

Abstract
Cancer is the leading disease-related cause of death in children in developed countries. Arising in the context of actively growing tissues, childhood cancers are fundamentally diseases of dysregulated development. Childhood cancers exhibit a lower overall mutational burden than adult cancers, and recent sequencing studies have revealed that the genomic events central to childhood oncogenesis include mutations resulting in broad epigenetic changes or translocations that result in fusion oncoproteins. Here, we will review the developmental origins of childhood cancers, epigenetic dysregulation in tissue stem/precursor cells in numerous examples of childhood cancer oncogenesis and emerging therapeutic opportunities aimed at both cell-intrinsic and microenvironmental targets together with new insights into the mechanisms underlying long-term sequelae of childhood cancer therapy.
Childhood cancers represent the leading cause of disease-related morbidity and mortality in childhood, second only to accidents as a cause of pediatric death1 in the United States and other developed countries. Childhood cancers encompass leukemias, lymphomas, central nervous system tumors, sarcomas of bone and soft tissue, neuroblastoma, retinoblastoma, rhabdoid tumors, liver tumors, renal tumors, germ cell tumors and additional rare cancers. Revolutionary advances in next-generation sequencing technology together with rapidly increasing progress in chromatin and stem cell biology have ushered in a new molecular understanding of childhood cancer. Recent landmark sequencing studies have demonstrated that the mutational burden in most childhood cancers is substantially lower than that in adult cancers2,3. Fusion genes are more common than in adult cancers, and certain specific mutations found in pediatric cancers are rare in adult malignancies. Rather than numerous mutational ‘hits’ frequently observed in adult cancers, the emerging theme is that epigenetic dysregulation is central to many forms of childhood cancer. Furthermore, evidence from genetically engineered mouse models of childhood cancer suggest that many pediatric tumors originate from stem or progenitor cells during particular developmental time windows4–11. It is clear that these cells of origin must provide a transcriptional program permissive for the tumorigenic effects of a first genetic or epigenetic hit, which, as a consequence, distorts further cell divisions toward favoring self-renewal over differentiation. Thus, the developmental context of cells in which tumorigenic mutations occur and the microenvironment in which they form both underscore the unique biology of childhood cancer and the challenges of therapeutically targeting these cancers in actively developing tissues, requiring unique therapeutic approaches. This Review will discuss the principles of childhood cancer pathobiology and therapy, chiefly focusing on childhood leukemias, brain tumors, rhabdoid tumors and sarcomas to illustrate these principles.
Mechanisms of deregulation in childhood cancers
Common types of genetic alterations.
Not only is the rate of mutations and structural variants lower in childhood malignancies compared with adult cancer, but also the types of alterations and particular genes affected differ from adult cancers. Pediatric-cancer-driving point mutations are enriched in genes that encode epigenetic machinery and are largely specific to the diseases in which they arise2,3. Additionally, chromosomal fusion events that juxtapose oncogenes with gene partners that deregulate their proper activation or function are particularly prevalent among many types of childhood cancers (Supplementary Table 1). These fusion genes often activate genes crucial to development, such as the neurotrophic growth factor receptor family (NTRK) genes12,13. Another feature of pediatric cancers is that a rather high percentage of pediatric cancer patients (~8%)2 carry an unambiguous germline mutation that predisposes them to developing cancer. The extent to which cancer surveillance efforts may identify those at greater risk for childhood malignancy and whether any interventions could be used at the pre-malignant state remain under investigation.
Histone mutations.
Rather than the multiple genomic events that characterize oncogenesis in many adult cancers, a prominent feature of childhood cancers is epigenetic dysregulation, with malignancy attributed to broad dysregulation of gene expression. One striking example of this principle is the discovery of point mutations in histone genes that dysregulate histone post-translational modifications modulating gene expression14–16. The best-studied example of this is the histone-3 (H3)-K27M mutation that occurs in diffuse midline gliomas, such as diffuse intrinsic pontine glioma (DIPG) and thalamic and spinal cord gliomas of childhood17. In DIPG, the H3-K27M (rarely H3-K27I) mutation occurs in both canonical and non-canonical H3 genes encoding H3.3 and H3.1 and is found in ~80% of DIPG cases. While the mutation is always heterozygous, and so affects less than 15% of the total histone protein pool in a cell, a profound decrease in overall H3-K27 methylation is observed with the mutation18,19. The proposed mechanistic model is that the H3-K27M mutation leads to a global decrease in H3-K27 trimethylation by causing dysfunction of the methyltransferase activity of the catalytic enhancer of zeste homolog 2 (EZH2) subunit of polycomb repressive complex 2 (PRC2), preventing methylation of wild-type (WT) H3 at residue 27. The resulting derepression of gene expression then promotes gliomagenesis4,5,18,20 (Fig. 1). Oncogenesis is only thought to happen if the mutation occurs in the context of a cell in a susceptible transcriptional state, such as early neural or glial precursor cells4,5,20–22.
Fig. 1 |. Epigenetic dysregulation and therapeutic opportunities in midline gliomas with histone mutations.
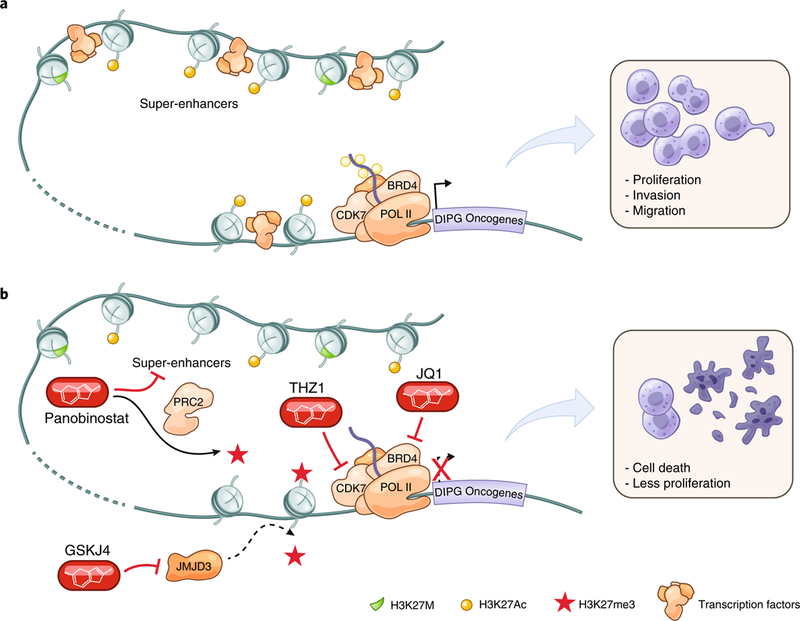
a, H3.3- or H3.1-K27M mutations (green histones) influence transcriptional regulation through global reduction of methylation (shown by red stars, which are absent in a) at position H3-K27 and global increases in acetylation (yellow circles) at H3-K27 (refs. 18,19). This alters local as well as distal gene regulation through super-enhancers and leads to de-repression of target oncogenes. b, Epigenetic modifiers panobinostat (HDAC inhibitor)87, GSKJ4 (histone lysine demethylase inhibitor)88 and JQ1 (bromodomain inhibitor)89,90 as well as transcriptional inhibitors like THZ1 (cyclin-dependent kinase 7 inhibitor)89 partially counteract the downstream effects of the histone mutation, including restoration of H3-K27 tri-methylation (red stars) by panobinostat and subsequent repression of target oncogenes87. POL II, RNA polymerase II. BRD4, bromodomain-containing protein 4. CDK7, cyclin-dependent kinase 7. PRC2, polycomb repressive complex II.
