

Graphical abstract
Keywords: Microbiome, Contamination, Amplicon sequence variant, Quantitative profiling, Active microbiota, Open data
Highlights
-
•
Sample storage and nucleic acid isolation influence microbiota compositions.
-
•
Error-corrected amplicon sequence variants (ASVs) improve 16S rRNA analysis.
-
•
Contamination and host cells confound and complicate microbiota analysis.
-
•
Quantitative and active microbiota analyses can complement existing methods.
-
•
Open data and protocol sharing increases transparency and reproducibility.
Abstract
Microbiome research has transformed the scientific landscape, as reflected by the exponential increase in microbiome-related publications from many different disciplines. Host-associated microbial communities play a role for almost all aspects of human, animal and plant biology and health. Consequently, there are tremendous expectations for the development of new clinical, agricultural and biotechnological applications of microbiome research. However, the field continues to be largely shaped by descriptive studies, the mechanistic understanding of microbiome functions for their hosts remains fragmentary, and direct applications of microbiome research are lacking. The aim of this review is therefore to provide a general introduction to the technical opportunities and challenges of microbiome research, as well as to make experimental and bioinformatic recommendations, i.e. (i) to avoid, reduce and assess the confounding effects of sample storage, nucleic acid isolation and microbial contamination; (ii) to minimize non-microbial contributions in host-associated microbiome samples; (iii) to sharpen the focus on physiologically relevant microbiome features by distinguishing signals from metabolically active and inactive or dead microbes and by adopting quantitative methods; and (iv) to enforce open data and protocol policies in order increase the transparency, reproducibility and credibility of the field.
Introduction
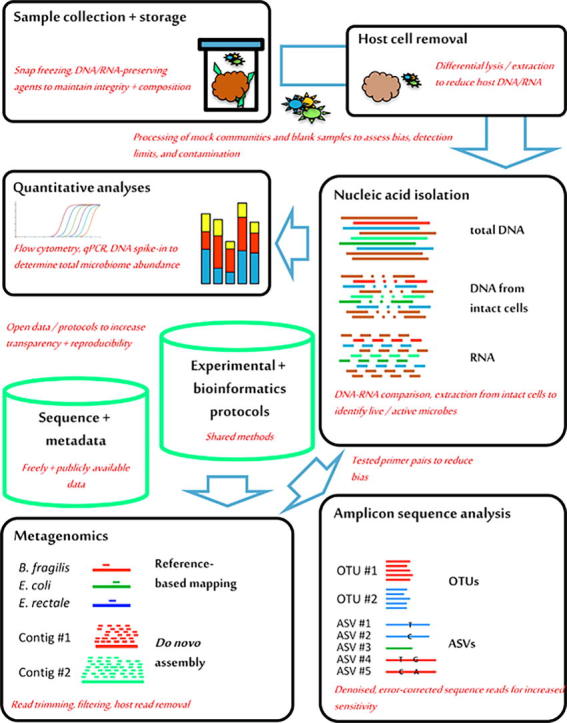
Most microbiome projects today apply large-scale parallel sequencing to taxonomically and functionally characterize previously described and not-yet-cultivated, uncharacterized microorganisms. The widespread application of high-throughput genomic approaches has been afforded by next-generation sequencing platforms that are easy to install and maintain. In addition, widely established experimental and bioinformatic protocols exist for sample processing, nucleic acid isolation, sequence target amplification, library preparation, sequence data processing and statistical analysis. Other high-throughput methods for system-wide microbiome analyses, such as metaproteomics or metabolomics/metabonomics [1], are less well established and widely used but are often successfully combined with genomics for systems-level approaches to simultaneously study different aspects of the microbiome. Cultivation-based isolation and characterization of individual microorganisms from microbiome samples can further complement nucleic acid sequencing-based and other 'omic approaches [2]. In the following, the microbiota will be referred to as the 'assemblage of microorganisms present in a defined environment' and the microbiome as the 'entire habitat, including the microorganisms … , their genomes … , and the surrounding environmental conditions' [1]. As sequencing-based microbiome analysis continues to be the most popular technique across the field, this review focuses on the discussion of experimental and bioinformatic aspects of this approach to highlight current problems and pitfalls as well as future chances and possibilities (Fig. 1).
Fig. 1.

Overview of recommendations for improved sequence-based microbiome analysis. Important technical components of typical laboratory and bioinformatic microbiome analysis projects (black boxes) and the bioinformatic resources that are generated in these projects (green columns) are shown, together with specific recommendations to expand and improve existing protocols (in red). Abbreviations: qPCR, quantitative real-time PCR; OTUs, operational taxonomic units; ASVs, amplicon sequence variants.
Genomics and bioinformatics techniques of microbiome analysis
Sequencing-based characterizations of entire microbial communities, as well as their individual components and functions in unprecedented detail, is largely afforded by two main techniques: amplicon sequencing and metagenomics. The first method generates taxonomic compositional microbiota profiles at relatively moderate costs that allow even small research groups to run large-scale bacterial microbiota analysis projects. The latter method generally affords a more comprehensive, but also more costly, taxonomic and functional analysis of the entire viral, bacterial and eukaryotic microbiota [3]. Both approaches have been scaled up to include thousands of samples in a single study. Best practice recommendations for microbiome analysis, including laboratory and bioinformatic procedures are available, for example, from the U.S. Microbiome Quality Control [4] project.
