing steps included reduction and alkylation of cysteine residues with tris(2- carboxyethyl)phosphine and iodoacetamide, respectively, sequential digestion with endoprotease Lys-C (Wako) and trypsin (Promega), clean-up using solid-phase extraction (Waters Sep-Pak tC18 cartridges) and fractionation of the purified digests by size exclusion chromatography (SEC; GE Superdex Peptide PC 3.2/300). SEC fractions were analyzed by liquid chromatography-tandem mass spectrometry on a Thermo Easy-nLC 1000 system coupled to a Thermo Orbitrap Elite system as described in detail elsewhere (Greber et al., 2014). MS data analysis was performed using xQuest/xProphet (Walzthoeni et al., 2012) using databases consisting of the TRiC and PFD sequences (including tags) of the respective complexes and contaminant proteins, if applicable. Decoy databases were generated as described in (Walzthoeni et al., 2012), and the results were filtered to a false discovery rate of 5%.
PFD–TRiC affinity
Human PFD (8 μM) and Human TRiC (0.5 μM) in ATPase buffer (30 mM Tris pH 7.4, 120 mM KCl, 5 mM MgCl2 and 1 mM dithiothreitol added fresh) were serial diluted and mixed 1:1 with TRiC yielding a final concentration of 0.25 μM TRiC and 0.063 to 4 μM PFD. Binding was allowed to equilibrate for approximately 30 min at room temperature and samples were run out on clear Native PAGE (either 4–12% or 4–16%). Protein was transferred to nitrocellulose and probed with Rabbit serum SU-230 (1:2000) developed against recombinant human PFD (Cocalico Biologicals). For the lower concentrations of PFD, the signal for free and TRiC-bound PFD was summed and the signal/uM calculated for each blot. This value was then used to calculate the amount of PFD bound to TRiC at each concentration. The amount of free PFD was calculated by subtracting the amount bound from the starting concentration. Binding was done in triplicate, and error bars represent SEM.
PFD–TRiC anisotropy
TRiC (400 nM) was prepared in 1XATPase buffer leaving room (30% volume) for nucleotide addition (closed)/H2O (open) 1/10 volume 10 mM ATP, 10 mM AlNO3, and 60 mM NaF were each added in quick succession to close TRiC for 30 min at 37 °C. Human Alexa647 labeled PFD was prepared at 50 nM in 1XATPase buffer, and mixed 1:1 with open TRiC, closed TRiC, and 1XATPase for final concentrations of 200 nM TRiC and 25 nM PFD, and allowed to equilibrate at room temperature. Anisotropy was measured using a CLARIOstar plate reader from BMG Tech with the PFD alone sample normalized to 100 prior to readings.
Actin folding assays
Endpoint assays:
TRiC variants (bovine TRiC, WT hTRiC, C1-GFP hTRiC, and C6-GFP hTRiC) were diluted to 0.25 μM in ATPase buffer. [35S]actin, prepared as described,in (Meyer et al., 2003; Thulasiraman et al., 2000) in 6 M Guanidnium-HCl, 100 mM HEPES pH 7.4 was rapidly diluted 1:100 with TRiC variants or with ATPase buffer alone. Actin was allowed to bind for 30 min on ice. To remove aggregated Actin, samples were spun 10 min at 19000xg. Supernatants were moved and layered onto either water or ATP to a final concentration of 1 mM. Samples were incubated 1 h at 37 °C. DNAseI was added to each sample (5 μg) to bind and compact folded Actin bands for native gel analyses. Samples were incubated 10 min on ice to allow binding, then large aggregates removed by centrifugation for 10 min at 19,000XG. Samples were run out on clear native gels 4–16%, gels were coomassie stained, dried and exposed to a phosphorscreen. Phosphorscreens were imaged using a Typhoon scanner.
Kinetic analyses:
0.13 μM [35S]actin in 6 M Guanidnium-HCl, 100 mM HEPES pH 7.4 was rapidly diluted 1:100 with TRiC at a concentration of 0.25 μM with or without PFD at 2.5 μM in ATPase buffer with fresh 1 mM DTT. Samples were incubated 30 min on ice to allow [35S]actin binding. To remove aggregated actin, samples were spun 10 min at 19000 G. Supernatents were moved into fresh tubes, and the TRiC alone sample split into (2) aliquots. When indicated, the TRiC-[35S]actin was further purified by ion-exchange chromatography on a MonoQ column as described (Frydman and Hartl, 1996; Reissmann et al., 2007). PFD was added to one aliquot of TRiC alone to a final concentration of 2.5 μM (equal volumes of ATPase buffer was added to the other samples). ATP was added to 1 mM and the folding reactions initiated by moving samples into a 37 °C water bath. Samples were taken at 0,2,4,8,16,32,48,64 and 128 min and the reactions stopped by addition of 0.2 Units of apyrase and moving samples to ice (the apyrase should deplete all ATP is less than 1 minute, and TRiC ATPase activity is negligible at 4 °C). DNAseI was added to each time point (5 μg) to bind folded Actin and compact bands in native gels. Samples were incubated 10 min on ice to allow binding, then large aggregates removed by centrifugation for 10 min at 19000xg. Timepoints were run out on clear native gels 4–16%, gels were coomassie stained, dried and exposed to a phosphorscreen. Phosphorscreens were imaged using a Typhoon scanner. The amount of Actin folding was quantified using image quant, and each experiment was normalized to the final three timepoints (48, 64, and 128 min) of TRiC alone folding which was set as 1 arbritrary unit (AU). Experiments were done in triplicate and error bars represent SEM.
