

Abstract
Lipoprotein(a) [Lp(a)], discovered in 1963, has been associated with atherosclerotic cardiovascular disease (ASCVD) independent of other traditional risk factors, including LDL cholesterol. Lp(a) is an apolipoprotein B (apoB)-containing lipoprotein, which contains an LDL-like particle. Unlike LDL, which is a primary therapeutic target to decrease ASCVD, current guidelines recommend measuring Lp(a) for risk assessments because there is no clear evidence demonstrating the clinical benefit of decreasing Lp(a) using classical drugs such as niacin. However, recent Mendelian randomization studies indicate that Lp(a) causally correlates with ASCVD. In addition, novel drugs, including PCSK9 inhibitors, as well as antisense oligonucleotide for apo(a), have exhibited efficacy in decreasing Lp(a) substantially, invigorating a discussion whether Lp(a) could be a novel therapeutic target for further ASCVD risk reduction. This review aims to provide current understanding, and future perspectives, of Lp(a), which is currently considered a mere biomarker but may emerge as a novel therapeutic target in future clinical settings.
Keywords: Lipoprotein(a), Aortic valve stenosis, Atherosclerotic cardiovascular disease, LDL
Introduction
Atherosclerotic cardiovascular disease (ASCVD), including coronary artery disease and stroke, is the leading cause of mortality worldwide. Despite advancements in ASCVD prevention through LDL-lowering therapies, using statins and various other agents, so-called “residual risk” remains a significant challenge1–3). Of several biomarkers shown as residual risks, lipoprotein(a) [Lp(a)], an apolipoprotein B (apoB)-containing lipoprotein containing a LDL-like particle, has been reported as a causal risk factor for ASCVD by Mendelian randomization studies, as well as genome-wide association studies (GWAS)4–6). Conversely, the incidence of aortic valve stenosis, based on calcific aortic valvulopathy, where no effective option exists for its progression, is growing among industrialized countries because of aging societies7, 8). Moreover, Lp(a) is considered a causal factor in calcific aortic valvulopathy development9, 10), making Lp(a) a potential therapeutic target to decrease calcific aortic valvulopathy progression.
This review aims to provide a current understanding, and future perspectives, of Lp(a), which is currently considered a mere biomarker but may emerge as a novel therapeutic target in future clinical settings.
What is Lp(a)?
Lp(a) is a particle containing two different elements (Fig. 1). The first element is an LDL-like particle containing an apoB-100 particle, which is insoluble in water. Reportedly, the LDL-like particle in Lp(a) is larger in size and higher in lipid content, with a density marginally lower than the LDL particle isolated from the same individual11). The second element is a hydrophilic glycoprotein called apo(a) that shares homology with plasminogen, giving the particle atherogenic properties. Plasminogen has five kringles (KI–KV); apo(a) does not contain KI–KIII but has 10 subtypes of KIV (with KIV1, and KIV3–KIV10 have one copy, and KIV2 has one to > 40 copies), and one copy of KV. The apo(a) isoform size and the Lp(a) concentration correlate inversely since smaller isoforms can be made in larger quantities compared to larger isoforms. Both components, LDL-like particle and apo(a), are linked by a single disulfide bond that was localized in one of apo(a) (kringle IV9)'s repeat structures12).
Fig. 1.
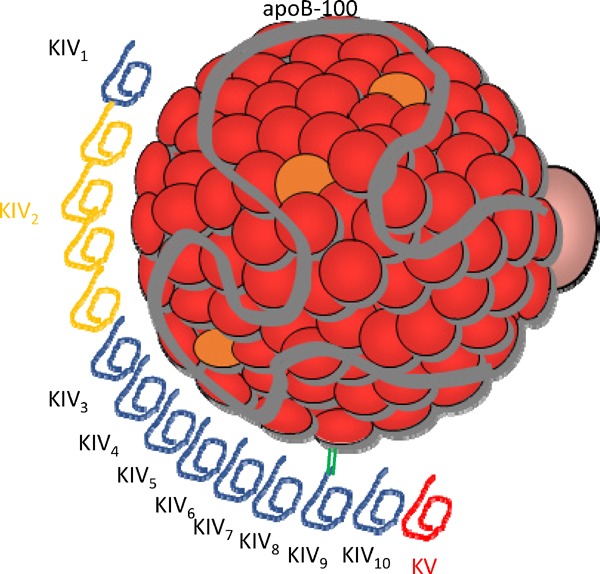
The structure of lipoprotein [Lp(a)]
Lp(a) is composed of an LDL-like particle, including apolipoprotein B-100 (apoB-100) and apolipoprotein(a) [apo(a)], which has kringle IV (KIV) and KV. Apo(a) contains 10 subtypes of KIV repeats, containing one copy each of KIV1, multiple copies of KIV2 (yellow colored), and one copy of KIV3-10, KV.