Taxonomic microbiome profiling by amplicon sequencing
Amplicon sequencing methods rely on the selective binding of universal primer pairs to highly conserved regions within the genomes of specific microbiome members of interest and the sequencing of the resulting PCR products, which encompass taxon-specific hypervariable regions [5]. The most commonly used target amplicon for microbiome analysis is the bacterial 16S rRNA gene, but universal primer pairs have also been described for archaeal and eukaryotic small subunit ribosomal RNA genes, internal transcribed spacers (ITS) of the fungal and other ribosomal RNA operons and other conserved genomic loci [6]. Within the bacterial 16S rRNA gene numerous primer combinations have been proposed to amplify different hypervariable regions and to generate PCR products of variable lengths suitable for different sequencing platforms (e.g., Pacific Biosciences vs. Illumina) [5]. However, even “universal” primers can preferentially bind specific bacterial taxa, leading to compositional study biases that vary between microbiome types (e.g. gut vs. vaginal microbiome) and should be considered in the project planning phase [7], [8].
Sequence variations in 16S and 18S rRNA genes, ITS regions and other metagenomic loci contain phylogenetic information that can be used to infer the taxonomic relationships of their microbial hosts. However, natural genetic variations are not easily distinguishable from sequencing errors, which even on the relatively accurate Illumina sequencing platform affects ∼0.1% of all sequenced nucleotides [9]. Given the scale of current microbiome studies, bioinformatic protocols therefore have to account for millions of wrong base calls per project.
For amplicon sequencing-based microbiota analysis, sequences are traditionally clustered into operational taxonomic units (OTUs) based on arbitrarily defined thresholds of sequence similarity. For example, 16S rRNA gene fragments of >97% sequence identity are clustered into separate OTUs that reflect the phylogenetic boundaries of distinct bacterial species. Sequence clustering can be guided by bacterial reference genomes, yet common methods often also include de novo clustering to identify previously unknown species [10]. OTU picking assigns similar, but slightly different sequences to the same taxon, assuming a shared biological origin. Clustering therefore diminishes the impact of technical variation on the analysis results, but at the expense of reduced sensitivity in detecting biological variation. Fungal microbiota analysis by ITS amplicon sequencing follows similar principles as bacterial 16S rRNA analysis but sequence clustering and classification are complicated by inconsistent amplicon lengths and varying sequence similarities between fungal species [11]. The UNITE project represents an effort to generate a resource to represent the growing, known diversity of ITS sequence data [12], similar to the well-established SILVA database for pro- and eukaryotic small and large subunit rRNA genes [13].
To differentiate between biological and technical sequence variations, reference-free statistical denoising methods such as Deblur or Dada2 [14], [15] have recently been implemented in QIIME2, a popular open-source software package for 16S rRNA analysis [16]. These tools generate error profiles of amplicon sequence datasets, which are then used to resolve sequencing errors and achieve single-nucleotide resolution for each amplicon sequence. Compared to OTU-based approaches, analysis of the resulting amplicon sequence variants (ASVs) provides improved sensitivity and specificity and reduces the problem of inflated microbiota datasets due to falsely identified distinct OTUs originating from mis-clustered sequences [17]. In addition, OTU clustering results are bound by the specific sequence data from which they were inferred and are therefore non-reproducible with modified or expanded datasets. The latest denoising algorithms overcome this limitation by recovering independent biological sequences as ASVs, fostering the reproducibility and comparability of amplicon-based microbiome analysis [18].
Taxonomic and functional profiling of the entire microbiome by metagenomics
Metagenomics uses the whole-genome shotgun approach to fragment and sequence the entire DNA of a microbiome sample instead of 16S rRNA gene fragments or other target amplicons alone. Correspondingly, the generated reads can originate from phages, viruses, bacteria, archaea, fungi and other eukaryotes and include plasmids and other extra-chromosomal elements as well as host, chloroplast and mitochondrial DNA. Compared to 16S rRNA analysis, this method needs significantly more data to obtain the sequencing depth that is required to identify and characterize rare microbiota members, often reaching several terabases per study and increasing costs and bioinformatic demands. However, as metagenomics potentially allows for functional microbiota characterization and, in theory, affords taxonomic resolution down to the level of individual microbial strains, it has become increasingly popular in microbiome research [19].
Quality control measures for metagenomic shotgun sequencing with new tools, such as KneadData, combine quality-based metagenomic read trimming and filtering with the bioinformatic detection and removal of human, plant and other eukaryotic host DNA (http://huttenhower.sph.harvard.edu/kneaddata). Metagenomic sequence data are typically analysed either by de novo assembly or by comparing reads individually to reference databases in a mapping-based process [20]. The de novo assembly of microbial genomes can help identify and comprehensively characterize previously unknown members of the microbiota [21]. However, because assembly requires substantial sequencing depth, assembly-based methods are typically restricted to the genomic reconstruction of highly abundant microbiome members. Marker gene-based sequence mapping with tools such as MetaPhlAn2 can be used for taxonomic profiling of entire microbial communities, including rare microbiome members [22].
