

Introduction
Piebaldism is a rare autosomal dominant disorder characterized by congenital patches of depigmented skin and hair that lack melanocytes (MIM172800).
The KIT gene, mapped to chromosome 4q12, is one of the common causative genes in autosomal dominant piebaldism and accounts for 75% of patients with piebaldism.1 Various studies in mice found that mutations involving the murine KIT locus led to dominant white spotting and abnormalities in hematopoiesis and germ cell development. More than 47 mutations have been described that are associated with piebaldism.2, 3, 4
Here we describe a Saudi nonconsanguineous family with an autosomal dominant form of inheritance, with marked phenotypic and genotypic variability. All the affected individuals had widespread depigmented patches involving more than 70% to 80% body surface area with almost total poliosis. Although all these features presented since their birth, the father noticed heterochromia iridis as an adult. We used next-generation sequencing with 214 dermatologic gene panels (DGP)5 to identify pathogenic mutations in the KIT gene that underlies the autosomal dominant form of piebaldism in this family.
Case report
A Saudi family comprising 4 affected individuals with widespread depigmentation of skin and poliosis since birth were enrolled in an approved research project (RAC#2140013) with full informed consent (Fig 1, A). All patients underwent a thorough physical examination including a full skin examination. Results of baseline laboratory workup including complete blood counts, hepatic profile, renal profile, blood sugar and thyroid function tests were normal. Because the patients had additional features besides the clinical signs of piebaldism, a provisional diagnosis of atypical piebaldism was made (Table I; Fig 2).
Fig 1.
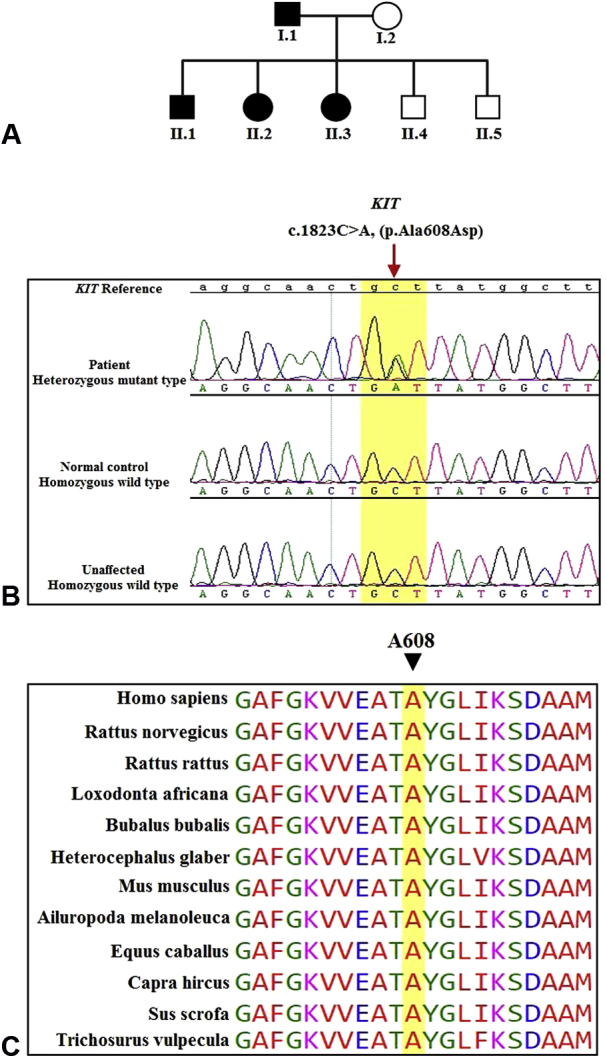
A, Pedigree of the described Family 1. Parents (I.1 and I.2) of the individuals (II.1, II.2, II.3, II.4, and II.5). Filled boxes indicate individuals affected by piebaldism, and open boxes indicate phenotypically normal individuals. B, Sanger sequencing analysis of the KIT gene identified a heterozygous variant (c.1823C>A; p.Ala608Asp) in the affected individual (upper panel), homozygous wild type in the normal control (middle panel) and unaffected sibling (lower panel). C, Evolutionary conservation of the alanine at position 608 of KIT1.
Table I.