Aberrant patterns of H3-K27 methylation also occur in subgroups of pediatric ependymomas and medulloblastoma, although at a low frequency for the latter24,25. This convergence of genetic and epigenetic alterations of K27 in H3 variants in tumors of the hindbrain may point toward an important role of H3-K27 during normal development and tumorigenesis in the hindbrain. This point is further exemplified by posterior fossa ependymomas, discussed in more detail below.
Extending the role of ‘oncohistones’ to tumors outside of the hindbrain, the oncogenic histone mutations H3-K27M and H3-K27L are also found occasionally in acute myeloid leukemia (AML) in adolescents and adults23. Other histone mutations further illustrate spatio-temporal and tumor-type specificity: exclusive H3.3- H3.3-G34R and H3.3-G34V mutations occur in hemispheric pediatric glioblastomas, while H3-K36M mutations occur in chondroblastomas of young adulthood and human papillomavirus (HPV)-negative head and neck squamous cell carcinomas in older adults, and H3-G34W and H3-G34L mutations occur in giant cell tumors of the bone, chiefly affecting young adults26–28.
Epigenetic modifiers.
In addition to oncohistones, mutations in epigenetic modifiers such as histone modifying enzymes (for example, SETD2 and KDM6A) and chromatin complexes (for example, the BRG1-associated factors (BAF) complex and polycomb repressive complex 2 (PRC2)) have emerged as the most common and largest group of mutated genes in a recent pan-cancer analysis of pediatric cancers2,3. Notably, all types of pediatric leukemias (T cell acute lymphoblastic leukemia (T-ALL), B cell ALL and AML) exhibited prominent somatic mutations in genes with roles in epigenetic modification (present in 53.7%, 38.7% and 34%, respectively). Frequently mutated genes include those encoding histone methyl transferases SETD2 and MLL1, histone demethylase KDM6A, histone deacetylase HDAC2, chromatin complex BAF member ARID1A, histone deacetylase interactors BCORL1 and BCOR, and isocitrate dehydrogenase 1 and 2 (IDH½—when mutated, this produces abnormal metabolite 2-hydroxyglutarate, which inhibits histone and DNA demethylases)2,3,29–31.
CpG methylation.
In two large studies of pediatric tumors representing two dozen molecularly defined tumor types across over 2,500 cases2,3 analyzed by whole-genome sequencing (WGS), 10% of all pediatric tumors did not have any underlying mutation or structural/copy-number variant2,3. This suggests an additional, non-genetic layer of epigenetic dysregulation at the root of tumorigenesis. One example is a subtype of ependymoma occurring in young children with a very low rate of recurring DNA mutations32. These ‘genetically quiet’ ependymomas of the posterior fossa (posterior fossa group A ependymomas (PFAs)) exhibit CpG-island hypermethylation compared with other forms of ependymoma, implicating broad epigenetic alterations in their pathogenesis32,33. Intriguingly, PFAs also have global histone hypomethylation at H3-K27 (ref. 33), analogous to the abovementioned pediatric midline gliomas with a H3-K27M mutation. While the cause and mechanisms of this largely non-genetic regulation of DNA and histone methylation are not yet known, a recent study reported H3-K27M mutations in 4.2% of PFAs24, suggesting the convergence of pathogenic mechanisms involving H3-K27-regulated gene expression in hindbrain tumors.
PFA ependymomas might be an extreme illustration of locked-in developmental stages at the center of tumorigenesis. Recent work has demonstrated that altered chromatin landscapes and super-enhancers in ependymomas activate associated genes on which tumor cells depend for proliferation34 and underscores the need for new epigenetic drugs targeting these molecular alterations. The absence of genomic alterations in most cases of ependymomas also exemplifies the need for including novel molecular platforms (see Box 1), like DNA methylation array testing, in the diagnoses and stratification of these highly diverse subtypes of pediatric brain tumors35. DNA methylation arrays can also be used to precisely classify undifferentiated tumors, especially sarcomas36.
Box 1 |. Epigenetic diagnostics in the clinical lab
DNA sequencing and fusion testing of genes involved in epigenetic regulation (widely used in academic centers)
H3-K27M immunohistochemistry (widely used)
Histone posttranslational modification testing; e.g., K27me3 IHC33 (widely used)
CpG island methylation32 (less commonly used)
Large-scale DNA-methylation-based classification35 (currently used in select academic centers in Europe and the United States; becoming more common)
Box 2 |. Challenges in clinical trials in childhood cancer
Relatively rare diseases make enrollment of a sufficient number of subjects difficult at any single institution and geographical location, requiring multi-institutional collaboration
Randomized controlled trials are ideal, but given the small numbers of patients, this can be difficult; defining small but significant incremental improvements in outcome requires a prohibitively large number of patients
Trial designs often do not take heterogeneous molecular characteristics into account; cutting-edge molecular diagnostics are increasingly required for appropriate classification and subtype stratification
Detailed pharmacokinetic and pharmacodynamic assessments are often missing owing to small sample size (or not done as in blood-brain barrier penetrability assessment)
Clinical trials in pediatrics are generally limited to age 3–18 years or 3–21 years; however, pharmacokinetics, tolerability, metabolism, immune system and microenvironment can differ dramatically within this age range
Therapeutics are often developed for adult indications and re-purposed for pediatric diseases; therapeutics developed for the often unique pathophysiology of childhood cancers are in need
Monitoring cognitive and other long-term sequelae of childhood cancer therapies through development and into adulthood are particularly important for childhood cancer clinical studies
Developmental origins of childhood cancers
Leukemias.
Evidence indicates that the cellular context in which oncogenic mutations occur are developmental-stage-specific. The first demonstration that the initial genetic hit for a childhood cancer can arise in a cell of fetal origin was in childhood B-ALL. This cancer of the B lymphoid line of blood cells is the most common cancer of childhood. Chimeric fusion genes (lysine lethyltransferase 2A (KMT2A, also known as mixed-lineage leukemia 1 (MLL)) fusions to variety of partner genes and TEL-AML1 fusions) were found in monozygotic twins with concordant leukemia, while additional chromosomal abnormalities were subclonal and distinct in each twin37,38. Direct evidence for the prenatal origin of childhood leukemia was then provided by retrospectively examining clonal fusion gene sequences in archived neonatal blood spots—previously used for testing of metabolic disease and known as Guthrie cards—from individuals who later developed leukemia during childhood39. At the time of diagnosis, the corresponding archived neonatal blood spots had the same genetic fusion years before leukemia emerged. This was demonstrated in subjects who developed childhood ALL and carried the chimeric transcription factor fusion gene ETV6-RUNX1 or the chimeric chromatin modifier fusion gene MLL-AF4, and in about half of patients who developed childhood AML with chimeric transcription factor fusion gene AML1-ETO39–43. In an independent study of unselected cord blood of healthy children, the incidence of the ETV6-RUNX1 fusion was about 100-fold higher than the incidence of ETV6-RUNX1 fusion-positive B-ALL in children, suggesting that additional genetic events or a specific micro-environmental context are necessary to turn preleukemic cells into overt leukemia44.