Apical domain assays
PFD binding:
Apical domains and yPFD/hPFD were each diluted to 2 μM in ATPase buffer. Samples were mixed 1:1 and allowed to bind at room temperature for ~10 min. Samples were run out on a 15% SDS PAGE for a loading control, and on a clear Native PAGE 4–16%. for mobility shift analysis. Protein from native page gels was transferred to nitrocellulose membranes and antibodies raised against recombinant yeast PFD/human PFD at 1:2000 were used to detect proteins
Competitive inhibition of PFD binding TRiC:
hPFD/yPFD at 2 μM was mixed 1:1 with Apical domain 4 serially diluted from 40 μM to 1.25 μM and allowed to equilibrate ~ 10 min at room temperature. These samples were then mixed 1:1 with 0.5 μM TRiC in ATPase buffer and allowed to equilibrate for 10 min at room temperature. Samples were run out on a 15% SDS PAGE for a loading control, and on a clear Native PAGE 4–16%. for mobility shift analysis. Protein from native page gels was transferred to nitrocellulose membranes and antibodies raised against recombinant yeast PFD/human PFD at 1:2000 were used to detect PFDs.
ATPase measurements
ATPase rates were determined as described in. (Reissmann et al., 2007). Briefly, ATP hydrolysis by 0.25 μM hTRiC or 0.25 μM hTRiC + 2.5 μM hPFD was measured at 37 °C in ATPase buffer, in the presence of 188 to 1500 M [α−32P]ATP. After 5 min of preincubation, the reaction was started by mixing 6 l [α−32P]ATP solution with 34 l 1.176-fold concentrated reaction mix. At the indicated time points, 2- l samples were taken and transferred onto PEI-cellulose F thin-layer chromatography plastic sheets (EMD Chemicals). The plates were developed in a solvent system containing 1 M LiCl and 0.5 M formic acid in H2O, air-dried and exposed to a phosphorimager (Kodak). After the screen was scanned in a Typhoon 9410 imager, the amount of [α−32P]ATP was quantified using ImageQuant 5.2, amount of [α−32P]/ATP was determined from a standard curve from a dilution set on the thin- layer chromatography sheet.
TRiC–PFD CryoEM specimen preparation
Purified 20 μM hPFD was diluted 10-fold into MQ buffer containing 1 μM purified hTRiC, resulting in a 2:1 molar ratio of hPFD to hTRiC. The complex mixture was incubated for 30 min in room temperature and Octyl glucoside (OG) was added to a concentration of 0.1 % before grid placement to increase the yield of side views in electron micrographs. The resulting samples were applied to 200-mesh R1.2/1.3 holey-carbon grids (Quantifoil) and vitrified using a Gatan CP3 (Gatan). We loaded grids into a JEM3200FSC (300 KeV) electron microscope with an in- column omega filter (25 eV energy slit). We recorded images at 1.5–3 m underfocus on a K2 Summit direct electron detector (Gatan) in super-resolution mode at nominal 20K magnification, corresponding to a sampling of 1.7 Å/pixel (super-resolution sampling: 0.85 Å/pixel).
Image processing
We initially binned each movie stack by two and then corrected for drift and radiation damage using DE_process_frames.py (Direct Election Ltd). The first three frames were ignored during movie processing. We used EMAN2 to automatically select particle images (Bell et al., 2016). Contrast transfer-function (CTF) parameters were estimated internally based on the boxed particles (e2ctf.py). We then performed two-dimensional reference-free averaging with ~2,000 particle images using the default parameters in EMAN2 and generated an initial three- dimensional model based on the two-dimensional class averages, with no symmetry imposed. We converted a set of CTF-corrected particles to a RELION-compatible format using E2refinetorelion3d.py. We performed all further refinements using RELION 1.4 (Scheres, 2012), starting with maps that were low-pass filtered to 50 Å from the initial model generated by EMAN2. At first, we perform three-dimensional classification (K=3) with C1 symmetry to exclude non-TRiC-like particles. Then, we subsequently refined the remaining particles using the “auto_refine” command, which resulted in a 6.3 Å map based on a gold-standard Fourier shell correlation (FSC) at 0.143. In this step we were identified noisy density around TRiC’s apical domains, which stems from conformational heterogeneity of PFD.
Focused classification and localized reconstruction of individual subunits
The overall workflow using RELION 1.4 for the focused classification (Bai et al., 2015) and localized reconstruction (Ilca et al., 2015; Roh et al., 2017) of TRiC–PFD is described in Figure S4. We split the 6.5 Å map into two TRiC ring maps by masking out one ring at a time (Figure S4A,a–b). We then applied a soft mask around each map of TRiC ring and used the resulting masked map for subtraction of the signal from each raw particle image. This process generated two sub-particles from each raw particle image, which corresponds to either cis or trans ring of TRiC (Figure S4A,c). Using these sub-particles, we performed three-dimensional classification (K=5) with C1 symmetry and excluded 15% of particles with sub-optimally resolved features (Figure S4A,d). Then, we subjected the remaining 85% sub-particles for auto-refinement with a mask for single TRiC ring to determine the best orientation for each single TRiC ring (Figure S4A,e). This resulted in a 6.4 Å map containing only 8 subunits of TRiC. We next performed a focused 3D classification (K=8) on the putative PFD binding area with a spherical mask without orientation search (Figure S4A,f). This operation resulted in three groups of unbound TRiC and five groups with extra density around apical domains of TRiC (Figure S4A,g). Three unbound groups were combined as an unbound class. Therefore, one unbound class (Class1) and each of five bound classes (Class2–6) was respectively reconstructed with corresponding original two ring particles, while keeping the particle orientation from the single ring refinement (Figure S4A,h). In final, each map was sharpened using auto B-factor in RELION post processing step.