Phenotypic spectrum of the affected individuals
| Skin | Hair | Other features | |
|---|---|---|---|
| Patient I.1 (father) 57/male | Widespread depigmented patches involving about 70%-80% body surface area with few small patches of normally pigmented skin within the affected areas. | Completely white (total poliosis) | Heterochromia iridis |
| Patient II.1 (son) 18/male | Widespread depigmented patches involving about 70%-80% body surface area with few small patches of normally pigmented skin within the affected areas. | Completely white (total poliosis) | Impaired hearing right ear (sensorineural deafness). Left ear is normal. |
| Patient II.2 (daughter) 26/female | Widespread depigmented patches involving about 70%-80% body surface area with few small patches of normally pigmented skin within the affected areas. | Completely white (total poliosis) | None |
| Patient II.3 (daughter) 28/female | Widespread depigmented patches involving about 70%-80% body surface area with few small patches of normally pigmented skin within the affected areas. | Completely white (total poliosis) | None |
Fig 2.

A, Heterochromia iridis in patient I.1. B, Widespread depigmented patches with total poiliosis in patient II.1. Note: Both the patients have dyed their hair.
Genomic DNA was extracted from peripheral blood using the standard salt precipitation method. To identify the causal mutation we carried out next-generation sequencing with DGP and validated by Sanger sequencing. Further Insilco analysis was performed to establish the pathogenicity of the identified mutation (Supplemental Data).
The resultant variant file from DGP identified several heterozygous variants that were absent from the dbSNP (Single Nucleotide Polymorphism Database), the 1,000 Genomes Project, and our in-house database. Sanger sequencing identified and validated a potential causative variant as a heterozygous change c.1823C>A; p.Ala608Asp in exon 12 of KIT (NM_000222) (Fig 1, B). The novel variant was vetted for familial segregation based on autosomal dominant pattern of inheritance. Further, Sanger sequencing of 600 ethnically matched normal controls did not reveal this variant. Multiple sequence alignment showed that Ala608 was evolutionarily conserved across various species (Fig 1, C). Moreover, this missense mutation was predicted to have a deleterious impact on protein function by different prediction programs (SIFT, PolyPhen, MutationTaster) and map to tyrosine kinase domains of the KIT gene. The phenotypic spectrum of the patients is described in Table I and Fig 2.
Discussion
We identified a novel missense mutation in the KIT gene in all 4 patients consistent with autosomal dominant pattern of inheritance. The alterations in the KIT gene are consistent with the classical phenotype of piebaldism. Besides having the features of piebaldism, our patients presented some additional features in the form of sensorineural deafness in patient II.1 and heterochromia iridis in patient I.1. Although the combination of piebaldism-like features with additional abnormalities have been described in several syndromes, they are rarely associated with the abnormalities in KIT gene. These include Waardenburg syndrome (autosomal dominant) characterized by a congenital white forelock, lateral displacement of the medial canthi, a hypertrophic nasal root, partial or total heterochromia of the iris, and sensorineural deafness.6, 7 Mutations in multiple genes cause the various forms of Waardenburg syndrome. The 6 genes, with different frequencies, involved are PAX3 (encoding the paired box-3 transcription factor), MITF (microphthalmia-associated transcription factor), EDN3 (endothelin 3), EDNRB (endothelin receptor type B), SOX10 (encoding the Sry bOX10 transcription factor), and SNAI2 (snail homolog 2). Ziprkowski et al8 described an X-linked recessive disorder characterized by hypomelanosis, deafness, and mutism in a Jewish Israeli family of Sephardic origin. It has now been included in the albinism-deafness syndrome with an unidentified gene abnormality localized to Xq24-q26.9 Piebaldism, in association with congenital deafness was first reported by Woolf in 2 Hopi Indian brothers in Arizona.10 The Tietz syndrome was first described as a congenital generalized depigmentation and profound congenital sensorineural deafness, transmitted as an autosomal dominant trait with full penetrance, and attributed to a mutation in the microphthalmia-associated transcription factor (MITF) gene in descendants of the same family.11