Indeed, when the ETV6-RUNX1 fusion gene was introduced into human cord blood cells and transplanted into immune-compromised mice, a pool of pre-leukemic stem cells emerged that self-renewed and survived45, but this fusion gene alone was not sufficient to cause full leukemic transformation. All these findings have led to the development of a multihit model for ALL, in which gene fusions like ETV6-RUNX1 create a pre-leukemic pool when introduced in hematopoetic stem cells, but secondary cooperating events are required to develop overt leukemia. Recent work shows that this pre-leukemic cell pool can also be generated by introducing ETV6-RUNX1 into a developmentally restricted B cell progenitor unique to early embryonic life46.
Another disease for which a prenatal cell of mutation has been demonstrated is transient myeloproliferative disease (TMD) and the related acute megakaryoblastic leukemia in patients with Down syndrome/trisomy 21 (Down syndrome-AMKL or Down syndrome-AML D7). TMD is a disorder of the megakaryocyte lineage and occurs in about 5–10% of neonates with Down syndrome/trisomy 21. In most children, TMD resolves spontaneously; however, about 20–30% of these affected individuals present a few years later with DS-AMKL, an acute leukemia of the megakaryocyte linage47,48. Both of these diseases are thought to arise because of mutations in the hematopoetic transcription factor GATA binding protein-1 (GATA1) cooperating with as yet unknown genes on chromosome 21. One mouse study elegantly demonstrated that a mutated GATA1 allele leads to hyperproliferation of embryonic megakaryocyte precursor cells that first appear in the yolk sac and then move to the fetal liver. This hyperproliferation was only seen when the GATA1 mutation was introduced into prenatal cells, but the same mutation did not affect the proliferation of adult bone marrow megakaryocyte progenitors6. These findings might account for the restriction of both TMD and DS-AMKL to infants and young children, as mutations and cooperating overexpression of trisomy 21 genes might only be oncogenic during a short window of developmentally regulated transcriptional states.
The developmental origins of pediatric AML have also been described, especially for AMLs with fusions of histone methyl-transferase MLL. Initial work in mice has demonstrated that both self-renewing hematopoetic stem cells (HSCs) as well as short-lived myeloid progenitors could be successfully transduced with the highly conserved MLL fusion gene to result in rapid onset AML, supporting the evidence that cancer stem cells do not necessarily need to overlap with multipotent stem cells8. Other oncogenic fusions, like the MOZ-TIF2 fusion between the histone acetyltransferase MOZ and the nuclear receptor transcriptional activator TIF2, also induced overt leukemia when introduced into either HSCs or more committed progenitor cells in mice7.
The exact cell type in which a mutation occurs can influence the phenotype of the resulting malignancy. Gene expression analyses have demonstrated that leukemia stem cells generated from HSCs are more chemotherapy resistant and are more similar to poor-prognostic human AMLs as compared with leukemia stem cells generated from more committed progenitors, demonstrating that the exact cell of mutation can influence the phenotype of the resulting leukemia49. This suggests that the transcriptional/epigenetic state of the cell in which the initial mutation occurs can impact the overall clinical behavior of the resulting leukemia49, a principle that likely extends to other pediatric malignancies. Moreover, DNA methylation patterns differed in preclinical models of leukemias derived from HSCs versus committed progenitors and matched different human AML subtypes, again highlighting that the epigenetic state of the cell of mutation contributes to the final gene expression signature and phenotype of the resulting leukemia49. Additional work has also demonstrated that the MLL gene fusion partner results in cell type–preferential effects on the resulting lymphoid and myeloid malignancies50. This again suggests that different oncogenic hits require specific developmental and transcriptional cellular backgrounds to induce malignancies.
Another study extended this line of inquiry and demonstrated that HSC and myeloid progenitors can only be genetically transformed to leukemic blasts if the cells are able to transition to a more committed stage of differentiation after the introduction of the driver mutations in mouse models. If this differentiation step is genetically ablated, leukemogenic mutations no longer transform HSCs and myeloid progenitors. This elegantly demonstrates that the cells with the initial mutation might be either HSCs or myeloid progenitor cells, but full transformation appears to require progression to a more permissive, committed stage of differentiation, at least for the MLL-AF9 and MOZ-TIF2 fusion oncogenic programs51. Thus, the cell of tumorigenesis can be distinct from the cell in which the mutation occurs, i.e., the mutation occurs in a developmental state pre-tumorigenesis, a principle that appears to apply in many childhood cancers (Fig. 2). Like in ALL, the sequential order of mutations in defined cellular states appear to have an important role in AML: founder mutations in genes such as DNMT3A provide a clonal advantage to pre-leukemic HSCs, but need subsequent mutations, such as in NPM1, in downstream progenitor cells to drive progression to overt AML52.
Fig. 2 |. Developmental origins of childhood cancer.
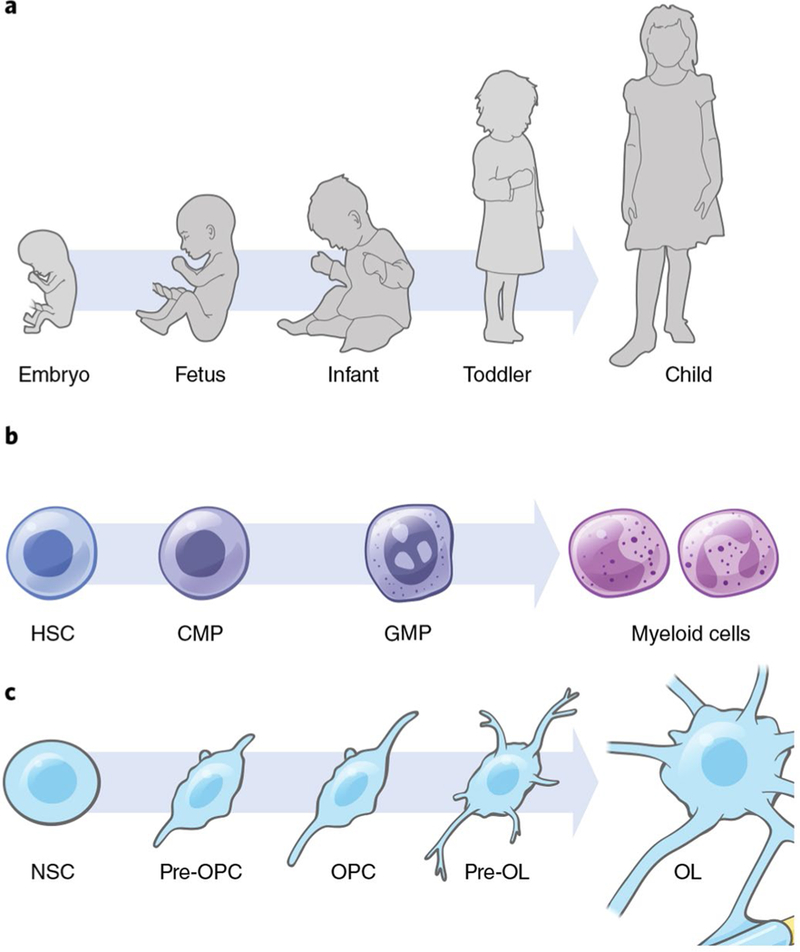
a, Progression of human development from embryo to childhood. Oncogenic mutations may occur in the prenatal period, but cancer only emerges at a later developmental time point. This principle has been well described in childhood leukemias. b, Myeloid differentiation, from HSC, to common myeloid progenitor (CMP), granulocyte monocyte precursor (GMP) and differentiated myeloid cells. The cell of initial mutation for AML is an earlier stem cell, while the AML cell of origin is a more differentiated precursor cell. c, Oligodendrocyte maturation, from neural stem cell (NSC) to OPC and mature myelinating oligodendrocyte (OL). Childhood gliomas may arise from precursors in the oligodendroglial lineage (pre-OPC or OPC). While the timing and cell of mutation for each pediatric glioma subtype is not yet clear, some evidence suggests that the key oncogenic mutations may occur at an earlier developmental time point .