Microbiome sample handling and processing
Maintaining microbiome integrity during sample collection and storage
Among many other factors, the accuracy of sequencing-based microbiota analysis depends on how well the original structure of the microbial community can be preserved between the time of sample collection and processing. Distinct members of the human, plant and environmental microbiota respond differently to extended periods of sample storage by dying or by suspending, retaining or increasing metabolic activity. Problematic artefacts for taxonomic or functional microbiota analysis can also arise from unintended disruption of the sample environment due to freeze–thaw cycles; exposure to oxygen, UV light, or osmotic stress; storage buffer components, etc. As a consequence, storage conditions can affect microbiome analysis and lead to biased results [23].
Snap freezing of microbiome samples in liquid nitrogen and their long-term storage at −80 °C are generally considered as the gold standard for sample preservation [24]. However, commercial nucleic acid-preserving reagents and sampling kits that are used to maintain sample integrity in studies involving the collection of environmental samples or self-collected human specimens outside of the laboratory environment have generally been reviewed favourably [23]. Studies have suggested that temperature shifts alone have minor effects on taxonomic compositions and inter-individual differences in human gut microbiota analyses [24]. Chu et al. (2017) found the living bacterial microbiota of faecal samples to be most strongly affected by oxygen exposure, rather than by other factors, even repeated freeze–thaw cycles [25]. The same accounts for fungal microbiome samples, which are commonly stored with nucleic acid-preserving agents [26]. As mycorrhizal soil fungi colonize plant root tissues, the disruption of root connections after sampling can reduce mycorrhizal mycelial abundance and subsequently, induce the growth of mycelium-dependent other fungal opportunists, highlighting a specific protential problem for plant-associated fungal microbiota analysis [27].
Avoiding selective enrichment and depletion of microbes during nucleic acid isolation
Obtaining personalized gut microbiome analysis results from consumer microbiome testing services, journalist Tina Saey was surprised to receive substantially different results, particularly with respect to the relative abundance of the two dominant bacterial gut phyla Firmicutes and Bacteroidetes [28]. While numerous confounding factors might account for these observed variations, differences between nucleic acid isolation protocols have been known to introduce biases in taxonomic microbiota analysis. Even widely used commercial kits for DNA and RNA isolation differ in their efficiency in lysing specific microbes, including Gram-positive and Gram-negative bacteria, such as Firmicutes and Bacteroidetes, respectively [29], [30]. Host-associated and environmental microbiome samples typically contain heterogeneous mixtures of viral, archaeal and eukaryotic microorganisms, including live and dead, active and inactive, vegetative and sporulated cells; cellular debris; free nucleic acids and other macromolecules. Microbial lysis protocols differ in their capacity to break open these different types of microbial components for nucleic acid isolation. Humic acids, melanin, polysaccharides, polyphenols and other sample components can interfere with DNA and RNA isolation and downstream applications, such as nucleic acid amplification or concentration determination [31].
Most microbiome analysis protocols include combinations of physical and enzymatic disruptions of microbial cells for nucleic acid isolation [4], which can be amended based on project-specific requirements, e.g., by adding specific polysaccharide-degrading enzymes such as lyticase for fungal microbiome analysis projects [32]. However, protocol variations lead to study-specific biases, which is one reason for the scarcity of meta-analyses of microbiome data [33], [34], [35]; these meta-analyses have had trouble with, for example, the identification of universal, disease-specific biomarkers across separate human microbiome studies. Depending on the microbiome sample type and specific microbial taxa of interest, testing and evaluating different nucleic acid extraction protocols on mock communities of diverse, defined microbial composition should be part of the early project planning phase. But project-specific technical biases are difficult to completely avoid, and consistency of the applied methods within specific microbiome studies might be most useful and practical.
Reducing, assessing and characterizing microbiome contamination
The interpretation of microbiome data can be complicated by contamination from sources other than the original sample [36]. The high sensitivity of sequencing-based microbiome analysis, particularly 16S rRNA gene amplicon sequencing, in detecting previously unknown, rare, and often non-cultivable microbiome members can also be problematic when contamination leads to false positive results. Laboratory consumables, reagents and even DNA extraction kits contain trace amounts of microbial DNA, and to some extent, sample collection, handling and processing always lead to low-level contamination [37], [38]. Salter et al. (2014) ran microbiome analyses on serial dilutions of the same clonal culture of Salmonella bongori and identified a diverse microbiome that included both environmental and host-associated bacteria from the human skin and gut [37]. Importantly, the relative abundance of bacterial signals from contamination was positively correlated with the dilution factor of the original culture, demonstrating that the microbiome signal from contamination becomes more significant with decreasing amounts of sample starting material. Thus, contamination is less relevant for the analysis of faecal or soil samples of high microbial density than for host-associated human or plant microbiome studies of low microbial biomass, such as skin and vaginal swabs, tissue biopsies, urine, and the phyllosphere [39], [40].