Model building
We first fit the crystal structure of yeast TRiC (4V94) (Leitner et al., 2012) using Chimera (Pettersen et al., 2004) in closed conformation to the CryoEM map of the open conformation; the model matches the map only in the equatorial domain. Then, molecular dynamics flexible fitting (MDFF) (Trabuco et al., 2008) was used on the fitted model and map. This method applies forces at atom position in the direction of the density gradient (density gradient weight of 0.3), while performing a molecular dynamics (MD) simulation at a given temperature (300K). After ~105 MD steps, the model matched the map much better, especially in the apical domains. This procedure thus opened the closed conformation of yeast TRiC to the open conformation of TRiC seen in the 6.3Å cryoEM map. We then threaded the sequence of each subunit in the Human TRiC through the open state of the yeast TRiC model based on sequence similarity. We performed a final round of model optimization with Phenix.real_space_refine for global minimization (Afonine et al., 2012). Since at this resolution the density does not constrain the structure tightly, we used probabilistic modeling to estimate the uncertainty at atomic positions using ProMod (Pintilie et al., 2016). Uncertainty values are stored in the Bfactor column of the PDB/model.
For Human PFD model, two copies of Archeal PFD crystal structures (2ZDI) (Sahlan et al., 2010) fitted into the extra density in Class 6, considering the right arrangement from XL/MS results (Figure 5). The PFD structure fitted well around the helical tips of TRiC, and contacts are clearly seen. We then threaded the sequence of each subunit of the Human PFD through the Archeal PFD model based on sequence similarity. We rigid-body fitted this human PFD model into each extra density in Class 2–6 and then combined with TRiC model, which generated previously. The tips of CCT were manually adjusted to the density to avoid overlapping with PFD. Each combined model was flexible fitted again to respective map by MDFF (Trabuco et al., 2008). Each resulting model was optimized into each map using Phenix.real_space_refine for global minimization (Afonine et al., 2012), and again probabilistic modeling was used to measure uncertainties at atomic positions (Afonine et al., 2012).
yTRiC pulldowns with CNBr-yPFD
One liter of WT or EEE yeast strains were grown to ~ 0.5 OD600 at 37 °C (permissive temperature for EEE mutant) and harvested by pelleting at 4000XG for 10 min. Pellets were resuspended in PBS with PMSF, pelleted for 5 min at 4000xg, and then resuspended in equal volume lysis buffer (100 mM HEPES-KOH pH 7.4, 50 mM KCl, 5 mM MgCl2, 2 mM DTT, 20% glycerol, 0.25% NP40 and complete protease inhibitors Roche). Pellet was then frozen by slowly dripping into liquid nitrogen for lysis. Cells were lysed using a cryogrinder for 1 minute at 20 Hertz. Lysate was cleared by centrifugation at 19000*G for 10 min at 4 °C, and supernatent was isolated. Protein concentration for each cleared lysate was determined using a bradford assay, and all cultures were diluted to 2 mg/ml using lysis buffer. 1 ml of cleared lysate was mixed with 200 μl of CNBr-yPFD resin and incubated for 2 hr at 4 °C. Lysate/CNBr-yPFD mixtures were split into three tubes of 300 μl. Resin was pelleted by centrifugation 22*G for 1 minute and unbound protein/supernatent was removed. Resin was washed 3 times by pelleting and resuspension with 500 μl of column buffer (100 mM HEPES-KOH pH 7.4, 50 mM KCl, 5 mM MgCl2, 2 mM DTT, 20% glycerol, 0.25% NP40) supplemented with an additional 50 mM, 150 mM or 350 mM KCl for final concentrations of 100, 200, and 400 mM KCl respectively. Resin was moved to a fresh tube after final wash, and bound protein was eluted with 1X Lamealli buffer.
TRiC closure assay
Yeast lysates were prepared as in the yTRiC pulldowns. Cleared lysates were diluted to 2 mg/ml to a final concentration of 30 mM HEPES-KOH pH 7.4, 100 mM KCl, 5 mM MgCl2, 20% glycerol and 1 mM DTT. Samples were prepared leaving 30% volume for addition of ATP, AlNO3, and NaF. 35 μl of each sample was brought to 30 °C, and either 15 μl of H2O (Open) or 5 μl of 10 mM ATP, 5 μl of 10 mM AlNO3, and 5 μl of 60 mM NaF in quick succession (closed) was added. Samples were incubated for 15 min to allow TRiC to fully close. TRiC was separated using 4% Native-PAGE. Protein from native page gels was transferred to nitrocellulose membranes and an antibody raised against yTRiC apical domains (Bio277) was used to detect TRiC.
Drop tests
Yeast strains were grown to log phase (~ 0.6 OD600) at 37 °C. Cells were diluted back to an OD600 of 0.08, and then serially diluted 1:10 three times. Cells were plated onto YPD and incubated at 37 °C and 30 °C for 48 h, and 23 °C for 72 h and imaged.