Evolution of the Apo(a) Gene
In primates, the apo(a) gene arose from a plasminogen gene ∼40 million years ago during evolution12). Remarkably, apo(a) is present in humans, nonhuman primates, and Old World monkeys, but not in prosimians, or lower mammals.
Lp(a) Concentration and its Related Factors
The distribution of Lp(a) concentration in most populations is highly positively skewed13). Moreover, there are high differences in Lp(a) concentrations across populations; Africans have higher Lp(a) than Caucasian or Asian populations (Fig. 2)13,14). Although the Lp(a) concentration is primarily evaluated by common, single-nucleotide polymorphisms in the LPA gene15) and is only minimally affected by environmental factors, several other conditions, including renal dysfunction16) and familial hypercholesterolemia (FH), have been reported to affect its level14, 17).
Fig. 2.
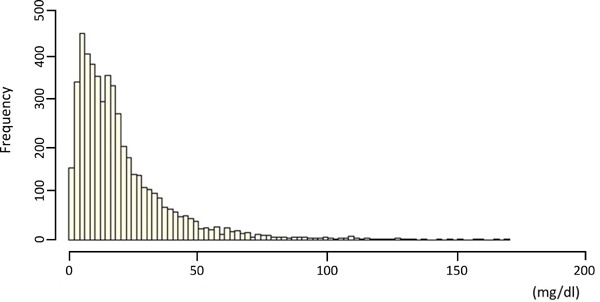
The distribution of the lipoprotein [Lp(a)] frequency
The X-axis represents the serum Lp(a) level, while the Y-axis represents the frequency.
Lp(a) Catabolism
The serum Lp(a) concentration is primarily measured by the rate of apo(a) synthesis, rather than the apo(a) degradation18, 19). In addition, the liver has been reported as a major site of apo(a) synthesis, evidenced by studies of patients undergoing therapeutic liver transplantation20). The Lp(a) assembly is a two-step process that begins with docking apo(a) to an LDL particle, followed by creating a disulfide bond between the kringle structure and apoB-100; this has been reported to occur at the hepatocyte cell surface, rather than at the endoplasmic reticulum or Golgi21). Conversely, an Lp(a) catabolism pathway remains unclear. The liver is now considered the main organ that clears Lp(a) from the circulation21), and some studies using mice models have also suggested the kidneys are contributors22, 23). Moreover, kinetic studies in humans have revealed that Lp(a) catabolism was slower than that of LDL, independent of the Lp(a) concentration24, 25). Those results suggested that synthesis, rather than catabolism, determines the Lp(a) concentration. Likewise, a plasmapheresis study reported similar results26).
Measurements of Lp(a)
Some assays of Lp(a) measurements are reportedly affected by the number of KIV2 repeats27). Nevertheless, several studies using methods sensitive to the apo(a) size reported significant correlations between Lp(a) and the ASCVD risk, consistent with those using methods independent of apo(a) size variations. Accordingly, we intend to highlight that the critical question of whose Lp(a) should be measured is more pertinent than how to measure Lp(a).
Pathological and Physiological Roles of Lp(a)
Lp(a) and/or apo(a) have been correlated with prothrombotic properties through interfering with reactions in fibrinolysis regulation, including plasminogen binding to fibrinogen, fibrin, and tetranectin, plasminogen activation by tissue plasminogen activator (t-PA), and augmentation of plasminogen activator inhibitor-1 (PAI-1) activity28–30). Besides those interactions with the fibrinolytic system, other functional properties have been explained as its pathophysiology, including the release of monocyte chemotactic activity from endothelial cells31), inhibition of the plasma catalyzed activation of transforming growth factor-β (TGF-β)32), enhanced proliferation and migration of smooth muscle cells33), proliferation of endothelial cells34), as well as mesangial cells35), and stimulation of the expression of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin36, 37).