A prominent example of a controversially discussed microbiome finding concerns the placenta [41]. While several prominent publications reported on the presence of a unique placental microbiome in clinically asymptomatic women [42], [43], these reports have been challenged as contradicting the paradigm of a tightly immune-controlled sterile womb and the practice of surgically removing sterile mouse pups from pregnant mice to generate germ-free mice [41]. Lauder et al. (2016) compared human placenta samples with vaginal swabs and experimental controls, including sterile and 'air swabs', and found the bacterial density and taxonomic composition of the healthy placental samples to be indistinguishable from those of microbiome-negative controls [44].
A three-tiered approach has been proposed to address the contamination problem [36]: First, good laboratory practice measures can reduce the chance of contamination when handling and preparing microbiome samples. This includes using purified, DNA-free reagents and kits, whenever possible, as well as spatially separating sample processing and DNA isolation, PCR setup and subsequent steps in the lab. Besides bacterial cells and genomic DNA from environmental sources, amplified PCR products can pose an important laboratory source of contamination for 16S rRNA analysis [37]. Second, the extent of contamination should be assessed by including technical replicates and internal controls in every step of the sample preparation protocol. Negative, microbiome-free, extraction controls and positive controls of microbial mock communities in defined concentrations can be used to determine the upper and lower limits of detection. Third, contamination controls should be sequenced and analysed together with the biological samples to characterize the influence of contamination on analysis results. For example, similarities between microbiome profiles of biological samples and negative controls can be quantified to compare the effect sizes of biological findings against contamination signals. However, the general exclusion of putative contamination signals from the analysis, by removing taxa from negative controls, can also distort microbiome analysis results and should be avoided. As contamination often originates from the laboratory environment, it can be directly influenced by related projects and include microbial signals that are similar to those from the original samples [37].
Reducing the impact of host DNA
Non-microbial DNA from human, animal or plant hosts is another major concern for sequencing-based microbiome analysis. Inadequate removal of host DNA can significantly increase the cost of host-associated microbiome projects or even make them practically impossible if the sequencing effort to obtain sufficient coverage of the microbial metagenome becomes prohibitively large. Healthy human faeces typically contain <10% human DNA, but up to 90% of sequence reads from low-microbial biomass samples such as saliva, nasopharyngeal, skin and vaginal swabs can be assigned to the human host [40]. While bacterial concentrations in urine increase during bladder infection, the concomitant increase in host DNA from epithelial cell damage can complicate microbiome analyses [Fricke, unpublished data]. As chloroplast and mitochondrial genomes from eukaryotic cells also carry 16S rRNA genes, host DNA can be problematic for 16S rRNA analysis, especially for food or plant microbiome projects [45]. Finally, host sequence removal may be mandatory before newly generated sequence data can be released in public databases to secure the privacy and confidentiality of human study participants, as required by most journals and funding agencies prior to publication, or to protect proprietary information from genetically modified or patented crops.
The relative level of host DNA can be reduced experimentally, either by removing host cells before DNA extraction or by selectively enriching microbial DNA after DNA extraction, or host DNA can be deleted bioinformatically by identifying and removing host reads from resulting sequence data, as described above. To remove host cells before DNA extraction, differential lysis can be used to selectively release and degrade host DNA before microbial (bacterial and fungal) DNA is isolated since mammalian cells are less robust than most microbial cells [46]. Density gradient centrifugation has also been used to separate host tissue from bacterial cells in plant samples [47]. However, microbiome samples, such as human faeces, also contain free microbial DNA from dead bacteria or cells that were disrupted during sample collection or storage, and certain microbes may be more susceptible to eukaryotic lysis regimens than others. Therefore, differential lysis protocols can reduce total yields of isolate nucleic acids [48] and bias subsequent compositional microbiota analyses towards specific taxa such as hard-to-lyse gram-positive bacteria [49]. Commercial solutions have become available to detect and remove vertebrate DNA by binding methylated CpG sequence motifs, which are abundant in eukaryotic but rare in microbial genomic DNA [50]. The latter method has been used to enrich bacterial and protist DNA for subsequent analysis of human and fish samples [50]. As an alternative approach to reduce the number of host-derived, non-bacterial PCR products, Lundberg et al. (2013) developed synthetic oligomers that bind as peptide nucleic acid (PNA) PCR clamps specifically to plant chloroplast and mitochondrial 16S rRNA gene sequences and block them from amplification [45]. In a similar approach, Agler et al. (2016) used specific nested primers, or “blocking oligos”, inside the 16S rRNA gene of unwanted plant organelle DNA, to avoid amplification of the full-length PCR product for subsequent analyses [51].