Malignant rhabdoid tumors
Malignant rhabdoid tumors (MRTs) are highly aggressive tumors of the kidneys, soft tissues and central nervous system (CNS) that occur in young children and are largely without mutations—genetically quiet—except for germline or somatic mutations of SNF5 (also known as Ini1/BAF47/Smarcb1)53–55. SNF5 is part of the SWI-SNF chromatin remodeling complex and functions as a tumor suppressor in a broad range of developing tissues. While the homozygous deletion of SNF5 is embryonically lethal in mice, heterozygous deletions lead to early onset sarcomas, but not CNS tumors56–58. Consequently, the generation of rhabdoid tumors in the developing brain (also termed atypical teratoid/rhabdoid tumors (AT/RT)) seems to require deletion of both copies of SNF5 in more specific cell types and developmental contexts: when both copies of Snf5 were ablated in the population of mouse neural progenitor cells that express nestin, all mice died prenatally. However, when Snf5 was ablated in the population of neural progenitor cells marked by expression of GFAP instead, mice developed neurodegeneration, migration defects and seizures. Only when Snf5 deletion was combined with TP53 loss in the former population of neural precursor cells did mice develop highly aggressive brain tumors displaying many histopathological and clinical features of AT/RTs in this mouse model9.
A subsequent study expanded these findings by conditionally inactivating SNF5 at various time points in mice from early embryonic stages of development into adulthood. The timing of the inactivation had dramatic effects on the type and location of resulting cancers. Intra- and extra-cranial sarcomas and lymphomas developed in a manner dependent on the developmental timing of SNF5 inactivation. Furthermore, only early embryonic SNF5 inactivation between E6 and E10 induced fully penetrant, mainly intracranial rhabdoid tumors that closely resembled all transcriptional subclasses of human AT/RTs10, whereas SNF5 inactivation during slightly later embryonal stages (embryonic day 12 (E12)) did not lead to tumor formation10. TP53 inactivation was not required in this model and is not prevalent in the human disease. These findings suggest that the SNF5 mutation is tumorigenic only during a rather limited window during early embryonal brain development. These findings modeling rhabdoid tumors were further confirmed in a recent study11, in which SNF5 loss only induced rhabdoid tumor formation if deleted in neural crest cells during early embryogenesis (E9.5), but not when inactivated at slightly later time points on E12.5 or E13.5. As with the childhood leukemias discussed above, the mutational event and tumorigenesis appear to be temporally distinct in malignant rhabdoid tumors, with mutation occurring in the early prenatal stage and delayed tumor manifestation in the early postnatal period, at least as evident in mouse models. For further review of the literature on the prenatal origins of pediatric cancers, please see refs. 59,60.
Gliomas.
High-grade gliomas (HGG) of childhood and adolescence exhibit a striking spatio-temporal and spatio-molecular pattern of incidence. As discussed above, midline gliomas of the thalamus, pons and spinal cord exhibit a highly recurrent and specific mutation in H3 genes and occur chiefly in younger children. In contrast, high-grade gliomas of the cerebral lobes occur in adolescents and young adults, and, intriguingly, a subset exhibit a different specific mutation in the same H3 gene26,61. Broadly, these spatio-temporal patterns of gliomagenesis map onto developmental waves of myelination in the human central nervous system62,63; for detailed review, see Gibson et al.64. This correlation is concordant with the mounting evidence that high-grade gliomas of childhood may arise from precursor cells in the oligodendroglial lineage20,21,65. While midline tumors emerge during this period of active midline structure myelin development, it is not yet fully clear when the mutation(s) occur, or in what cell type.
Like other childhood cancers discussed above, the cell of mutation and the developmental period in which that mutation occurs may be distinct from the cell of cancer origin. Indeed, recent evidence from genetic mouse models suggests that the classic genomic alterations associated with midline gliomas only induce tumors in the postnatal period if introduced during prenatal brain development5, and, while neural stem cells can be transformed by introduction of these mutations in vitro, the resultant tumors do not recapitulate the typical histological appearance of diffuse midline gliomas when transplanted into the mouse brain4. A precedent exists in the adult glioblastoma (GBM) literature to support this model of tumorigensis in which the mutation occurs in the neural stem cell resulting in cancer originating from the more restricted neural precursor cell. In one genetic mouse model, GBM-associated mutations introduced into a neural stem cell give rise to a tumor only when the stem cell differentiates into an oligodendrocyte precursor cell (OPC), the neural precursor cell that gives rise to myelinating oligodendrocytes66. Further work suggests that GBMs can arise from both NSCs and OPCs, but the molecular subtype is determined by the cell of origin67. To fully elucidate the cell of mutation and cell of origin for diffuse midline glioma, future studies in which the mutations are introduced that characterize diffuse midline gliomas at various developmental time points and in various cellular contexts, from neural stem cell to pre-OPC to oligodendrocyte precursor cell, will be required.
Another interesting spatiotemporal aspect of pediatric gliomas are non-brainstem HGGs arising in infants and young children less than 3 years old. A large percentage of these tumors have a lower mutational burden than other pediatric HGGs; however, these infant HGGs frequently harbor fusions of different genes with the kinase domains of neurotrophic tyrosine kinase receptors 1–3 (NTRK 1–3). These fusions, though not unique to infant HGGs, showed strong oncogenic activity in experimental settings12. Underscoring their unique biology, the resulting infant tumors are associated with a significantly better prognosis than HGGs in older children68.