Lp(a) as a Causal Factor for the ASCVD Development: A Standpoint from Genetic Studies
Since the initial results from a GWAS focusing on seven major diseases using 2000 cases and 2000 controls38), several GWAS have been conducted to determine novel loci related to various diseases. Of these, correlations between common variants in LPA loci and cardiovascular disease, including coronary artery disease and aortic valve stenosis, have been reported often39, 40). Notably, common variants in an LDL receptor markedly correlated with ASCVD, whereas these were not related to calcific aortic valvulopathy outcome40). Conversely, common variants in the LPA gene markedly correlate with ASCVD, as well as calcific aortic valvulopathy outcomes as well. Such correlations between genetic variants, resulting in an increase/decrease of a particular biomarker and an outcome, could be considered a proxy of a randomized controlled trial using a particular inhibitor; these are known as “Mendelian randomization studies”41). Of note, Mendelian randomization studies could be useful for validating, as well as estimating, the effects/side effects of particular drugs targeting molecule “X,” as demonstrated in multiple lipid-modifying drugs42–44). Accordingly, Lp(a) could be a causal factor for ASCVD and related diseases, including coronary heart disease, stroke, chronic kidney disease, calcific aortic valvulopathy, heart failure, and peripheral vascular disease45). Overall, meta-analyses of epidemiological and genetic studies have demonstrated that elevated Lp(a) levels correlated with an increased risk for ASCVD (Fig. 3).
Fig. 3.
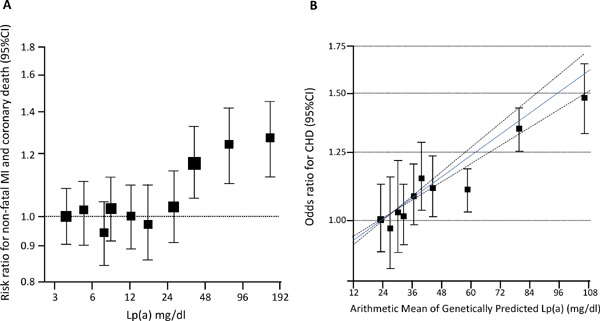
The evidence for lipoprotein [Lp(a)] as an independent and causal risk factor for atherosclerotic cardiovascular disease (ASCVD)
A, A meta-analysis of epidemiological studies, adjusted for usual levels of systolic blood pressure, smoking status, history of diabetes, body mass index, and total cholesterol. The X-axis represents the serum Lp(a) level, while the Y-axis represents the risk ratio for non-fatal MI and coronary death. B, the correlation between genetically predicted Lp(a) and CHD risk. The X-axis represents the serum Lp(a) level, while the Y-axis represents the odds ratio for the CHD risk. The blue solid line represents the best-fitting fractional polynomial to model the dose-dependent relationship; the dotted lines show the 95% confidence intervals for the relationship.
Lp(a) and Calcific Aortic Valvulopathy
Calcific aortic valvulopathy, characterized by calcium deposition and thickening of the aortic valve, correlates with aortic valve stenosis. In addition, epidemiological studies have reported several risk factors, including classical coronary risk factors, such as age, male, body mass index, hypertension, diabetes, smoking, renal dysfunction, and LDL cholesterol related to calcific aortic valvulopathy, indicating that treating or preventing those risk factors might decrease the risk of developing aortic valve stenosis7). Under these hypotheses, a randomized controlled trial (RCT) was conducted to determine whether further reduction of LDL cholesterol, using ezetimibe on the top of statins, could effectively slow the progression of aortic valve stenosis46). Nevertheless, no medical treatment, thus far, has been reported to affect disease progression in patients with calcific aortic valvulopathy. Accordingly, Lp(a) has emerged as a “causal” risk factor based on genetic associations, which could be potential therapeutic targets to prevent calcific aortic valvulopathy development. Furthermore, elevated Lp(a) levels enhance the calcific aortic valvulopathy progression and, thus, the need for aortic valve replacement (Fig. 4)47).
Fig. 4.
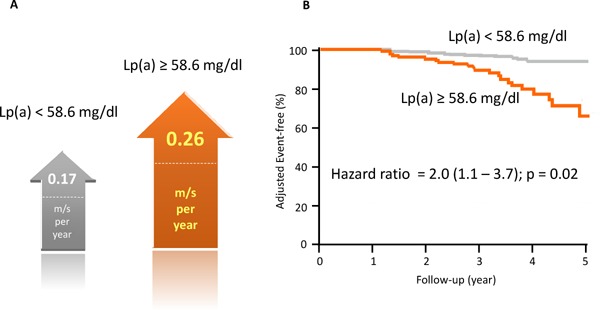
Lipoprotein [Lp(a)] and progression of aortic valve stenosis, as well as the incidence of aortic valve replacement
A, both arrows indicate the predicted progression of preexisting mild-to-moderate calcific aortic valve stenosis (CAVS) on echocardiography in meters/second/year in the ASTRONOMER trial. The gray arrow shows the progression of aortic valve stenosis among patients whose Lp(a) < 58.6 mg/dL; the orange arrow shows the progression of aortic valve stenosis among patients whose Lp(a) ≥ 58.6 mg/dL. B, the event-free survival curve divided into two groups based on the Lp(a) level. The gray line represents patients whose Lp(a) < 58.6 mg/dL; the orange arrow represents patients whose Lp(a) ≥ 58.6 mg/dL.