New perspectives: Quantitative analysis and identification of active microbes
Adopting methods for quantitative microbiome profiling
Without accounting for potential differences in absolute microbial abundance between samples, the vast majority of microbiome projects today aim to characterize microbial communities based on compositional data [52]. These studies typically determine fractions of an unknown total number of microbial species, 16S rRNA gene copies, and other taxon-specific genes or functional gene categories. Unfortunately, compositional data tend to be misinterpreted as suggesting absolute shifts, reductions or increases in specific microbial taxa, gene functions or other microbiome parameters. Changes in absolute abundance of microbiome features can be biologically and clinically relevant, e.g. in small intestinal bacterial overgrowth (SIBO) [53], but tend to be ignored in standard microbiota projects. Vandeputte et al. (2017) found the bacterial load of human faeces to vary between healthy people and in individuals over time and bacterial density correlated with faecal enterotype [54]. Moreover, the authors demonstrated that quantitative microbiota profiling can change clinical perspectives. In this case, compared to previous reports based on relative faecal microbiota profiling, different bacterial taxa could be identified as specific biomarkers for inflammatory bowel diseases [54].
Different experimental approaches have been proposed to gather quantitative microbiome information, including cell counting by flow cytometry [54], quantitative or real-time PCR of the universal bacterial 16S rRNA gene [55] and normalization of bacterial relative abundances based on defined cell numbers that are spiked into the samples before nucleic acid isolation [56]. While the first approach is technically more demanding, commercial kits have become available to easily integrate quantitative analyses into microbiome project workflows. Importantly, sequencing depth, i.e., the number of reads assigned to each sample after 16S rRNA gene amplicon sequencing, should not be used to infer quantitative information, as inconsistent read counts between samples are typically technical artefacts that do not reflect quantitative differences [54]. However, the sequencing depth per sample does affect the alpha- and beta-diversity parameters of the microbiota and should be controlled, e.g., by bioinformatically rarefying read counts to equal numbers prior to statistical analysis [57].
Differentiating between total and active microbes
Sequencing-based microbiome studies typically rely on DNA as sole evidence for the existence of a microbiota in a sample. However, DNA from dying cells or spores or cell-free DNA in a sample may be evidence for microbial contact, but it does not necessarily indicate microbial life and an active microbiota in the sample. For example, the existence of a blood microbiome remains controversial, despite PCR-based evidence for bacterial 16S rRNA genes in blood DNA extracts from non-septic individuals, as attempts to culture bacteria from the same samples have mostly been unsuccessful [58]. While bacterial adaptation to the harsh conditions of the stomach has been demonstrated, metabolically active microbes in the stomach are difficult to distinguish from ingested, inactive microbes from other, adjacent body sites or food using DNA-based microbiota surveys alone [59]. To address this problem, a number of experimental and bioinformatic approaches have recently been proposed to identify metabolically active microbes reflective of a thriving microbiota.
Propidium monoazide (PMA) intercalates into double-stranded DNA, preventing it from being amplified by PCR and has been used by Chu et al. (2017) to remove free DNA from dead microbes prior to 16S rRNA gene amplicon sequencing [25]. Several groups have shown that 16S rRNA-based taxonomic microbiota compositions differ between RNA and DNA fractions isolated from the same sample [59]. This has been used to differentiate between transcriptionally active bacteria, which are identified on the basis of RNA evidence, and all other bacteria, which are identified on the basis of DNA evidence. Moreover, if DNA- and RNA-based analyses are combined with quantitative microbiota profiling, the ratio of 16S rRNA transcript-to-gene copies can be used to quantify transcriptional activity and stratify bacterial taxa [59]. However, recent studies on soil bacteria also found 16S rRNA transcripts to remain stable for extended periods of time [60] and 16S rRNA gene and transcript compositions to be indistinguishable [61], suggesting that RNA-based methods to measure metabolic activity do not work equally well for all microbiome types. Importantly, experimental protocols need to support the simultaneous isolation of DNA and RNA from the same sample and extracted RNA should be carefully controlled for contamination with trace amounts of DNA, in order to avoid selectively enriching specific microbial taxa with separate lysis protocols or erroneously interpreting DNA-based signals as indicators of transcriptional activity, respectively [59].
An interesting approach to bioinformatically infer microbial growth rates from metagenome sequence data has been proposed by Korem et al. (2015) [62]. The authors demonstrated a positive correlation between bacterial growth and replication activity in vitro that is reflected by relatively increased concentrations of DNA from genomic regions around the origin compared to that from the terminus of replication. By mapping metagenomic sequence reads to bacterial reference genomes, a 'peak-to-trough' coverage ratio was calculated by comparing the origin and terminus DNA concentrations for each individual genome. This ratio was then used to stratify gut bacteria according to replicational activity and to statistically associate specific active bacteria with diseases such as inflammatory bowel disease and type II diabetes [62].
Release of published microbiome data and protocols
Microbiome research benefits from the availability of research data and protocols, and efforts should be made to establish and maintain open data and protocol policies across the entire field of microbiome research [63]. Progress in human microbiome research is increasingly driven by large, multi-centre studies based on the processing, sequencing and analysis of thousands of samples, often using custom laboratory and bioinformatic protocols to generate a statistical basis to detect subtle microbiome phenotypes. As a consequence, newly generated raw data and metadata, tools and protocols represent a substantial scientific resource to the broader research community that allows others to reproduce and expand published findings, recombine datasets for meta-analyses and develop new analytical approaches. For this reason, raw sequence and other omics data, associated sample metadata, and experimental and bioinformatic protocols for sample processing and analysis from published studies need to be made fully, freely and easily accessible. Accurate, detailed and complete bioinformatics analysis protocols should all scripts and precise commands that are needed to allow for full reproduction of raw data processing, data analysis and the generation of published figures. Although most funding agencies and journals in theory have set formal policies for data availability, access can be complicated in practice due to incomplete or inconsistent datasets, missing metadata information, and simple technical difficulties. Authors can be reluctant to comply with formal requirements that journals and funding agencies are struggling to enforce. Universal mandatory data and protocol release before manuscript submission would facilitate and improve peer review and allow journals to check for data availability as part of the submission process.