Proteostat/aggregate staining
Yeast strains were grown to approximately 0.2 OD600 at 37 C and then shifted to either 37 °C or 30 °C. Cells were allowed at least (2) doubling times (3 h) at the respective temperatures. Cells were fixed by addition of paraformaldehyde to a final concentration of 4% (1.5 mls culture, 0.5 mls 16% paraformaldehyde) for 15 min. Cells were pelleted and washed 2 times with PBS to remove paraformaldehyde. Cells were resuspended in proteostat buffer (PBS with 5% BSA and 0.1% Tween20) for 30 min. Proteostat was prepared by 1:2000 dilution in the buffer, and each pellet was resuspended with 250 μl and incubated for 30 min at 4 °C. Cells were washed 3X using proteostat buffer, and 2X with PBS. Cells were adhered to concanavalin A coverslips using Prolong antifade with DAPI and imaged with a Ziess LSM700 with the 100X oil immersion lens.
Aggregate purifications
Yeast strains were grown overnight at 30 °C and diluted in the morning into 100 mls of YPD grown to ~ 0.6 OD after at least two doublings and harvested by pelleting cultures at 3000 G. Pellets were resuspended in 1 ml PBS with PMSF, pelleted and snap frozen in liquid N2 and stored at – 80 C until further processing. Cell lysates were prepared by resuspending each pellet in 1 ml of lysis buffer (50 mM Na-phosphate, pH 6.8, 50 mM Potassium Acetate, 10 mM DTT, 1 mM EDTA, 1 mM PMSF, complete protease inhibitor cocktail (Roche), 3 mg/ml zymolyase and 1.25 U/ml benzonase) and incubation at room temperature for 20 min. Chilled samples were treated with tip sonication (3 s on, 7 s off, 25% power) for 1 min using a Qsonica Q500 sonicator. Lysates were cleared at 200*G for 20 min, and concentrations determined by bradford assay. Each lysate was adjusted to 1 mg/ml with lysis buffer. Aggregates were pelleted at 16000*G for 20 min, resuspended in 1 ml of wash buffer (50 mM Na-phosphate, pH 6.8, 50 mM Potassium Acetate, 10 mM DTT, 1 mM EDTA, 1 mM PMSF, 2% NP40) and tip sonicated (3 s on, 7 s off, 25% power), and pelleted again at 16000*G for 20 min. Wash step was repeated, and pellets were resuspended in SDS sample buffer and analyzed by SDS-PAGE followed by coomassie staining/western blot analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification for bands in Figure 1J, S1F, 2HI, S2E, 3C–F, and S3B,C were all performed using imagequant software. Data in figures for 2HI, S2E, 3C–F, and S3B,C represent the mean ± standard error of the mean (SEM) of three independent experiments (n = 3). For Figure S7D, the enriched proteins from each sample were subjected to pathway analysis to search for enriched GO categories using the DAVID database. Shown are categories using p < 0.05 using Fisher’s exact test followed by Benjamini-Hochberg multiple testing correction.
Supplementary Material
Figure S1. A recombinant system for human TRiC, Related to Figure 1 (A) Location of GFP insertion for CCT1 and CCT6 (B) Cell Growth test for WT vs GFP- insertions in indicated subunits at indicated temperatures. (C) Representative micrograph and Gold Standard FSC at 0.143 for each TRiC-CCT1-GFP and TRiC- CCT6-GFP. Local resolution maps (ResMap) (D) and overall architecture (E) of TRiC-CCT1-GFP and TRiC-CCT6-GFP showing psuedo-two-fold symmetry and the location of a protruding subunit (CCT1 indicated by red arrows). (F) Autoradiogram from native PAGE of denatured [35S]actin folding assay and quantification plot of folded actin vs time, comparing actin folding kinetics of bovine TRiC and hTRiC.
Figure S6. TRiC–PFD interaction site mediated by an electrostatic pivot point on CCT4, Related to Figure 6. (A) Purified yApical domains stabilized in a split-GFP construct were mixed with yPFD and analyzed by Native-PAGE followed by immunoblot for yPFD. Only the apical domain of yCCT4 caused a dramatic shift in PFD mobility. (B) The apical domain of yCCT4 was mixed with yPFD at varying concentrations to look for competitive inhibition of binding to TRiC and analyzed by Native-PAGE followed by immunoblot, binding is inhibited at near equimolar concentrations to PFD, and binding inhibition correlates with a shift in PFD mobility. (C) Electrostatic surface display (unwrapped TRiC and PFD) of TRiC and PFD. Circled numbers indicate the location of respective PFD subunit on TRiC. (D) Cartoon showing that PFD is recruited to TRiC through the electrostatic interaction to align the substrate binding chambers of these two complexes.
Figure S7. Loss of TRiC–PFD electrostatic interaction results in proteostasis defects, Related to Figure 7 (A) Outline of aggregation purification protocol. (B) Total (T), soluble (S) and aggregate pellet (P) fractions from indicated yeast strains, separated by SDS-PAGE and stained with coomassie, red dots added to emphasize multiple species present in the TRiC CCT4-EEE strain. (C) Immunoblot of above fractions for TRiC and alpha-Tubulin, note loss of alpha-Tubulin in CCT4- EEE strain. (D) Plot of the functional enrichment of proteins found in CCT4-EEE aggregates separated by GO categories, enriched classes are consistent with general proteostasis disruption.
Movie S1. Pivoting trajectory of PFD on TRiC (Density maps) Related to Figure 4. Conformational trajectory between the Classes (2→3→4→5→6), showing a pivoting mechanism of PFD on TRiC.