Lp(a) as One of the Residual Factors of Statin Therapies
Several biomarkers are considered so-called “residual risk of statin therapies;” of those, the evidence level obtained from sub-analyses, using RCTs, could be considered higher than those obtained from single-center observational studies. Thus, only a few biomarkers, including triglycerides48, 49) and Lp(a)50, 51), have been explicitly “established” as the residual risk of statin therapies through RCT investigations. Remarkably, both biomarkers have been projected to be causal factors for ASCVD development through Mendelian randomization studies. These facts motivate us to lower those biomarkers, especially among patients with ASCVD under statin therapies.
Lp(a)-Lowering Therapies
For a long time, no satisfactory therapeutic approach existed to lower Lp(a) levels. We, among other groups, have reported Lp(a) levels among FH patients were caused by LDL receptor mutations14, 17), resulting in the estimation that LDL-lowering therapies, such as statins, resins, and ezetimibe, could be useful for this purpose. However, studies using those drugs have recurrently reported almost no effect on decreasing Lp(a) levels52, 53). Conversely, recently approved proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, which elevate LDL receptor levels by inhibiting its degradation, have been reported to lower Lp(a) levels as much as 30%, with the extent of Lp(a) lowering correlating with the LDL reduction54–56). The mechanism underlying this effect was recently the subject of a comprehensive investigation. Another option could be mipomersen, an apoB inhibitor, which could correlate with the reduction of Lp(a) ∼30%57); however, it almost always causes fatty liver since it blocks the secretion of apoB-containing lipoproteins from the liver. Another emerging option could be antisense oligonucleotide (ASO) targeted to apolipoprotein(a). The first-generation drug called IONIS-APO(a)Rx has been reported as a tolerable, potent therapy for decreasing Lp(a) concentrations54). Recently, AKCEA-APO(a)-LRx (ISIS 681257), a second-generation, N-acetyl-galactosamine-conjugated, ASO targeted to apolipoprotein(a), was reported to lower the mean plasma Lp(a) levels by 92%58).
Conclusions and Perspectives
Lp(a), an old molecule, has long been considered a vital causal factor of ASCVD, including calcific aortic valvulopathy. Now, specific therapies reducing Lp(a) quite effectively are projected to become available soon in clinical practice. We would witness whether such emerging novel approaches could tamp down the residual risk, as well as the progression, of calcific aortic valvulopathy.
Acknowledgments and Notice of Grant Support
None.
Conflicts of Interest
None.
References
- 1). Reith C, Armitage J. Management of residual risk after statin therapy. Atherosclerosis, 2016; 245: 161-170 [DOI] [PubMed] [Google Scholar]
- 2). Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM, IMPROVE-IT Investigators Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med, 2015; 372: 2387-2397 [DOI] [PubMed] [Google Scholar]
- 3). Suwa S, Ogita M, Miyauchi K, Sonoda T, Konishi H, Tsuboi S, Wada H, Naito R, Dohi T, Kasai T, Okazaki S, Isoda K, Daida H. Impact of Lipoprotein (a) on Long-Term Outcomes in Patients with Coronary Artery Disease Treated with Statin After a First Percutaneous Coronary Intervention. J Atheroscler Thromb, 2017; 24: 1125-1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol, 2013; 61: 1146-1156 [DOI] [PubMed] [Google Scholar]
- 5). Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M, PROCARDIS Consortium Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med, 2009; 361: 2518-2528 [DOI] [PubMed] [Google Scholar]