Conclusions and future perspectives
Microbiome research continues to excite both the scientific community and the public at large. However, the field has also been blamed for overselling findings and not producing reliable, applicable results [64]. While the mechanistic understanding of microbiota functions may yet remain too fragmentary to allow for the immediate development of diagnostic and therapeutic applications, there is little doubt about the general importance of human, animal and plant microbiomes for their hosts. To foster successful microbiome research in the future, it will be important for researchers, authors, reviewers, journals and funding agencies to (i) push the field towards the more widespread application of carefully controlled protocols for sample storage, nucleic acid isolation, contamination, amplification, sequencing and bioinformatic analysis; (ii) develop, optimize and standardize appropriate, improved analysis protocols; (iii) adopt and combine new experimental techniques, such as DNA- and RNA-based, relative and quantitative microbiota profiling; and (iv) increase the transparency and outreach of microbiome research by releasing data, metadata and protocols from published studies (Fig. 1).
Acknowledgments
Conflict of interest
The authors have declared no conflict of interest.
Compliance with Ethics Requirement
This article does not contain any studies with human or animal subjects.
Acknowledgements
Daniel Podlesny is supported by funds from the German Research Foundation (DFG) under project number 316130265.
Biographies

Alena M. Fricker is a PhD student in the group of Dr. Fricke, in the Department of Microbiome Research and Applied Bioinformatics at the University of Hohenheim, Stuttgart, Germany. She completed her M.S. in Molecular Nutritional Sciences at the University of Hohenheim and is now pursuing a Ph.D. project in human microbiome research focusing on culture- and sequencing-based methods to study the influence of dietary interventions on the gut microbiota.

Daniel Podlesny completed a M.S. in Molecular Nutritional Sciences at the University of Hohenheim and is now a PhD student in Dr. Fricke's group at the University of Hohenheim, Stuttgart, Germany. His work involves the development of new bioinformatic tools for strain-level metagenomic sequence analysis to study the microbiome dynamics in response to fecal transplantation.

W. Florian Fricke is Professor for Microbiome Research and Applied Bioinformatics at the University of Hohenheim in Stuttgart, Germany and Adjunct Assistant Professor at the Institute for Genome Sciences, University of Maryland School of Medicine in Baltimore, Maryland, USA. He received his PhD at the University of Goettingen, Germany, and was a post-doctoral fellow at The Institute for Genomic Research (TIGR), Rockville, Maryland. His group uses microbial genomics and applied bioinformatics to study the dynamics of the human gastrointestinal microbiota in the context of health and disease. His research focuses on the stomach microbiota, fecal microbiota transplantation and the development of functional biomarkers for host-microbe interactions.
Footnotes
Peer review under responsibility of Cairo University.
References
- 1.Marchesi J.R., Ravel J. The vocabulary of microbiome research: a proposal. Microbiome. 2015;3:31. doi: 10.1186/s40168-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clavel T., Gomes-Neto J.C., Lagkouvardos I., Ramer-Tait A.E. Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol Rev. 2017;279(1):8–22. doi: 10.1111/imr.12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franzosa E.A., Hsu T., Sirota-Madi A., Shafquat A., Abu-Ali G., Morgan X.C. Sequencing and beyond: integrating molecular 'omics' for microbial community profiling. Nat Rev Microbiol. 2015;13(6):360–372. doi: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sinha R., Abu-Ali G., Vogtmann E., Fodor A.A., Ren B., Amir A. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol. 2017;35(11):1077–1086. doi: 10.1038/nbt.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Amore R., Ijaz U.Z., Schirmer M., Kenny J.G., Gregory R., Darby A.C. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics. 2016;17:55. doi: 10.1186/s12864-015-2194-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kittelmann S., Seedorf H., Walters W.A., Clemente J.C., Knight R., Gordon J.I. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS One. 2013;8(2) doi: 10.1371/journal.pone.0047879. e47879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z., Hui P.C., Hui M., Yeoh Y.K., Wong P.Y., Chan M.C.W. Impact of preservation method and 16S rRNA hypervariable region on GutMicrobiota profiling. mSystems. 2019;4(1) doi: 10.1128/mSystems.00271-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graspeuntner S., Loeper N., Kunzel S., Baines J.F., Rupp J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci Rep. 2018;8(1):9678. doi: 10.1038/s41598-018-27757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schirmer M., Ijaz U.Z., D'Amore R., Hall N., Sloan W.T., Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015;43(6) doi: 10.1093/nar/gku1341. e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rideout J.R., He Y., Navas-Molina J.A., Walters W.A., Ursell L.K., Gibbons S.M. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2 doi: 10.7717/peerj.545. e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halwachs B., Madhusudhan N., Krause R., Nilsson R.H., Moissl-Eichinger C., Hogenauer C. Critical issues in mycobiota analysis. Front Microbiol. 