Movie S2. Pivoting trajectory of PFD on TRiC (Model ribbon diagram) Related to Figure 4. Conformational trajectory between the Classes (2→3→4→5→6), showing a pivoting mechanism of PFD on TRiC.
Table S1. CryoEM Data Collection, 3-D Reconstruction, and Model Refinement, Related to Figures 1, 4, 5, S1 and S4. Summary table includes parameters for CryoEM map reconstruction and model building.
Table S2: Crosslinking/Mass spectrometry of TRiC, PFD, and TRiC–PFD samples, Related to Figures 1, 2, 5, S2, and S5. Table includes identified intra and inter crosslinks for hTRiC, hTRiC CCT1-GFP, hTRiC with hPFD, hTRiC CCT1-GFP with hPFD, hPFD alone, yPFD alone, and yPFD with yTRiC. See table legend for description of column headers.
Table S3: Mass spectrometry analysis of aggregates from wild type and CCT4 EEE yeast strains, Related to Figures 7, S7. Aggregates were run out on a short SDS-PAG, and extracted for analysis. Table includes Log2 MS values for WT and EEE aggregates, and Fisher’s test for functional annotation enrichment of proteins detected in EEE sample, relative to entire proteome.
Table S4: Oligonucleotides used in this study Related to STARS methods.
Figure S2. PFD XL maps and TRiC–PFD binding, Related to Figure 2 Top and side views of yPFD (A), and hPFD (B) with XLs obtained from DSS crosslinking and MS mapped onto homology models refined using density from hTRiC-hPFD structure. (C) Histograms of Cα-Cα distance of XLs mapped onto the homology model of yPFD for the two subunit arrangements that satisfied the most constraints. (D) SDS-PAGE of hPFD variants with PFD1 or PFD4 tagged with GFP, migration shift allows identification of the respective subunit. (E) Full data points of binding data from hTRiC-hPFD binding experiments show a second TRiC–PFD binding at high concentrations. (F) Raw fluorescence polarization table of Alexa647- hPFD in the absence or presence of TRiC in the open (no nucelotide), and closed (ATP–AlF4) conformation, polarization in the absence of TRiC is normalized to 100.
Figure S3. Actin transfer between hPFD and hTRiC ATPase rates, Related to Figure 3 (A) [35S]actin transfer from PFD to TRiC, and from TRiC to PFD in the absence of ATP, and folding after ATP addition allows identification of all Actin bands from autoradiography experiments. (B) Plots of ATP hyrdolyzed/TRiC vs time for TRiC alone, and TRiC+PFD at 0.75 mM [ATP]. (C) ATPase rates for TRiC and TRiC+PFD at [ATP] from 0.188 to 1.5 mM. Measurements done in triplicate, error bars represent SEM, and ATPase rates are determined from linear fits to the data.
Figure S4. CryoEM map reconstructions of TRiC–PFD complex, Related to Figure 4 (A) data analysis workflow using local reconstruction and focused classification strategy. (B) Reference-free 2D class averages after 3D classification. Yellow circled area indicates the locations of PFD binding in each class. (C) Euler angle distribution of the particles that were included in the 3D reconstruction indicating overall isotropic coverage of orientations. Inside of sphere, blue map (TRiC/CCT) is shown at default contour (0.1). Green (GFP) and red (PFD) densities are shown as a difference map between the original map and lowpass filtered map (20Å). (D) Gold standard FSC for TRiC–PFD maps of all particle, half-ring and classes. (E) Local resolution map for each class shows structural heterogeneity around apical tips and PFD bound.
Figure S5. TRiC and PFD interact in a subunit specific manner. Related to Figure 5 (A) Schematics of the two possible mirror image arrangements of yPfd on yTRiC. Obtained XLs were mapped onto TRiC–PFD models from CryoEM, histograms of Cα-Cα distances establish the correct arrangement of the PFD subunits. (B) TRiC contact points for PFD identified by CryoEM reconstructions of the different classes highlighted in yellow. (C) Surface display and (D) histogram to show Cα RMSD analysis among six models from the class1–6. (E) Overall and zoom-in view to the contact of CCT4 to PFD (4/6) in Class 6 (F) Top and side view of TRiC- PFD complex as a space filling model (Class 6) showing ~40Å fenestration between anchoring subunit (CCT4) and protruding subunit (CCT1).
Highlights.
Generation and characterization of active recombinant hTRiC and hPFD
Cryo–EM, XL–MS and modelling reveal the structure of TRiC/CCT–PFD complex
PFD pivots around a conserved electrostatic interface with TRiC/CCT
PFD acts on TRiC/CCT-substrate complex to enhance the rate of the folding reaction
Direct interactions between two chaperonins allow them to feed folding substrates bi-directionally between active sites, preventing aggregation and promoting proteostasis.