Neither spatio-temporal and spatio-molecular patterns in pediatric gliomas are yet fully understood. Specificity to the midline is also observed in other childhood cancers. In germ cell tumors, midline location is thought to be due to the midline route of germ cell progenitors during embryogenesis, and in chordoma due to the midline location of the embryonic notochord. While the developmental origins of germ cell tumors and chordoma are well explained by the anatomical position of their developmental origins, no such explanation is yet apparent for midline gliomas. Insights into the heterogeneity of oligodendroglial and other neural precursor cells may shed some light on these patterns of gliomagenesis. Alternatively, the patterns may be explained by an as-of-yet undetermined intersection of region-specific micro-environmental influences with these cell-intrinsic vulnerabilities. Future work in both normal neurodevelopment and glioma pathobiology will be required to elucidate this enigmatic pattern of childhood glioma incidence.
The microenvironment of actively developing tissues
Microenvironmental influences critically regulate cancer growth, and this principle is applicable to the rich growth environment of actively developing tissues. Intercellular interactions governing tissue development and growth are subverted to promote malignancy in childhood cancers that arise from the very tissue stem/precursor cell populations these mechanisms are meant to support. A clear example of this principle is found in the influence of neuronal activity on the proliferation of both oligodendrocyte precursor cells and pediatric glioma cells. Neuronal activity promotes robust proliferation of OPCs and pre-OPCs69 as part of an adaptive response of myelin-forming cells that influences myelin development70–72 and represents a mechanism of experience-dependent neural plasticity. Myelin plasticity in the healthy nervous system has implications for neurological functions such as motor performance69 and learning73 (for review, see ref. 74). In glial cancers, such as pediatric glioblastoma and H3-K27M-driven diffuse midline glioma, neuronal activity similarly promotes glioma proliferation, growth and progression through activity-regulated secretion of both known (brain-derived neurotrophic factor (BDNF)) and novel (neuroligin-3 (NLGN3)) growth factors75,76. Glial cancers exhibit a striking growth dependency on one of these activity-regulated mechanisms of glioma growth—activity-regulated cleavage and shedding of neuroligin-3 (NLGN3) from synapses76. Neuroligin-3 is a potent mitogen for glioma cells owing to stimulation of numerous oncogenic signaling pathways upon binding, but the reasons for this dramatic dependency76 remain to be fully clarified.
Neuronal activity is a robust regulator of normal and neoplastic stem cell niche in numerous tissues, important for organogenesis77, homeostasis, regeneration and malignancy (for in-depth discussion, see ref. 78). Nervous system activity influences a number of adult cancers arising in diverse tissue types such as the brain75 and also prostate, pancreas, colon, gastric and skin cancers79–82. The extent to which innervation regulates non–central nervous system childhood cancers remains a largely unexplored and rich area for future study.
Therapeutic approach to childhood cancers
Among the challenges (see Box 2) in developing better therapies for childhood cancers are clinical trial execution for relatively rare diseases. Successful establishment of cooperative childhood cancer clinical trial consortia, such as the international Children’s Oncology Group (COG), the international Societe Internationale D’Oncologie Pediatrique (SIOP) and more specialized groups like the US-based Pediatric Brain Tumor Consortium (PBTC), have facilitated efficient trial execution, allowing for enrollment of sufficient subjects in a reasonable timeframe. Incentivizing legislation in the United States, such as the Creating Hope Act (21 U.S.C. 360ff) and the RACE for Children Act (21 US Code 355c), have increased interest from and collaboration with the pharmaceutical industry to advance new therapies for childhood cancers.
Distinct mechanisms of disease require dedicated therapeutic discovery effects.
Successes in treating childhood malignancies such as ALL with a combination of multiple chemotherapeutic drugs and Wilms tumor with a combination of surgery, chemotherapy and sometimes radiation therapy illustrate the power of combination approaches. Effective therapy for presently intractable childhood cancers will likely require a combination of targeting cell-intrinsic vulnerabilities, microenvironmental dependencies and tumor cell–enriched or tumor cell–specific antigens for immunotherapy. Appreciating the molecularly defined and functionally important subtypes of each disease will enable stratification of therapies to best target such vulnerabilities and dependencies. For each of these therapeutic approaches, childhood cancer targets may be non-overlapping with those in adult malignancies because of differences in the cellular and molecular context in which these cancers arise and the unique or differentially robust growth programs in the microenvironment of actively developing tissues.
There are many examples of differences in the response of childhood cancers that appear to histologically mirror their adult cancer counterparts to therapies that are effective in these counterparts. For example, temozolomide, an alkylating agent that has demonstrated survival benefit for adult high-grade gliomas83, exhibits no clinical benefit for H3K27M+ diffuse midline gliomas of childhood84–86. However, the pathophysiology of the H3-K27M oncohistone mutation creates disease-specific therapeutic opportunities. As discussed above, these cells have H3-K27M-induced H3-K27 hypomethylation and resulting transcriptional dysregulation, meaning that H3-K27 hypomethylation87,88 or transcriptional dependencies89,90 may be targeted for glioma therapy (Fig. 1). Alternatively, an immunotherapeutic approach may be taken in which aberrantly high levels of particular cell surface antigens can be targeted whose overexpression is driven by aberrant epigenetic regulation91.
Epigenetic approaches.
It is sometimes the case that drugs developed for adult indications can be repurposed for childhood cancers, but in these cases the mechanism of action may be disease-specific. For example, the histone deacetylase (HDAC) inhibitor panobinostat, a Food and Drug administration (FDA)-approved drug for the adult hematological malignancy multiple myeloma, exhibits preclinical activity against diffuse midline glioma cells with mutant H3-K27M because, at least in part, of the restoration of H3-K27 methylation and subsequent normalization of gene expression87. This is unexpected as histone H3-K27 methylation represses gene expression, and histone deacetylation does also. The normalization of H3-K27 methylation is thought to occur possibly through steric hindrance of the interactions between the mutant methionine of H3-K27M-mutant histones and the PRC2 complex attributable to polyacetylation of nearby residues in the histone tail92, thereby preventing PRC2 dysfunction and enabling H3K27 methylation of wild-type histones in the cell87. This mechanism of action, unique to cancer cells with the H3-K27M mutation, is serendipitously achieved by a drug developed for an adult indication. Panobinostat is presently in phase I clinical trials for DIPG (NCT02717455, NCT03566199, NCT03632317) (Fig. 1). As our knowledge of childhood cancer biology increases, therapeutic development efforts can be increasingly focused on the unique pathophysiologies of childhood cancers.
Targeting fusion gene products.
Early clinical trials have demonstrated a promising response to targeted inhibition of fusion gene products that involve dysregulation of a kinase. Larotrectinib, a kinase inhibitor targeting gene fusions involving NTRK1, NTRK2 or NTRK3, has demonstrated safety and efficacy in treating NTRK-fusion-positive childhood cancers in a phase I/II clinical trial (NCT02637687) and has recently received FDA approval for use in NTRK-fusion-positive malignancies, regardless of tissue type or patient age. Targeting BRAF fusions using MEK inhibitors appears similarly promising in pediatric clinical trials93,94. More challenging to therapeutically target are oncogenic gene fusions involving transcription factors. For example, ~95% of Ewing’s sarcoma cases exhibit a fusion gene involving an RNA binding protein EWS and the ETS transcription factor FLI1 (ref. 95). Recent advances in the preclinical literature have identified a promising compound to target the EWS-FLI1 fusion gene product96, which may advance to the clinic in the future.