- 6). CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, König IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikäinen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, DIAGRAM Consortium. CARDIOGENICS Consortium. Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Müller-Nurasyid M, MuTHER Consortium. Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schäfer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJ, Wellcome Trust Case Control Consortium. Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrières J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kähönen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Trégouët DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvänen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimäki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O'Donnell C, Reilly MP, März W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet, 2013; 45: 25-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Owens DS, O'Brien KD. Clinical and genetic risk factors for calcific valve disease. In: Otto CM, Bonow RO, eds. Valvular Heart Disease. A Companion to Branwald's Heart Diseqase. 4th ed. Philadelphia: Elsevier-Saunders; 2013: 53-62 [Google Scholar]
- 8). Bossé Y, Mathieu P, Pibarot P. Genomics: the next step to elucidate the etiology of calcific aortic valve stenosis. J Am Coll Cardiol, 2008; 51: 1327-1336 [DOI] [PubMed] [Google Scholar]
- 9). Rogers MA, Aikawa E. A Not-So-Little Role for Lipoprotein(a) in the Development of Calcific Aortic Valve Disease. Circulation, 2015; 132: 621-623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, Tam JW, Teo KK, Mahmut A, Yang X, Witztum JL, Arsenault BJ, Després JP, Pibarot P, Tsimikas S. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J Am Coll Cardiol, 2015; 66: 1236-1246 [DOI] [PubMed] [Google Scholar]
- 11). Fless GM, ZumMallen ME, Scanu AM. Isolation of apolipoprotein(a) from lipoprotein(a). J Lipid Res, 1985; 26: 1224-1229 [PubMed] [Google Scholar]
- 12). McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature, 1987; 330: 132-137 [DOI] [PubMed] [Google Scholar]
- 13). Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol, 2017; 69: 692-711 [DOI] [PubMed] [Google Scholar]
- 14). Tada H, Kawashiri MA, Yoshida T, Teramoto R, Nohara A, Konno T, Inazu A, Mabuchi H, Yamagishi M, Hayashi K. Lipoprotein(a) in Familial Hypercholesterolemia With Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Gain-of-Function Mutations. Circ J, 2016; 80: 512-518 [DOI] [PubMed] [Google Scholar]
- 15). Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest, 1992; 90: 52-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Lin J, Reilly MP, Terembula K, Wilson FP. Plasma lipoprotein(a) levels are associated with mild renal impairment in type 2 diabetics independent of albuminuria. PLoS One, 2014; 9: e114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17). Langsted A, Kamstrup PR, Benn M, Tybjærg-Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol, 2016; 4: 577-587 [DOI] [PubMed] [Google Scholar]
- 18). Krempler F, Kostner GM, Bolzano K, Sandhofer F. Turnover of lipoprotein (a) in man. J Clin Invest, 1980; 65: 1483-1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Rader DJ, Cain W, Zech LA, Usher D, Brewer HB., Jr Variation in lipoprotein(a) concentrations among individuals with the same apolipoprotein (a) isoform is determined by the rate of lipoprotein(a) production. J Clin Invest, 1993; 91: 443-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Kraft HG, Menzel HJ, Hoppichler F, Vogel W, Utermann G. Changes of genetic apolipoprotein phenotypes caused by liver transplantation. Implications for apolipoprotein synthesis. J Clin Invest, 1989; 83: 137-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21). White AL, Lanford RE. Cell surface assembly of lipoprotein(a) in primary cultures of baboon hepatocytes. J Biol Chem, 1994; 269: 28716-2823 [PubMed] [Google Scholar]
- 22). Hrzenjak A, Frank S, Wo X, Zhou Y, Van Berkel T, Kostner GM. Galactose-specific asialoglycoprotein receptor is involved in lipoprotein (a) catabolism. Biochem J, 2003; 376: 765-771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Cain WJ, Millar JS, Himebauch AS, Tietge UJ, Maugeais C, Usher D, Rader DJ. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res, 2005; 46: 2681-2691 [DOI] [PubMed] [Google Scholar]