Even though our patients had an atypical presentation of piebaldism with some additional findings as described above, all the affected individuals have an abnormality in KIT gene. Mutations in this gene have been reported in a patient with piebaldism with associated sensorineural deafness.12 The presence of varied morphologic features, besides the usual features of piebaldism, point toward the concept of an expanded syndrome as proposed by Spritz and Beighton12 The genotype-phenotype relationship for the severity of piebaldism is already known, with milder morphologic variants associated with mutations in the extracellular domains of the transmembrane receptor tyrosine kinase (TK) (receptor KIT), and severe piebaldism has an association with dominant-negative inhibition caused by a missense mutation in the cytoplasmic part of the receptor TK domain. KIT belongs to the family of TK transmembrane receptors that undergoes dimerization within the cell membrane upon stem cell factor binding to its extracellular domain, thereby activating the intracellular TK. The wild-type residue A608WT is located in protein kinase domain and buried in the core of the protein that is important for binding of other molecules, whereas the mutant-type residue A608DMT is bigger and less hydrophobic than the wild-type residue A608WT causing the loss of hydrophobic interactions in the core of the protein. Because of the loss of the hydrogen bond, the mutant residue introduces a bigger residue at this position, unable to fit in the core, and might disturb and abolish the function of tyrosine kinase. Dominant-negative inhibition of the kit receptor, because of missense mutations, preserve approximately 25% of the KIT function, resulting in characteristically severe piebald phenotypes. Mutations in the KIT gene result in decreased receptor tyrosine kinase, signaling impaired migration of melanoblasts to the skin during embryologic development and a decrease in melanogenesis.13, 14, 15
Our patients have a severe morphologic variant that correlates with the site of mutation present in the TK domain. Protein modelling of this missense variant (A608D) supports the hypothesis that the mutated residues might potentially interfere by disrupting the KIT protein and thus abolish its functions, suggesting that the mutation in KIT results in the atypical presentation of piebaldism phenotype in our families. This study highlights the clinical utility of next-generation sequencing dermatologic panel for the rapid identification of the heterogeneous genetic etiology thus affecting the diagnostic and scientific interpretation.
Acknowledgments
The patients and their family members are warmly acknowledged for their participation. We greatly thank Genotyping and Sequencing Core Facilities at King Faisal Specialist Hospital & Research Center for their support.
Footnotes
Funding: No external (noninstitutional) funding was used for this study.
Acknowledgements: The authors gratefully acknowledge the use of Saudi Human Genome Project resources, funded by the King Abdulaziz City for Science and Technology, Riyadh, Saudi Arabia.
Conflicts of interest: None disclosed.
Supplemental Material
Next-Generation Sequencing Library preparation
The paired-end library preparation was performed with an AmpliSeq HiFi mix according to the manufacturer’s instructions (Thermo Fisher, Carlsbad, CA). The sequencing panel included genes reported in the Online Mendelian Inheritance in Man (OMIM) database on dermatologic phenotypes; genes with recurrent mutations suspected in dermatologic manifestations5 were used. The Ampliseq library was obtained by treating a DNA sample with DGP primer pools and amplifying it with Ampliseq mix. After digestion, amplicons were ligated, purified, and quantified using quantitative polymerase chain reaction (PCR) and normalized. These normalized libraries were then ligated with Ion Xpress barcode adapters for emulsion PCR on the Ion OneTouch System and further enriched using Ion OneTouch EX enrichment system according to manufacturer’s instructions (Thermo Fisher). Finally, the template Ion Sphere particles were processed for sequencing on the Ion Proton instrument. The probe set was composed of more than 4833 probes, which targeted exons of the 214 genes in the DGP. Finally, the readings were mapped and aligned on read files (FASTQ) generated from the sequencing platform via the manufacturer’s proprietary software and using human genome (hg19/b37) with the Burrows-Wheeler Aligner (BWA) package,16 version 0.6.1. For the variant calling and analysis workflow, the aligned file involved the exclusion of deep intronic variants (more than 20 bases far from the exon terminals), UTRs, and non–frameshift indels. Then variants that were frequent in public databases (the 1000 Genomes database) with minor allelle frequency (MAF) greater than 1% were excluded. Variants that were also frequent in our in-house database are also excluded (MAF >1%). The remaining variants were then filtered based on a score (>100) computed by Torrent Suite (Thermo Fisher Scientific, Waltham, MA) according to different criteria, such as confidence of base calls, depth, and context bases. The final step involved filtering based on zygosity (if the variants were heterozygous), and causative variants within the possible candidate disease genes were considered.