Immunotherapeutic approaches.
A shining example of successful development of a new and effective therapeutic targeting the unique biology of a childhood malignancy is the CD19-directed chimeric antigen receptor (CAR) T cell therapy, which has recently been approved by the FDA for refractory childhood leukemia97. This potentially transformational therapy is the result of focused development efforts for childhood leukemias, which, unlike adult leukemias, express high levels of CD19, and represents only the fourth agent to gain FDA approval for a pediatric cancer indication in US history. This clinical success, together with the exponentially increasing understanding of childhood cancer-specific pathophysiology, will hopefully encourage and enable similar development efforts in the biotechnology industry.
Microenvironmental therapies.
As noted above, nervous system activity can exert a robust growth-promoting influence on pediatric gliomas and other forms of cancer. Blocking the cleavage and release of the synaptic adhesion protein neuroligin-3 (NLGN3) into the tumor microenvironment causes a profound growth inhibition in preclinical models of a range of glioma types76, and this represents a promising new strategy to explore for therapy of these lethal childhood brain cancers. A clinical trial targeting NLGN3 release into the pediatric glioma microenvironment will be forthcoming. Similarly, rhabdomyosarcoma metastases in lung require receptive perivascular stromal cells and extracellular matrix factors for establishment98, highlighting microenvironmental targets to limit the spread of this childhood sarcoma and other cancers. A deeper understanding of tumor-stromal cell interactions that enable and promote cancer progression will uncover additional microenvironmental targets for childhood cancer therapy. Parallel studies of microenvironmental interactions mediating normal tissue development and plasticity will elucidate potential toxicities of such strategies and potentially ways to overcome unintended effects on healthy tissue development and homeostasis.
The effects of cancer therapy in developing tissues.
Both traditional and targeted cancer therapies induce important, lasting toxicities in normal tissues, and this is particularly true when administered during periods of active development and growth. Many cancer therapeutic modalities, such as radiation and cytotoxic chemotherapy, are designed to kill proliferating cells and hence induce lasting damage to diverse tissue-specific stem and progenitor cell populations that are necessary for growth, regeneration and plasticity. It is not surprising that therapies targeting cancers that originate from and molecularly resemble normal precursor cell populations would also be affected by cancer therapies. The lasting effects of therapy on normal tissues are multifactorial and include both direct effects on tissue stem/progenitor cells and indirect effects via disruption of the normal tissue microenvironment and through influences on the endocrine system. In the musculoskeletal system, long-term effects of childhood exposure to cancer therapies on bone growth and muscle development can cause short stature, reduced muscle bulk and skeletal deformities. These musculoskeletal effects are mediated both by direct effects of radiation, chemotherapy and oncological surgeries on developing bone, muscle and connective tissues and by indirect effects on these tissues through treatment-induced disruption of the endocrine system (for review, see ref. 99).
Neurological effects of childhood cancer therapy are particularly common and debilitating, and a syndrome characterized by impaired attention, concentration, memory function, multitasking and fine motor function affects a large fraction of children treated for childhood cancer100–102. This cancer therapy-related cognitive impairment syndrome is particularly severe following brain radiation, but occurs after systemic chemotherapy exposure as well.
The cellular and molecular underpinnings of this lasting neurological dysfunction are beginning to come to light. Radiation and chemotherapy cause an acute depletion of neural stem and progenitor cell populations103–105, cell populations broadly important for neural development, homeostasis, regeneration and plasticity. Following radiation of the hippocampus, a site of ongoing neurogenesis throughout life106–108, the neural stem cell compartment regenerates to baseline levels of precursor cell numbers in the subacute period, but the long-term self-renewal capacity of neural precursor cells (NPCs) is diminished and NPC function is severely impaired owing to radiation-induced perturbation of the neurogenic microenvironment109. Central to this dysfunction of the neurogenic microenvironment of the hippocampus is radiation-induced microglial inflammation (Fig. 3), which inhibits proper neuronal differentiation through inflammatory cytokine secretion107,109,110. Microglial depletion using a small molecule CSF1R inhibitor rescues memory function in a mouse model of radiation-induced cognitive impairment111,112. As microglial inflammation has recently been shown to induce a neurotoxic activation state of astrocytes113, intercellular interactions mediating neurological dysfunction after radiation exposure may be even more complex, as has been demonstrated after methotrexate chemotherapy114, discussed below (Fig. 3). Both microglia and astrocytes contribute to pruning synapses115,116, and decreased dendritic spine density—potentially indicating exuberant synapse engulfment (Fig. 3)—has been observed following radiation111 and is rescued by microglial depletion112. Adding an additional layer of complexity, recent work indicates that bone marrow-derived monocytes contribute to repair mechanisms after radiation, so craniospinal radiation and chemotherapy-induced injury to hematological precursor cell populations and the bone marrow niche may further exacerbate radiation-induced neurological injury117.
Fig. 3 |. Microglial inflammation is central to childhood cancer-therapy-related cognitive impairment.
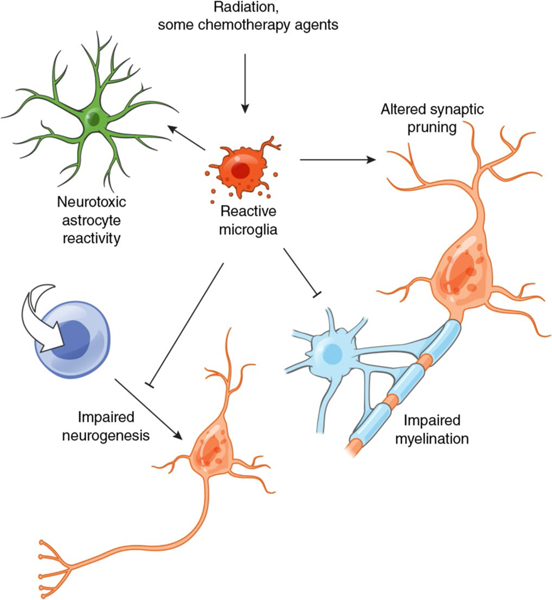
Childhood cancer therapies, such as cranial radiation107,109,110, methotrexate114 and cyclophosphamide122, induce persistent microglial reactivity that in turn induces neurotoxic astrocyte reactivity114, disrupts oligodendroglial lineage dynamics and impairs myelination114, blocks neuronal differentiation of hippocampal stem cells and thereby disrupts hippocampal neurogenesis107,109,110 and contributes to aberrantly exuberant pruning of synapses111,123. Strategies to modulate microglia, such as CSF1R inhibition111,112,114, represent an important therapeutic opportunity for cancer-therapy-related cognitive impairment.