- 24). Rader DJ, Cain W, Ikewaki K, Talley G, Zech LA, Usher D, Brewer HB., Jr The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest, 1994; 93: 2758-2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech LA, Hoeg JM, Davignon J, Lupien P, Grossman M, et al. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest, 1995; 95: 1403-1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26). Armstrong VW, Schleef J, Thiery J, Muche R, Schuff-Werner P, Eisenhauer T, Seidel D. Effect of HELP-LDLapheresis on serum concentrations of human lipoprotein(a): kinetic analysis of the post-treatment return to baseline levels. Eur J Clin Invest, 1989; 19: 235-240 [DOI] [PubMed] [Google Scholar]
- 27). Marcovina SM, Albers JJ, Scanu AM, Kennedy H, Giaculli F, Berg K, Couderc R, Dati F, Rifai N, Sakurabayashi I, Tate JR, Steinmetz A. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem, 2000; 46: 1956-1967 [PubMed] [Google Scholar]
- 28). Palabrica TM, Liu AC, Aronovitz MJ, Furie B, Lawn RM, Furie BC. Antifibrinolytic activity of apolipoprotein(a) in vivo: human apolipoprotein(a) transgenic mice are resistant to tissue plasminogen activator-mediated thrombolysis. Nat Med, 1995; 1: 256-259 [DOI] [PubMed] [Google Scholar]
- 29). Edelberg JM, Gonzalez-Gronow M, Pizzo SV. Lipoprotein(a) inhibition of plasminogen activation by tissue-type plasminogen activator. Thromb Res, 1990; 57: 155-162 [DOI] [PubMed] [Google Scholar]
- 30). Edelberg JM, Reilly CF, Pizzo SV. The inhibition of tissue type plasminogen activator by plasminogen activator inhibitor-1. The effects of fibrinogen, heparin, vitronectin, and lipoprotein(a). J Biol Chem, 1991; 266: 7488-7493 [PubMed] [Google Scholar]
- 31). Poon M, Zhang X, Dunsky KG, Taubman MB, Harpel PC. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation, 1997; 96: 2514-2519 [DOI] [PubMed] [Google Scholar]
- 32). Kojima S, Harpel PC, Rifkin DB. Lipoprotein (a) inhibits the generation of transforming growth factor beta: an endogenous inhibitor of smooth muscle cell migration. J Cell Biol, 1991; 113: 1439-1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Grainger DJ, Kemp PR, Liu AC, Lawn RM, Metcalfe JC. Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature, 1994; 370: 460-462 [DOI] [PubMed] [Google Scholar]
- 34). Yano Y, Seishima M, Tokoro Y, Noma A. Stimulatory effects of lipoprotein(a) and low-density lipoprotein on human umbilical vein endothelial cell migration and proliferation are partially mediated by fibroblast growth factor-2. Biochim Biophys Acta, 1998; 1393: 26-34 [DOI] [PubMed] [Google Scholar]
- 35). Morishita R, Yamamoto K, Yamada S, Matsushita H, Tomita N, Sakurabayashi I, Kaneda Y, Moriguchi A, Higaki J, Ogihara T. Stimulatory effect of lipoprotein (a) on proliferation of human mesangial cells: role of lipoprotein (a) in renal disease. Biochem Biophys Res Commun, 1998; 249: 313-320 [DOI] [PubMed] [Google Scholar]
- 36). Takami S, Yamashita S, Kihara S, Ishigami M, Takemura K, Kume N, Kita T, Matsuzawa Y. Lipoprotein(a) enhances the expression of intercellular adhesion molecule-1 in cultured human umbilical vein endothelial cells. Circulation, 1998; 97: 721-728 [DOI] [PubMed] [Google Scholar]
- 37). Allen S, Khan S, Tam Sp, Koschinsky M, Taylor P, Yacoub M. Expression of adhesion molecules by lp(a): a potential novel mechanism for its atherogenicity. FASEB J, 1998; 12: 1765-1776 [DOI] [PubMed] [Google Scholar]
- 38). Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 2007; 447: 661-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M, PROCARDIS Consortium Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med, 2009; 361: 2518-2528 [DOI] [PubMed] [Google Scholar]
- 40). Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O'Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang SJ, Liu Y, Sjogren M, van der Pals J, Kälsch H, Mühleisen TW, Nöthen MM, Cupples LA, Caslake M, Di Angelantonio E, Danesh J, Rotter JI, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O'Donnell CJ, Post WS, CHARGE Extracoronary Calcium Working Group Genetic associations with valvular calcification and aortic stenosis. N Engl J Med, 2013; 368: 503-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Tada H, Kawashiri MA, Yamagishi M. Clinical Perspectives of Genetic Analyses on Dyslipidemia and Coronary Artery Disease. J Atheroscler Thromb, 2017; 24: 452-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42). Myocardial Infarction Genetics Consortium Investigators. Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, Fontanillas P, Gupta N, Duga S, Goel A, Farrall M, Saleheen D, Ferrario P, König I, Asselta R, Merlini PA, Marziliano N, Notarangelo MF, Schick U, Auer P, Assimes TL, Reilly M, Wilensky R, Rader DJ, Hovingh GK, Meitinger T, Kessler T, Kastrati A, Laugwitz KL, Siscovick D, Rotter JI, Hazen SL, Tracy R, Cresci S, Spertus J, Jackson R, Schwartz SM, Natarajan P, Crosby J, Muzny D, Ballantyne C, Rich SS, O'Donnell CJ, Abecasis G, Sunaev S, Nickerson DA, Buring JE, Ridker PM, Chasman DI, Austin E, Kullo IJ, Weeke PE, Shaffer CM, Bastarache LA, Denny JC, Roden DM, Palmer C, Deloukas P, Lin DY, Tang ZZ, Erdmann J, Schunkert H, Danesh J, Marrugat J, Elosua R, Ardissino D, McPherson R, Watkins H, Reiner AP, Wilson JG, Altshuler D, Gibbs RA, Lander ES, Boerwinkle E, Gabriel S, Kathiresan S. N Engl J Med, 2014; 371: 2072-2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Nomura A, Won HH, Khera AV, Takeuchi F, Ito K, McCarthy S, Emdin CA, Klarin D, Natarajan P, Zekavat SM, Gupta N, Peloso GM, Borecki IB, Teslovich TM, Asselta R, Duga S, Merlini PA, Correa A, Kessler T, Wilson JG, Bown MJ, Hall AS, Braund PS, Carey DJ, Murray MF, Kirchner HL, Leader JB, Lavage DR, Manus JN, Hartze DN, Samani NJ, Schunkert H, Marrugat J, Elosua R, McPherson R, Farrall M, Watkins H, Juang JJ, Hsiung CA, Lin SY, Wang JS, Tada H, Kawashiri MA, Inazu A, Yamagishi M, Katsuya T, Nakashima E, Nakatochi M, Yamamoto K, Yokota M, Momozawa Y, Rotter JI, Lander ES, Rader DJ, Danesh J, Ardissino D, Gabriel S, Willer CJ, Abecasis GR, Saleheen D, Kubo M, Kato N, Ida Chen YD, Dewey FE, Kathiresan S. Circ Res, 2017; 121: 81-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, Amouyel P, Di Angelantonio E, Arveiler D, Assimes TL, Auer PL, Baber U, Ballantyne CM, Bang LE, Benn M, Bis JC, Boehnke M, Boerwinkle E, Bork-Jensen J, Bottinger EP, Brandslund I, Brown M, Busonero F, Caulfield MJ, Chambers JC, Chasman DI, Chen YE, Chen YI, Chowdhury R, Christensen C, Chu AY, Connell JM, Cucca F, Cupples LA, Damrauer SM, Davies G, Deary IJ, Dedoussis G, Denny JC, Dominiczak A, Dubé MP, Ebeling T, Eiriksdottir G, Esko T, Farmaki AE, Feitosa MF, Ferrario M, Ferrieres J, Ford I, Fornage M, Franks PW, Frayling TM, Frikke-Schmidt R, Fritsche LG, Frossard P, Fuster V, Ganesh SK, Gao W, Garcia ME, Gieger C, Giulianini F, Goodarzi MO, Grallert H, Grarup N, Groop L, Grove ML, Gudnason V, Hansen T, Harris TB, Hayward C, Hirschhorn JN, Holmen OL, Huffman J, Huo Y, Hveem K, Jabeen S, Jackson AU, Jakobsdottir J, Jarvelin MR, Jensen GB, Jørgensen ME, Jukema JW, Justesen JM, Kamstrup PR, Kanoni S, Karpe F, Kee F, Khera AV, Klarin D, Koistinen HA, Kooner JS, Kooperberg C, Kuulasmaa K, Kuusisto J, Laakso M, Lakka T, Langenberg C, Langsted A, Launer LJ, Lauritzen T, Liewald DCM, Lin LA, Linneberg A, Loos RJF, Lu Y, Lu X, Mägi R, Malarstig A, Manichaikul A, Manning AK, Mäntyselkä P, Marouli E, Masca NGD, Maschio A, Meigs JB, Melander O, Metspalu A, Morris AP, Morrison AC, Mulas A, Müller-Nurasyid M, Munroe PB, Neville MJ, Nielsen JB, Nielsen SF, Nordestgaard BG, Ordovas JM, Mehran R, O'Donnell CJ, Orho-Melander M, Molony CM, Muntendam P, Padmanabhan S, Palmer CNA, Pasko D, Patel AP, Pedersen O, Perola M, Peters A, Pisinger C, Pistis G, Polasek O, Poulter N, Psaty BM, Rader DJ, Rasheed A, Rauramaa R, Reilly DF, Reiner AP, Renström F, Rich SS, Ridker PM, Rioux JD, Robertson NR, Roden DM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sanna S, Sattar N, Schmidt EM, Scott RA, Sever P, Sevilla RS, Shaffer CM, Sim X, Sivapalaratnam S, Small KS, Smith AV, Smith BH, Somayajula S, Southam L, Spector TD, Speliotes EK, Starr JM, Stirrups KE, Stitziel N, Strauch K, Stringham HM, Surendran P, Tada H, Tall AR, Tang H, Tardif JC, Taylor KD, Trompet S, Tsao PS, Tuomilehto J, Tybjaerg-Hansen A, van Zuydam NR, Varbo A, Varga TV, Virtamo J, Waldenberger M, Wang N, Wareham NJ, Warren HR, Weeke PE, Weinstock J, Wessel J, Wilson JG, Wilson PWF, Xu M, Yaghootkar H, Young R, Zeggini E, Zhang H, Zheng NS, Zhang W, Zhang Y, Zhou W, Zhou Y, Zoledziewska M, Charge Diabetes Working Group. EPIC-InterAct Consortium. EPIC-CVD Consortium. GOLD Consortium. VA Million Veteran Program. Howson JMM, Danesh J, McCarthy MI, Cowan CA, Abecasis G, Deloukas P, Musunuru K, Willer CJ, Kathiresan S. Nat Genet, 2017; 49: 1758-176629083408 [Google Scholar]
