

SUMMARY
The heavy occupancy of transposons in the genome implies that existing organisms have survived from multiple, independent rounds of transposon invasions. However, how and which host cell types survive the initial wave of transposon invasion remains unclear. We show that the germline stem cells can initiate a robust adaptive response that rapidly endogenizes invading P-element transposons by activating the DNA-damage checkpoint and piRNA production. We find that temperature modulates the P-element activity in germline stem cells, establishing a powerful tool to trigger transposon hyper-activation. Facing vigorous invasion, Drosophila first shut down oogenesis and induce selective apoptosis. Interestingly, a robust adaptive response occurs in ovarian stem cells through activation of the DNA-damage checkpoint. Within 4 days, the hosts amplify P-element-silencing piRNAs, repair DNA damage, subdue the transposon, and reinitiate oogenesis. We propose that this robust adaptive response can bestow upon organisms the ability to survive recurrent transposon invasions throughout evolution.
INTRODUCTION
Repeated cycles of transposon invasion and silencing have allowed mobile genetic elements to accumulate in the genomes of nearly all organisms (Levin and Moran, 2011). From invertebrates to mammals, transposon invasion triggers catastrophic genomic instability, greatly reducing the viability or fertility of the invaded animals and leading to population crisis (Kidwell et al., 1977; O’Neill et al., 1998; Tarlinton et al., 2006). Given the abundance of transposons in the genome, all organisms are survivors from thousands, if not millions, of rounds of transposon invasions. With the unique role of maintaining the species during evolution, germ cells in existing organisms must have an adaptability to transposon invasions. However, we know very little about how host germ cells initially respond to the appearance of an invading element, which finally leads to transposon endogenization and species survival.
The P-element transposon rapidly swept through all wild type Drosophila melanogaster populations around eight decades ago (Engels, 1997). Vertical transmission of P-elements in wild Drosophila populations is a classic model to study transposon endogenization in the germline (Brennecke et al., 2008; Engels, 1997; Engels and Preston, 1979; Khurana et al., 2011; Kidwell et al., 1977; Kidwell and Novy, 1979). When male flies carrying P-elements mate with females lacking them, their progeny fail to silence P-elements, resulting in sterility (Kidwell et al., 1977). The reciprocal cross–a P-element-containing female mated with a P-element-devoid male–yields normal, fully fertile and P-element-silenced offspring, despite having the same genomic material as invaded flies. The directionality of dysgenic mating likely reflects the absence of maternal P-element-silencing piRNAs in the eggs of P-element-devoid mothers. These piRNAs, in eggs from the reciprocal cross, protect progeny from P-element activation (Brennecke et al., 2008). Although it appears that invaded progeny can ultimately silence P-elements by producing corresponding piRNAs (Khurana et al., 2011), it is still unclear which cells are responsible for the endogenization process and what is the initial response from those cells that leads to species survival. Compared with other developmental stages, oogenesis can be examined in great detail in adult flies (King et al., 1968; Xie and Spradling, 2000), making the adult ovary an ideal system to study the host response upon invasion. However, routinely modeling P-element invasion in the laboratory results in rudimentary ovaries in adult flies that contain very few, if any, germ cells (Brennecke et al., 2008; Khurana et al., 2011; Kidwell et al., 1977), impeding the progress on understanding the process of transposon endogenization.
For the invading cross, the environmental temperature influences the severity of sterility phenotypes (Engels and Preston, 1979; Kidwell and Novy, 1979). While invaded progeny reared at 25°C have rudimentary ovaries, progeny reared at 18°C possess fully developed, fertile ovaries. This phenomenon suggests that temperature modulates the activity of P-element, therefore it may serve as a powerful tool to adjust the intensity of P-element invasion. To establish this “tool box”, we precisely quantified the rates of P-element mobilization at different temperatures, and found that P-elements mobilize at a moderate rate at 18°C. This low rate of transposition does not affect oogenesis, and flies retain seemingly normal fertility. However, P-element increases its transposition rate by at least 7-fold in germline stem cells at 25°C and results in host sterility, clarifying the effect of temperature on the fertility of invaded offspring.
Based on the above findings, we used temperature switching as a tool to adjust the intensity of P-element invasion, and investigated how adult flies respond to vigorous transposon invasion in their ovaries, in which oogenesis can be examined in detail. We found that germline stem cells employ an adaptive response to rapidly tame invading elements by activating the DNA damage checkpoint and piRNA production. Upon robust invasion, undifferentiated germ cells activate the DNA damage checkpoint to arrest differentiation and promote apoptosis. Intriguingly, the arrest period, which is essential for adaptation, allows surviving germ cells to amplify P-element-silencing piRNAs. This, in turn, permanently silences the invading transposons and allows rapid resumption of oocyte production within four days. Mutations that remove the DNA damage checkpoint overcome germ cell arrest, but do not permit the invaded flies to endogenize invading transposons and regain fertility. We propose that the ability of germline stem cells to rapidly adapt and restore fertility via the activation of DNA damage signaling can bestow upon species the aptitude to resist the population crash caused by newly introduced transposons.