ACKNOWLEDGMENTS
We thank support of NIH (R01GM074074 to JF; F32GM103124 to DG; P41GM103832 and R01GM079429 to WC, P01NS092525 to WC and JF); the ERC (AdvG #233226 and AdvG #670821 to RA) and Seoul National University Settlement and NRF 2019R1C1C004598 (to S.-H.R). CryoEM data was collected at NCMI and processed at the CIBR at Baylor College of Medicine. We thank P. Aliaga and K. Goncalves for help in yeast strain production; K. Rainbolt and R. Samant for advice on Proteostat staining; E. Sontag for help with fluorescence microscopy; R. Aviner; K. Li and Al Burlingame for help with mass spectrometry; R. Andino and K. Dalton for discussions and comments on the manuscript and the Frydman lab for useful advice and discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND SOFTWARE AVAILABILITY
Model coordinates and densities maps from the cryoEM study have been deposited through PDB OneDep System; Class1 (PDB: 6MRA, EMD-0492), Class2 (PDB: 6NRB, EMD-0493), Class3 (PDB: 6NRC, EMD-0494), Class4 (PDB: 6NRD, EMD-0495), Class5 (PDB: 6NR9, EMD- 0491), Class6 (PDB: 6NR8, EMD-0490) and the cryoEM map with all particles (EMD-0496),
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abe A, Takahashi-Niki K, Takekoshi Y, Shimizu T, Kitaura H, Maita H, Iguchi-Ariga SM, and Ariga H (2013). Prefoldin plays a role as a clearance factor in preventing proteasome inhibitor-induced protein aggregation. J Biol Chem 288, 27764–27776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SH (2015). Sampling the conformational space of the catalytic subunit of human gamma-secretase. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Balchin D, Milicic G, Strauss M, Hayer-Hartl M, and Hartl FU (2018). Pathway of Actin Folding Directed by the Eukaryotic Chaperonin TRiC. Cell 174, 1507–1521 e1516. [DOI] [PubMed] [Google Scholar]
- Bell JM, Chen M, Baldwin PR, and Ludtke SJ (2016). High resolution single particle refinement in EMAN2.1. Methods 100, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty K, Chatila M, Sinha J, Shi Q, Poschner BC, Sikor M, Jiang G, Lamb DC, Hartl FU, and Hayer-Hartl M (2010). Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell 142, 112–122. [DOI] [PubMed] [Google Scholar]
- Comyn SA, Young BP, Loewen CJ, and Mayor T (2016). Prefoldin Promotes Proteasomal Degradation of Cytosolic Proteins with Missense Mutations by Maintaining Substrate Solubility. PLoS Genet 12, e1006184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. (2016). A global genetic interaction network maps a wiring diagram of cellular function. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreyra RG, and Frydman J (2000). Purification of the cytosolic chaperonin TRiC from bovine testis. Methods Mol Biol 140, 153–160. [DOI] [PubMed] [Google Scholar]
- Frydman J, and Hartl FU (1996). Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science 272, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Frydman J, Nimmesgern E, Ohtsuka K, and Hartl FU (1994). Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370, 111–117. [DOI] [PubMed] [Google Scholar]
- Gao Y, Thomas JO, Chow RL, Lee GH, and Cowan NJ (1992). A cytoplasmic chaperonin that catalyzes beta-actin folding. Cell 69, 1043–1050. [DOI] [PubMed] [Google Scholar]
- Geissler S, Siegers K, and Schiebel E (1998). A novel protein complex promoting formation of functional alpha- and gamma-tubulin. EMBO J 17, 952–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, and Ban N (2014). Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature 505, 515–519. [DOI] [PubMed] [Google Scholar]
- Hansen WJ, Cowan NJ, and Welch WJ (1999). Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J Cell Biol 145, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilca SL, Kotecha A, Sun X, Poranen MM, Stuart DI, and Huiskonen JT (2015). Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat Commun 6, 8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachimiak LA, Walzthoeni T, Liu CW, Aebersold R, and Frydman J (2014). The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159, 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaganovich D, Kopito R, and Frydman J (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Leitner A, Joachimiak LA, Bracher A, Monkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. (2012). The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 20, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A, Walzthoeni T, and Aebersold R (2014). Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat Protoc 9, 120–137. [DOI] [PubMed] [Google Scholar]
- Lopez T, Dalton K, and Frydman J (2015). The Mechanism and Function of Group II Chaperonins. J Mol Biol 427, 2919–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez V, Gonzalez-Peramato P, Suela J, Serrano A, Algaba F, Cigudosa JC, Vidal A, Bellmunt J, Heredero O, and Sanchez-Carbayo M (2013). Identification of prefoldin amplification (1q23.3-q24.1) in bladder cancer using comparative genomic hybridization (CGH) arrays of urinary DNA. J Transl Med 11, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Benito J, Boskovic J, Gomez-Puertas P, Carrascosa JL, Simons CT, Lewis SA, Bartolini F, Cowan NJ, and Valpuesta JM (2002). Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT. EMBO J 21, 6377–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville MW, McClellan AJ, Meyer AS, Darveau A, and Frydman J (2003). The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol Cell Biol 23, 3141–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AS, Gillespie JR, Walther D, Millet IS, Doniach S, and Frydman J (2003). Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell 113, 369–381. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pintilie G, Chen DH, Haase-Pettingell CA, King JA, and Chiu W (2016). Resolution and Probabilistic Models of Components in CryoEM Maps of Mature P22 Bacteriophage. Biophys J 110, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissmann S, Joachimiak LA, Chen B, Meyer AS, Nguyen A, and Frydman J (2012). A gradient of ATP affinities generates an asymmetric power stroke driving the chaperonin TRIC/CCT folding cycle. Cell Rep 2, 866–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissmann S, Parnot C, Booth CR, Chiu W, and Frydman J (2007). Essential function of the built-in lid in the allosteric regulation of eukaryotic and archaeal chaperonins. Nat Struct Mol Biol 14, 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh SH, Hryc CF, Jeong HH, Fei X, Jakana J, Lorimer GH, and Chiu W (2017). Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc Natl Acad Sci U S A 114, 8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russmann F, Stemp MJ, Monkemeyer L, Etchells SA, Bracher A, and Hartl FU (2012). Folding of large multidomain proteins by partial encapsulation in the chaperonin TRiC/CCT. Proc Natl Acad Sci U S A 109, 21208–21215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan OW, Poddar S, and Cate JH (2016). CRISPR-Cas9 Genome Engineering in Saccharomyces cerevisiae Cells. Cold Spring Harb Protoc 2016. [DOI] [PubMed] [Google Scholar]