2017;8:180. doi: 10.3389/fmicb.2017.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nilsson R.H., Larsson K.H., Taylor A.F.S., Bengtsson-Palme J., Jeppesen T.S., Schigel D. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47(D1):D259–D264. doi: 10.1093/nar/gky1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue) doi: 10.1093/nar/gks1219. D590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amir A., McDonald D., Navas-Molina J.A., Kopylova E., Morton J.T., Zech Xu Z. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2(2) doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J., Holmes S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopylova E., Navas-Molina J.A., Mercier C., Xu Z.Z., Mahe F., He Y. Open-source sequence clustering methods improve the state of the art. mSystems. 2016;1(1) doi: 10.1128/mSystems.00003-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callahan B.J., McMurdie P.J., Holmes S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11(12):2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garud N.R., Good B.H., Hallatschek O., Pollard K.S. Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol. 2019;17(1) doi: 10.1371/journal.pbio.3000102. e3000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nayfach S., Pollard K.S. Toward accurate and quantitative comparative metagenomics. Cell. 2016;166(5):1103–1116. doi: 10.1016/j.cell.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown C.T., Sharon I., Thomas B.C., Castelle C.J., Morowitz M.J., Banfield J.F. Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome. 2013;1(1):30. doi: 10.1186/2049-2618-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truong D.T., Franzosa E.A., Tickle T.L., Scholz M., Weingart G., Pasolli E. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 23.Choo J.M., Leong L.E., Rogers G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci Rep. 2015;5:16350. doi: 10.1038/srep16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bundgaard-Nielsen C., Hagstrom S., Sorensen S. Interpersonal variations in gut microbiota profiles supersedes the effects of differing fecal storage conditions. Sci Rep. 2018;8(1):17367. doi: 10.1038/s41598-018-35843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu N.D., Smith M.B., Perrotta A.R., Kassam Z., Alm E.J. Profiling living bacteria informs preparation of fecal microbiota transplantations. PLoS One. 2017;12(1) doi: 10.1371/journal.pone.0170922. e0170922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant S., Grant W.D., Cowan D.A., Jones B.E., Ma Y., Ventosa A. Identification of eukaryotic open reading frames in metagenomic cDNA libraries made from environmental samples. Appl Environ Microbiol. 2006;72(1):135–143. doi: 10.1128/AEM.72.1.135-143.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindahl B.D., de Boer W., Finlay R.D. Disruption of root carbon transport into forest humus stimulates fungal opportunists at the expense of mycorrhizal fungi. ISME J. 2010;4(7):872–881. doi: 10.1038/ismej.2010.19. [DOI] [PubMed] [Google Scholar]
- 28.Saey TH. The bizarre side of science [Internet]. Gory Details: Science News. 2014. Available from: https://www.sciencenews.org/blog/gory-details/here%E2%80%99s-poop-getting-your-gut-microbiome-analyzed.
- 29.Dineen S.M., Rt Aranda, Anders D.L., Robertson J.M. An evaluation of commercial DNA extraction kits for the isolation of bacterial spore DNA from soil. J Appl Microbiol. 2010;109(6):1886–1896. doi: 10.1111/j.1365-2672.2010.04816.x. [DOI] [PubMed] [Google Scholar]
- 30.Costea P.I., Zeller G., Sunagawa S., Pelletier E., Alberti A., Levenez F. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–1076. doi: 10.1038/nbt.3960. [DOI] [PubMed] [Google Scholar]
- 31.Tebbe C.C., Vahjen W. Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl Environ Microbiol. 1993;59(8):2657–2665. doi: 10.1128/aem.59.8.2657-2665.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leaw S.N., Chang H.C., Sun H.F., Barton R., Bouchara J.P., Chang T.C. Identification of medically important yeast species by sequence analysis of the internal transcribed spacer regions. J Clin Microbiol. 2006;44(3):693–699. doi: 10.1128/JCM.44.3.693-699.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duvallet C., Gibbons S.M., Gurry T., Irizarry R.A., Alm E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8(1):1784. doi: 10.1038/s41467-017-01973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sze M.A., Schloss P.D. Looking for a signal in the noise: revisiting obesity and the microbiome. MBio. 2016;7(4) doi: 10.1128/mBio.01018-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lozupone C.A., Stombaugh J., Gonzalez A., Ackermann G., Wendel D., Vazquez-Baeza Y. Meta-analyses of studies of the human microbiota. Genome Res. 2013;23(10):1704–1714. doi: 10.1101/gr.151803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenhofer R., Minich J.J., Marotz C., Cooper A., Knight R., Weyrich L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2018 doi: 10.1016/j.tim.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 37.Salter S.J., Cox M.J., Turek E.M., Calus S.T., Cookson W.O., Moffatt M.F. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiergeist A., Reischl U. Priority program intestinal microbiota consortium/ quality assessment p, Gessner A. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int J Med Microbiol. 