Sanger sequencing
To validate the next-generation sequencing results, mutational screening of filtered causative variants along with flanking introns of genes were carried out by PCR amplification using standard protocols. Primer sequences were designed using Primer3 (http://frodo.wi.Mit.edu/primer3/). PCR conditions are available upon request. PCR amplicons were purified and sequenced with ABI PRISM Big Dye Terminator v3.1 Cycle Sequencing Kit on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems Inc, Foster City, CA). Sequence analysis was performed using Lasergene (DNA Star Inc, Madison, WI) software package and then compared with the reference gene bank sequence (NM_000222). Numbering commenced with the A of the ATG initiation codon as +1. Segregation of the sequence variants with the disease phenotype was done for all the members in the family (Fig 1, B).
To predict the possible causality of the identified missense variants, we used MutationTaster, and Polymorphism phenotyping (Polyphen). For checking the evolutionary conservation of the mutated amino acids, the human KIT protein sequences with various species were aligned using multiple align and HOPE software.
References
- 1.Ezoe K., Holmes S.A., Ho L. Novel mutations and deletions of the KIT (steel factor receptor) gene in human piebaldism. Am J Hum Genet. 1995;56:58–66. [PMC free article] [PubMed] [Google Scholar]
- 2.Nishikawa S., Kusakabe M., Yoshinaga K. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit dependency during melanocyte development. EMBOJ. 1991;10:2111–2118. doi: 10.1002/j.1460-2075.1991.tb07744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chabot B., Stephenson D.A., Chapman V.M., Besmer P., Bernstein A. The proto oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 4.Geissler E.N., Ryan M.A., Housman D.E. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 5.Saudi Mendeliome Group Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol. 2015;16:134. doi: 10.1186/s13059-015-0693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janjua S., Khachemoune A., Guldbakke K.K. Piebaldism: A case report and a concise review of the literature. Cutis. 2007;80:411–414. [PubMed] [Google Scholar]
- 7.Read A.P., Newton V.E. Waardenburg's syndrome. J Med Genet. 1997;34:656–665. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziprkowski L., Krakowski A., Adam A., Costeff H., Sade J. Partial albinism and deaf-mutism due to a recessive sex-lnked gene. Arch Dermaol. 1962;86:530–539. doi: 10.1001/archderm.1962.01590100144027. [DOI] [PubMed] [Google Scholar]
- 9.Shiloh Y., Litvak G., Ziv Y. Genetic mapping of X-linked albinism-deafness syndrome (ADFN) to Xq26.3-Q27 I. Am J Hum Genet. 1990;47:20–27. [PMC free article] [PubMed] [Google Scholar]
- 10.Woolf C. Albinism among Indians in Arizona and New Mexico. Am J Hum Genet. 1965;17:23–25. [PMC free article] [PubMed] [Google Scholar]
- 11.Smith S.D., Kelley P.M., Kenyon J.B., Hoover D. Tietz syndrome (hypopigmentation/deafness) caused by mutations of MITF. J Med Genet. 2000;37:446–448. doi: 10.1136/jmg.37.6.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spritz R.A., Beighton P. Piebaldism with deafness: molecular evidence for an expanded syndrome. Am J Med Genet. 1998;75(1):101–103. doi: 10.1002/(sici)1096-8628(19980106)75:1<101::aid-ajmg20>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 13.Murakami T., Fukai K., Oiso N. New KIT mutations in patients with piebaldism. J Dermatol Sci. 2004;35:29–33. doi: 10.1016/j.jdermsci.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Murakami T., Hosomi N., Oiso N. Analysis of KIT, SCF, and initial screening of SLUG in patients with piebaldism. J Invest Dermatol. 2005;124:670–672. doi: 10.1111/j.0022-202X.2005.23637.x. [DOI] [PubMed] [Google Scholar]
- 15.Oiso N., Fukai K., Kawada A., Suzuki T. Piebaldism. J Dermatol. 2013;40:330–335. doi: 10.1111/j.1346-8138.2012.01583.x. [DOI] [PubMed] [Google Scholar]
- 16.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]