- Sahlan M, Zako T, Tai PT, Ohtaki A, Noguchi K, Maeda M, Miyatake H, Dohmae N, and Yohda M (2010). Thermodynamic characterization of the interaction between prefoldin and group II chaperonin. J Mol Biol 399, 628–636. [DOI] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, and Frydman J (2012). The Interplay Between the Chaperonin TRiC and N-terminal Region of Huntingtin Mediates Huntington’s Disease Aggregation and Pathogenesis In Protein Quality Control in Neurodegenerative Disease, Morimoto RI, and Christen Y, eds. (Springer Verlag; ). [Google Scholar]
- Siegers K, Waldmann T, Leroux MR, Grein K, Shevchenko A, Schiebel E, and Hartl FU (1999). Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J 18, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegert R, Leroux MR, Scheufler C, Hartl FU, and Moarefi I (2000). Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103, 621–632. [DOI] [PubMed] [Google Scholar]
- Sot B, Rubio-Munoz A, Leal-Quintero A, Martinez-Sabando J, Marcilla M, Roodveldt C, and Valpuesta JM (2017). The chaperonin CCT inhibits assembly of alpha-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci Rep 7, 40859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C, Miller EJ, McClellan AJ, and Frydman J (2006). Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol Cell 24, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Geller R, Spiess C, and Frydman J (2006). The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol 8, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S (2001). A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli. Protein Expr Purif 21, 224–234. [DOI] [PubMed] [Google Scholar]
- Tashiro E, Zako T, Muto H, Itoo Y, Sorgjerd K, Terada N, Abe A, Miyazawa M, Kitamura A, Kitaura H, et al. (2013). Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J Biol Chem 288, 19958–19972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman V, Ferreyra RG, and Frydman J (2000). Monitoring actin folding. Purification protocols for labeled proteins and binding to DNase I-sepharose beads. Methods Mol Biol 140, 161–167. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, and Schulten K (2008). Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, and Cowan NJ (1998). Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873. [DOI] [PubMed] [Google Scholar]
- Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Forster F, Beck M, and Aebersold R (2012). False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat Methods 9, 901–903. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhao J, Yang X, Guan S, Feng H, Han D, Lu J, Ou B, Jin R, Sun J, et al. (2015). PFDN1, an indicator for colorectal cancer prognosis, enhances tumor cell proliferation and motility through cytoskeletal reorganization. Med Oncol 32, 264. [DOI] [PubMed] [Google Scholar]
- Willmund F, del Alamo M, Pechmann S, Chen T, Albanese V, Dammer EB, Peng J, and Frydman J (2013). The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 152, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Her C (2013). VBP1 facilitates proteasome and autophagy-mediated degradation of MutS homologue hMSH4. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27, 4799–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin HT, Burlingame A, Gerstein M, and Frydman J (2008). Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 15, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zako T, Murase Y, Iizuka R, Yoshida T, Kanzaki T, Ide N, Maeda M, Funatsu T, and Yohda M (2006). Localization of prefoldin interaction sites in the hyperthermophilic group II chaperonin and correlations between binding rate and protein transfer rate. J Mol Biol 364, 110– 120. [DOI] [PubMed] [Google Scholar]
- Zang Y, Jin M, Wang H, Cui Z, Kong L, Liu C, and Cong Y (2016). Staggered ATP binding mechanism of eukaryotic chaperonin TRiC (CCT) revealed through high-resolution cryo- EM. Nat Struct Mol Biol 23, 1083–1091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A recombinant system for human TRiC, Related to Figure 1 (A) Location of GFP insertion for CCT1 and CCT6 (B) Cell Growth test for WT vs GFP- insertions in indicated subunits at indicated temperatures. (C) Representative micrograph and Gold Standard FSC at 0.143 for each TRiC-CCT1-GFP and TRiC- CCT6-GFP. Local resolution maps (ResMap) (D) and overall architecture (E) of TRiC-CCT1-GFP and TRiC-CCT6-GFP showing psuedo-two-fold symmetry and the location of a protruding subunit (CCT1 indicated by red arrows). (F) Autoradiogram from native PAGE of denatured [35S]actin folding assay and quantification plot of folded actin vs time, comparing actin folding kinetics of bovine TRiC and hTRiC.
Figure S6. TRiC–PFD interaction site mediated by an electrostatic pivot point on CCT4, Related to Figure 6. (A) Purified yApical domains stabilized in a split-GFP construct were mixed with yPFD and analyzed by Native-PAGE followed by immunoblot for yPFD. Only the apical domain of yCCT4 caused a dramatic shift in PFD mobility. (B) The apical domain of yCCT4 was mixed with yPFD at varying concentrations to look for competitive inhibition of binding to TRiC and analyzed by Native-PAGE followed by immunoblot, binding is inhibited at near equimolar concentrations to PFD, and binding inhibition correlates with a shift in PFD mobility. (C) Electrostatic surface display (unwrapped TRiC and PFD) of TRiC and PFD. Circled numbers indicate the location of respective PFD subunit on TRiC. (D) Cartoon showing that PFD is recruited to TRiC through the electrostatic interaction to align the substrate binding chambers of these two complexes.