Neural precursor cells are exquisitely sensitive to multiple forms of chemotherapy, and damage to precursor cells in the oligodendroglial lineage may be particularly severe105,118. Indeed, children suffering from chemotherapy-induced neurological dysfunction exhibit evidence of subtle white matter injury on diffusion tensor magnetic resonance imaging119,120. Oligodendroglial precursor cell populations normally exhibit robust regenerative capacity121, but recent work has demonstrated lasting depletion of white matter oligodendrocyte precursor cells long after chemotherapy exposure in both humans and a mouse model of methotrexate-chemotherapy-related cognitive impairment114. Following methotrexate, the oligodendroglial lineage is dysregulated, with depletion of both precursor cells and mature, myelinating oligodendrocytes and accumulation of oligodendroglial cells in an intermediate state of differentiation. Like radiation-induced neural precursor cell dysregulation, perturbation of the brain microenvironment after remote methotrexate chemotherapy underlies this persistent dysregulation of the oligodendroglial lineage114. Direct activation of white matter microglia by methotrexate is central to this pathophysiology (Fig. 3) and microglial depletion immediately following methotrexate exposure using a systemically delivered small-molecule CSF1R inhibitor normalizes neurotoxic astrocyte reactivity, oligodendroglial lineage dynamics and myelination and rescues cognitive behavioral impairment following methotrexate chemotherapy114. Future work is required to define the therapeutic window for microglial depletion strategies, the state of repopulating microglia following such depletion strategies and complementary or alternative therapeutic strategies. In addition, the similarities and differences in microglial reactivity following exposure to different chemotherapeutic and immunotherapeutic agents remain to be explored. Microglial activation has been reported in response to other chemotherapeutic agents, including cyclophosphamide122, but was not observed following 5-FU exposure118. Our understanding of the long-term impact of cancer therapy exposure on the gliogenic and neurogenic microenvironment and its lasting influence on precursor cell population dynamics is presently incomplete and requires further study. However, an emerging concept from numerous studies in diverse models indicates that therapy-associated microglial reactivity is central to therapy-induced neuropathophysiology (Fig. 3) and may represent an important target for strategies to mitigate or even prevent cancer-therapy-related cognitive impairment.
Conclusion and future directions
In summary, there are mechanistic themes unique to childhood cancer, regardless of the tissue of origin, which warrant distinct therapeutic approaches compared to adult cancer. The genomic landscape of childhood cancer is different, as mutational burden is lower and structural variants are less common in childhood cancer than in adult cancer. Additionally, driver point mutations most frequently occur in epigenetic modifiers, and, together with oncogenic fusion events, tend to be specific to individual cancer types (and sometimes even specific to anatomical locations). Epigenetic alterations not only appear to play an important role in terms of frequently found mutations but might also reflect a locked-in epigenetic state during development, which contributes to the final gene expression and phenotype of the resulting cancers. Moreover, the developmental contexts in which mutations occur and the exact timing of these mutations appear fundamentally important: the often-observed time delay between first mutations and tumor emergence, with certain initial events even occurring prenatally, points toward an important difference between the ‘cell of mutation’ and the ‘cell of origin.’ Inherent in this concept is the need to better understand the microenvironmental factors influencing cell state during development. Future efforts toward mechanistic understanding and ultimately therapy for childhood cancers will therefore need to consider various pre- and postnatal developmental stages and their specific microenvironmental and transcriptional states. Parallel efforts to understand the complex mechanisms through which childhood cancer therapies disrupt normal tissue development, homeostasis and plasticity will elucidate strategies to restore and prevent long-term sequelae of childhood cancer therapy.
Supplementary Material
Acknowledgements
The authors extend special thanks to S. Knemeyer for illustrations, to A. Groves for help with Supplementary Table 1 and to K. E. Warren for helpful input on Box 2, Challenges in clinical trials in childhood cancer. The authors acknowledge funding from the Career Award for Medical Scientist from Burroughs Wellcome Fund (M.G.E), Solving Kids’ Cancer (M.G.F.), The Cure Starts Now Foundation and DIPG Collaborative (M.G.F. and M.M.), National Institutes of Neurological Disorders and Stroke (R01NS092597 to M.M.), National Institutes of Health (DP1NS111132 to M.M.), Abbie’s Army (M.M.), McKenna Claire Foundation (M.M.), Unravel Pediatric Cancer Foundation (M.M.), Alex’s Lemonade Stand Foundation (M.G.F. and M.M.), Maternal and Child Health Research Institute at Stanford (M.M.) and the Anne T. and Robert M. Bass Endowed Faculty Scholarship in Pediatric Cancer and Blood Diseases (M.M.).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-019-0383-9.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutra with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Grobner SN et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Ma X et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Funato K, Major T, Lewis PW, Allis CD & Tabar V Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathania M et al. H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell. 32, 684–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z et al. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat. Genet. 37, 613–619 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Huntly BJ et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 6, 587–596 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Cozzio A et al. Similar MLL-associated leukemias arising from self- renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng JM et al. Generation of a mouse model of atypical teratoid/rhabdoid tumor of the central nervous system through combined deletion of Snf5 and p53. Cancer Res. 75, 4629–4639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han ZY et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 7, 10421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitte J, Gao F, Coppola G, Judkins AR & Giovannini M Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat. Commun. 8, 300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu G et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lannon CL & Sorensen PH ETV6-NTRK3: a chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 15, 215–223 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Wu G et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Khuong-Quang DA et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 124, 439–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louis DN et al. The2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Lewis PW et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bender S et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 24, 660–672 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monje M et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. NatlAcad. Sci. USA 108, 4453–4458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larson JD et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell. 35, 140–155.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehnertz B et al. H3 K27M/I mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 130, 2204–2214 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Pajtler KW et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubuc AM et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 125, 373–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sturm D et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 22, 425–437 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Behjati S et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 45, 1479–1482 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mar BG et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 5, 3469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson AK et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 25, 1570–1577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayton PM & Cleary ML Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 20, 5695–5707 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Mack SC et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayliss J et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med. 8, 366ra161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mack SC et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koelsche C et al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod. Pathol. 31, 1246–1256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ford AM et al. In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 363, 358–360 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Greaves MF, Maia AT, Wiemels JL & Ford AM Leukemia in twins: lessons in natural history. Blood 102, 2321–2333 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Gale KB et al. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl Acad. Sci. USA 94, 13950–13954 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiemels JL et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 354, 1499–1503 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Wiemels JL et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood 99, 3801–3805 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Hjalgrim LL et al. Presence of clone-specific markers at birth in children with acute lymphoblastic leukaemia. Br. J. Cancer 87, 994–999 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McHale CM et al. Prenatal origin of TEL-AML1-positive acute lymphoblastic leukemia in children born in California. Genes Chromosom. Cancer 37, 36–43 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Mori H et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl Acad. Sci. USA 99, 8242–8247 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong D et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 319, 336–339 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Boiers C et al. A human IPS model implicates embryonic B-myeloid fate restriction as developmental susceptibility to B acute lymphoblastic leukemia-associated ETV6-RUNX1. Dev. Cell 44, 362–377.