- 45). Emdin CA, Khera AV, Natarajan P, Klarin D, Won HH, Peloso GM, Stitziel NO, Nomura A, Zekavat SM, Bick AG, Gupta N, Asselta R, Duga S, Merlini PA, Correa A, Kessler T, Wilson JG, Bown MJ, Hall AS, Braund PS, Samani NJ, Schunkert H, Marrugat J, Elosua R, McPherson R, Farrall M, Watkins H, Willer C, Abecasis GR, Felix JF, Vasan RS, Lander E, Rader DJ, Danesh J, Ardissino D, Gabriel S, Saleheen D, Kathiresan S, CHARGE- Heart Failure Consortium. CARDIoGRAM Exome Consortium Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J Am Coll Cardiol, 2016; 68: 2761-2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46). Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Bärwolf C, Holme I, Kesäniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R, SEAS Investigators Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med, 2008; 359: 1343-1356 [DOI] [PubMed] [Google Scholar]
- 47). Capoulade R, Chan KL, Yeang C, Mathieu P, Bossé Y, Dumesnil JG, Tam JW, Teo KK, Mahmut A, Yang X, Witztum JL, Arsenault BJ, Després JP, Pibarot P, Tsimikas S. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J Am Coll Cardiol, 2015; 66: 1236-1246 [DOI] [PubMed] [Google Scholar]
- 48). Schwartz GG, Abt M, Bao W, DeMicco D, Kallend D, Miller M, Mundl H, Olsson AG. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol, 2015; 65: 2267-2275 [DOI] [PubMed] [Google Scholar]
- 49). Tada H, Kawashiri MA, Nomura A, Yoshimura K, Itoh H, Komuro I, Yamagishi M. Serum triglycerides predict first cardiovascular events in diabetic patients with hypercholesterolemia and retinopathy. Eur J Prev Cardiol, 2018; 25: 1852-1860 [DOI] [PubMed] [Google Scholar]
- 50). Albers JJ, Slee A, O'Brien KD, Robinson JG, Kashyap ML, Kwiterovich PO, Jr, Xu P, Marcovina SM. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol, 2013; 62: 1575-1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51). Khera AV, Everett BM, Caulfield MP, Hantash FM, Wohlgemuth J, Ridker PM, Mora S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation, 2014; 129: 635-642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Fieseler HG, Armstrong VW, Wieland E, Thiery J, Schütz E, Walli AK, Seidel D. Serum Lp(a) concentrations are unaffected by treatment with the HMG-CoA reductase inhibitor Pravastatin: results of a 2-year investigation. Clin Chim Acta, 1991; 204: 291-300 [DOI] [PubMed] [Google Scholar]
- 53). Awad K, Mikhailidis DP2, Katsiki N3, Muntner P4, Banach M5,6,7, Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs, 2018; 78: 453-62 [DOI] [PubMed] [Google Scholar]
- 54). Ginsberg HN, Stein EA. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol, 2014; 114: 711-715 [DOI] [PubMed] [Google Scholar]
- 55). Watts GF, Chan DC, Somaratne R, Wasserman SM, Scott R, Marcovina SM, Barrett PHR. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur Heart J, 2018; 39: 2577-2585 [DOI] [PubMed] [Google Scholar]
- 56). Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R, Karmally W, Nandakumar R, Fontanez N, Obunike J, Marcovina SM, Lichtenstein AH, Matthan NR, Matta J, Maroccia M, Becue F, Poitiers F, Swanson B, Cowan L, Sasiela WJ, Surks HK, Ginsberg HN. Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation, 2017; 135: 352-362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57). Nandakumar R, Matveyenko A, Thomas T, Pavlyha M, Ngai C, Holleran S, Ramakrishnan R, Ginsberg HN, Karmally W, Marcovina SM, Reyes-Soffer G. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J Lipid Res, 2018; 59: 2397-2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58). Tsimikas S. Potential Causality and Emerging Medical Therapies for Lipoprotein(a) and Its Associated Oxidized Phospholipids in Calcific Aortic Valve Stenosis. Circ Res, 2019; 124: 405-415 [DOI] [PMC free article] [PubMed] [Google Scholar]