2016;306(5):334–342. doi: 10.1016/j.ijmm.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Turner T.R., James E.K., Poole P.S. The plant microbiome. Genome Biol. 2013;14(6):209. doi: 10.1186/gb-2013-14-6-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marotz C.A., Sanders J.G., Zuniga C., Zaramela L.S., Knight R., Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. doi: 10.1186/s40168-018-0426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Munoz M.E., Arrieta M.C., Ramer-Tait A.E., Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. 2017;5(1):48. doi: 10.1186/s40168-017-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aagaard K., Ma J., Antony K.M., Ganu R., Petrosino J., Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collado M.C., Rautava S., Aakko J., Isolauri E., Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. 2016;6:23129. doi: 10.1038/srep23129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lauder A.P., Roche A.M., Sherrill-Mix S., Bailey A., Laughlin A.L., Bittinger K. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. 2016;4(1):29. doi: 10.1186/s40168-016-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lundberg D.S., Yourstone S., Mieczkowski P., Jones C.D., Dangl J.L. Practical innovations for high-throughput amplicon sequencing. Nat Methods. 2013;10(10):999–1002. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 46.Hunter S.J., Easton S., Booth V., Henderson B., Wade W.G., Ward J.M. Selective removal of human DNA from metagenomic DNA samples extracted from dental plaque. J Basic Microbiol. 2011;51(4):442–446. doi: 10.1002/jobm.201000372. [DOI] [PubMed] [Google Scholar]
- 47.Delmotte N., Knief C., Chaffron S., Innerebner G., Roschitzki B., Schlapbach R. Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci U S A. 2009;106(38):16428–16433. doi: 10.1073/pnas.0905240106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferretti P., Farina S., Cristofolini M., Girolomoni G., Tett A., Segata N. Experimental metagenomics and ribosomal profiling of the human skin microbiome. Exp Dermatol. 2017;26(3):211–219. doi: 10.1111/exd.13210. [DOI] [PubMed] [Google Scholar]
- 49.Horz H.P., Scheer S., Huenger F., Vianna M.E., Conrads G. Selective isolation of bacterial DNA from human clinical specimens. J Microbiol Methods. 2008;72(1):98–102. doi: 10.1016/j.mimet.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Feehery G.R., Yigit E., Oyola S.O., Langhorst B.W., Schmidt V.T., Stewart F.J. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0076096. e76096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agler MT, Mari A, Dombrowski N, Haquard S, Kemen EM. New insights in host-associated microbial diversity with broad and accurate taxonomic resolution. bioRxiv; 2016.
- 52.Gloor G.B., Wu J.R., Pawlowsky-Glahn V., Egozcue J.J. It's all relative: analyzing microbiome data as compositions. Ann Epidemiol. 2016;26(5):322–329. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Quigley E.M. Small intestinal bacterial overgrowth: what it is and what it is not. Curr Opin Gastroenterol. 2014;30(2):141–146. doi: 10.1097/MOG.0000000000000040. [DOI] [PubMed] [Google Scholar]
- 54.Vandeputte D., Kathagen G., D'Hoe K., Vieira-Silva S., Valles-Colomer M., Sabino J. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551(7681):507–511. doi: 10.1038/nature24460. [DOI] [PubMed] [Google Scholar]
- 55.Nadkarni M.A., Martin F.E., Jacques N.A., Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(Pt 1):257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 56.Stammler F., Glasner J., Hiergeist A., Holler E., Weber D., Oefner P.J. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome. 2016;4(1):28. doi: 10.1186/s40168-016-0175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5(1):27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Potgieter M., Bester J., Kell D.B., Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. 2015;39(4):567–591. doi: 10.1093/femsre/fuv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wurm P., Dorner E., Kremer C., Spranger J., Maddox C., Halwachs B. Qualitative and quantitative DNA- and RNA-based analysis of the bacterial stomach microbiota in humans, mice, and gerbils. mSystems. 2018;3(6) doi: 10.1128/mSystems.00262-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papp K., Mau R.L., Hayer M., Koch B.J., Hungate B.A., Schwartz E. Quantitative stable isotope probing with H2(18)O reveals that most bacterial taxa in soil synthesize new ribosomal RNA. ISME J. 2018;12(12):3043–3045. doi: 10.1038/s41396-018-0233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Papp K., Hungate B.A., Schwartz E. Microbial rRNA synthesis and growth compared through quantitative stable isotope probing with H2(18)O. Appl Environ Microbiol. 2018;84(8) doi: 10.1128/AEM.02441-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korem T., Zeevi D., Suez J., Weinberger A., Avnit-Sagi T., Pompan-Lotan M. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science. 2015;349(6252):1101–1106. doi: 10.1126/science.aac4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langille M.G.I., Ravel J., Fricke W.F. “Available upon request”: not good enough for microbiome data! Microbiome. 2018;6(1):8. doi: 10.1186/s40168-017-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanage W.P. Microbiology: Microbiome science needs a healthy dose of scepticism. Nature. 2014;512(7514):247–248. doi: 10.1038/512247a. [DOI] [PubMed] [Google Scholar]