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hitzler JK & Zipursky A Origins of leukaemia in children with Down syndrome. Nat. Rev. Cancer 5, 11–20 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Crispino JD GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr. Blood. Cancer 44, 40–44 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Krivtsov AV et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 27, 852–860 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W et al. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood 108, 669–677 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye M et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 17, 611–623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shlush LI et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sévenet N et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum. Mol. Genet. 8, 2359–2368 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Versteege I et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Judkins AR et al. INI1 protein expression distinguishes atypical teratoid/ rhabdoid tumor from choroid plexus carcinoma. J. Neuropathol. Exp.Neurol. 64, 391–397 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Guidi CJ et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol. Cell. Biol. 21, 3598–3603 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klochendler-Yeivin A et al. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 1, 500–506 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD & Orkin SH Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc. Natl Acad. Sci. USA 97, 13796–13800 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marshall GM et al. The prenatal origins of cancer. Nat. Rev. Cancer 14, 277–289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maris JM & Denny CT Focus on embryonal malignancies. Cancer Cell 2, 447–450 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Mackay A et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yakovlev PI The myelogenetic cycles of regional maturation of the brain in Regional Development of the Brain in Early Life (ed. Minkowski A) 3–70 (Blackwell Scientific Publications, Oxford, 1967). [Google Scholar]
- 63.Lebel C et al. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage 60, 340–352 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Gibson EM, Geraghty AC & Monje M Bad wrap: Myelin and myelin plasticity in health and disease. Dev. Neurobiol. 78, 123–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tate MC et al. Postnatal growth of the human pons: a morphometric and immunohistochemical analysis. J. Comp. Neurol. 523, 449–462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu C et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alcantara Llaguno SR et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 28, 429–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qaddoumi I, Sultan I & Gajjar A Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the Surveillance, Epidemiology, and End Results database. Cancer 115, 5761–5770 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gibson EM et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mensch S et al. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 18, 628–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baraban M, Mensch S & Lyons DA Adaptive myelination from fish to man. Brain Res. 1641, 149–161 (2016). Pt A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hines JH, Ravanelli AM, Schwindt R, Scott EK & Appel B Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci. 18, 683–689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKenzie IA et al. Motor skill learning requires active central myelination. Science 346, 318–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mount CW & Monje M Wrapped to adapt: experience-dependent myelination. Neuron 95, 743–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Venkatesh HS et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Venkatesh HS et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knox SM et al. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 329, 1645–1647 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Venkatesh H & Monje M Neuronal activity in ontogeny and oncology. Trends Cancer 3, 89–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Magnon C et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Stopczynski RE et al. Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res. 74, 1718–1727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hayakawa Y et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 31, 21–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peterson SC et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 16, 400–412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stupp R et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 84.Cohen KJ et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro-oncol. 13, 410–416 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jalali R et al. Prospective evaluation of radiotherapy with concurrent and adjuvant temozolomide in children with newly diagnosed diffuse intrinsic pontine glioma. Int. J. Radiat. Oncol. Biol. Phys. 77, 113–118 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Lashford LS et al. Temozolomide in malignant gliomas of childhood: a United Kingdom Children’s Cancer Study Group and French Society for Pediatric Oncology Intergroup Study. J. Clin. Oncol. 20, 4684–4691 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Grasso CS et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hashizume R et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 20, 1394–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagaraja S et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Piunti A et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mount CW et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 24, 572–579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brown ZZ et al. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J. Am. Chem. Soc. 136, 13498–13501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Banerjee A et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-oncol. 19, 1135–1144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kondyli M et al. Trametinib for progressive pediatric low-grade gliomas. J. Neurooncol. 140, 435–444 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Delattre O et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 331, 294–299 (1994). [DOI] [PubMed] [Google Scholar]
- 96.Zöllner SK et al. Inhibition of the oncogenic fusion protein EWS-FLI1 causes G2-M cell cycle arrest and enhanced vincristine sensitivity in ‘Ewing’s sarcoma. Sci. Signal. 10, eaam8429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Murgai M et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 23, 1176–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gawade PL et al. A systematic review of selected musculoskeletal late effects in survivors of childhood cancer. Curr. Pediatr. Rev. 10, 249–262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Green JL, Knight SJ, McCarthy M & De Luca CR Motor functioning during and following treatment with chemotherapy for pediatric acute lymphoblastic leukemia. Pediatr. Blood Cancer 60, 1261–1266 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Ellenberg L et al. Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology 23, 705–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pierson C, Waite E & Pyykkonen B A meta-analysis of the neuropsychological effects of chemotherapy in the treatment of childhood cancer. Pediatr. Blood Cancer 63, 1998–2003 (2016). [DOI] [PubMed] [Google Scholar]
- 103.Parent JM, Tada E, Fike JR & Lowenstein DH Inhibition of dentate granule cell neurogenesis with brain irradiation does not prevent seizure-induced mossy fiber synaptic reorganization in the rat. J. Neurosci. 19, 4508–4519 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tada E, Yang C, Gobbel GT, Lamborn KR & Fike JR Long-term impairment of subependymal repopulation following damage by ionizing irradiation. Exp. Neurol. 160, 66–77 (1999). [DOI] [PubMed] [Google Scholar]
- 105.Dietrich J, Han R, Yang Y, Mayer-Pröschel M & Noble M CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J. Biol. 5, 22 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eriksson PS et al. Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 (1998). [DOI] [PubMed] [Google Scholar]
- 107.Monje ML et al. Impaired human hippocampal neurogenesis after treatment for central nervous system malignancies. Ann. Neurol. 62, 515–520 (2007). [DOI] [PubMed] [Google Scholar]
- 108.Spalding KL et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Monje ML, Mizumatsu S, Fike JR & Palmer TD Irradiation induces neural precursor-cell dysfunction. Nat. Med. 8, 955–962 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Monje ML, Toda H & Palmer TD Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765 (2003). [DOI] [PubMed] [Google Scholar]
- 111.Feng X et al. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuro-inflammation. 13, 215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng X, Liu S, Chen D, Rosi S & Gupta N Rescue of cognitive function following fractionated brain irradiation in a novel preclinical glioma model. e Life. 7, e38865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gibson EM et al. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 176, 43–55.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schafer DP et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chung WS et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dietrich J et al. Bone marrow drives central nervous system regeneration after radiation injury. J. Clin. Invest. 128, 281–293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Han R et al. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J. Biol. 7, 12 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morioka S et al. Effects of chemotherapy on the brain in childhood: diffusion tensor imaging of subtle white matter damage. Neuroradiology 55, 1251–1257 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Edelmann MN et al. Diffusion tensor imaging and neurocognition in survivors of childhood acute lymphoblastic leukaemia. Brain 137, 2973–2983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hughes EG, Kang SH, Fukaya M & Bergles DE Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16, 668–676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Acharya MM et al. Stem cell transplantation reverses chemotherapy-induced cognitive dysfunction. Cancer Res. 75, 676–686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Andres AL, Gong X, Di K & Bota DA Low-doses of cisplatin injure hippocampal synapses: a mechanism for chemo’ brain? Exp. Neurol. 255, 137–144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.