Figure S7. Loss of TRiC–PFD electrostatic interaction results in proteostasis defects, Related to Figure 7 (A) Outline of aggregation purification protocol. (B) Total (T), soluble (S) and aggregate pellet (P) fractions from indicated yeast strains, separated by SDS-PAGE and stained with coomassie, red dots added to emphasize multiple species present in the TRiC CCT4-EEE strain. (C) Immunoblot of above fractions for TRiC and alpha-Tubulin, note loss of alpha-Tubulin in CCT4- EEE strain. (D) Plot of the functional enrichment of proteins found in CCT4-EEE aggregates separated by GO categories, enriched classes are consistent with general proteostasis disruption.
Movie S1. Pivoting trajectory of PFD on TRiC (Density maps) Related to Figure 4. Conformational trajectory between the Classes (2→3→4→5→6), showing a pivoting mechanism of PFD on TRiC.
Movie S2. Pivoting trajectory of PFD on TRiC (Model ribbon diagram) Related to Figure 4. Conformational trajectory between the Classes (2→3→4→5→6), showing a pivoting mechanism of PFD on TRiC.
Table S1. CryoEM Data Collection, 3-D Reconstruction, and Model Refinement, Related to Figures 1, 4, 5, S1 and S4. Summary table includes parameters for CryoEM map reconstruction and model building.
Table S2: Crosslinking/Mass spectrometry of TRiC, PFD, and TRiC–PFD samples, Related to Figures 1, 2, 5, S2, and S5. Table includes identified intra and inter crosslinks for hTRiC, hTRiC CCT1-GFP, hTRiC with hPFD, hTRiC CCT1-GFP with hPFD, hPFD alone, yPFD alone, and yPFD with yTRiC. See table legend for description of column headers.
Table S3: Mass spectrometry analysis of aggregates from wild type and CCT4 EEE yeast strains, Related to Figures 7, S7. Aggregates were run out on a short SDS-PAG, and extracted for analysis. Table includes Log2 MS values for WT and EEE aggregates, and Fisher’s test for functional annotation enrichment of proteins detected in EEE sample, relative to entire proteome.
Table S4: Oligonucleotides used in this study Related to STARS methods.
Figure S2. PFD XL maps and TRiC–PFD binding, Related to Figure 2 Top and side views of yPFD (A), and hPFD (B) with XLs obtained from DSS crosslinking and MS mapped onto homology models refined using density from hTRiC-hPFD structure. (C) Histograms of Cα-Cα distance of XLs mapped onto the homology model of yPFD for the two subunit arrangements that satisfied the most constraints. (D) SDS-PAGE of hPFD variants with PFD1 or PFD4 tagged with GFP, migration shift allows identification of the respective subunit. (E) Full data points of binding data from hTRiC-hPFD binding experiments show a second TRiC–PFD binding at high concentrations. (F) Raw fluorescence polarization table of Alexa647- hPFD in the absence or presence of TRiC in the open (no nucelotide), and closed (ATP–AlF4) conformation, polarization in the absence of TRiC is normalized to 100.
Figure S3. Actin transfer between hPFD and hTRiC ATPase rates, Related to Figure 3 (A) [35S]actin transfer from PFD to TRiC, and from TRiC to PFD in the absence of ATP, and folding after ATP addition allows identification of all Actin bands from autoradiography experiments. (B) Plots of ATP hyrdolyzed/TRiC vs time for TRiC alone, and TRiC+PFD at 0.75 mM [ATP]. (C) ATPase rates for TRiC and TRiC+PFD at [ATP] from 0.188 to 1.5 mM. Measurements done in triplicate, error bars represent SEM, and ATPase rates are determined from linear fits to the data.
Figure S4. CryoEM map reconstructions of TRiC–PFD complex, Related to Figure 4 (A) data analysis workflow using local reconstruction and focused classification strategy. (B) Reference-free 2D class averages after 3D classification. Yellow circled area indicates the locations of PFD binding in each class. (C) Euler angle distribution of the particles that were included in the 3D reconstruction indicating overall isotropic coverage of orientations. Inside of sphere, blue map (TRiC/CCT) is shown at default contour (0.1). Green (GFP) and red (PFD) densities are shown as a difference map between the original map and lowpass filtered map (20Å). (D) Gold standard FSC for TRiC–PFD maps of all particle, half-ring and classes. (E) Local resolution map for each class shows structural heterogeneity around apical tips and PFD bound.
Figure S5. TRiC and PFD interact in a subunit specific manner. Related to Figure 5 (A) Schematics of the two possible mirror image arrangements of yPfd on yTRiC. Obtained XLs were mapped onto TRiC–PFD models from CryoEM, histograms of Cα-Cα distances establish the correct arrangement of the PFD subunits. (B) TRiC contact points for PFD identified by CryoEM reconstructions of the different classes highlighted in yellow. (C) Surface display and (D) histogram to show Cα RMSD analysis among six models from the class1–6. (E) Overall and zoom-in view to the contact of CCT4 to PFD (4/6) in Class 6 (F) Top and side view of TRiC- PFD complex as a space filling model (Class 6) showing ~40Å fenestration between anchoring subunit (CCT4) and protruding subunit (CCT1).