RESULTS
P-element transposon invasion
For the progeny from the P-element invading cross, the effect of temperature on their fertility, which was reported four decades ago (Engels and Preston, 1979; Kidwell and Novy, 1979), suggests that temperature modulates the activity of P-element. We first replicated the reported transposon invasion experiments by mating w1 females (a strain lacking P-elements) with Harwich (Har) males (a strain that contains 128 potentially active P-elements, Table S1) to give birth to the dysgenic F1 progeny (henceforth termed “invaded progeny”; Figure 1A). The reciprocal cross, w1 males mated with Har females, served as the noninvasive control to produce genetically identical but P-element-silenced F1 reciprocal offspring (hereinafter termed “protected progeny”; Figure 1A). Because incubation at 18°C slows fly oogenesis, protected progeny lay one-fifth as many eggs per day as at 25°C (14 versus 61 eggs/female/day, respectively; Figure 1B). As previously reported (Engels and Preston, 1979; Kidwell and Novy, 1979), invaded progeny are sterile at 25°C (0.2 eggs/female/day) but fertile at 18°C (12 eggs/female/day). Eggs laid by invaded progeny at lower temperature hatched essentially as often as eggs from protected progeny (> 87%; Figure 1B). We conclude that, for invaded progeny, the severity of sterility phenotypes depends on temperature: while 18°C serves as a permissive temperature to give fully developed normal ovaries, the restrictive 25°C leads to rudimentary ovaries and low fertility.
Figure 1. P-element transposon invasion.
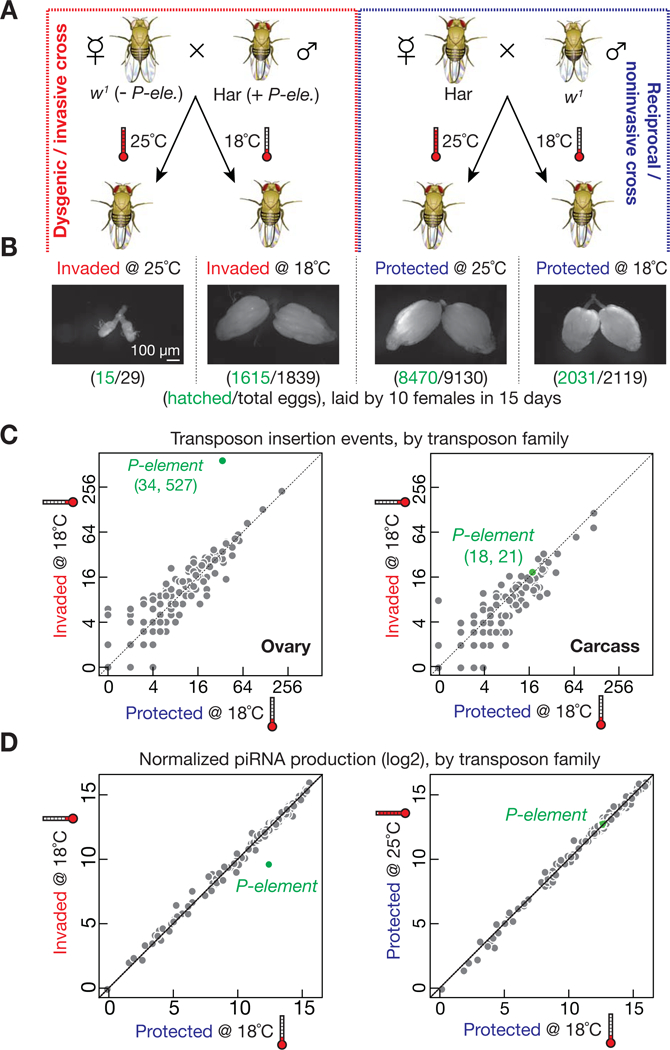
(A) Schematic diagram of experimental design. Hereinafter, the progeny from dysgenic/invasive cross are referred to as “Invaded”; the P-element-silenced offspring from noninvasive (reciprocal) cross are shortened as “Protected”.
(B) Ovarian morphology and fertility of invaded and protected progeny. Invaded flies have normal size ovaries at 18°C, but rudimentary ovaries at 25°C.
(C) Genome sequencing to globally probe new transposition events in invaded and protected progeny. Each dot represents one transposon family.
(D) Small RNA sequencing to detect the production of piRNAs mapped to transposon. Each dot represents one transposon family. Only piRNAs targeting P-elements (green dots) decreased in invaded progeny, compared with protected controls.
See also Figure S1.
It is possible that P-element activity is absent at 18°C, thus explaining how the flies maintain seemingly normal fertility. To test this, we quantified the P-element mobilization rate at 18°C, by employing paired-end genome sequencing and computational analysis to detect transposition events (Zhuang et al., 2014). We used DNA from early stage F1 embryos (0–3 hours after egg laying) as a reference to define how many new insertions occurred during the development of F1 adults (Figure S1A). In somatic cells, the P-element Somatic Inhibitor (PSI) protein blocks removal of intron 3 of the P-element, yielding an mRNA encoding an inactive transposase (Ignjatovic et al., 2005; Labourier et al., 2002; Siebel et al., 1995; Siebel et al., 1994). To estimate the rate of false-positive insertion, we also sequenced genomic DNA from the corresponding somatic carcasses that remained after ovary dissection (Figure S1A). With 20-fold genome coverage, we detected only 21 new insertions in the carcasses of invaded progeny and 18 in the protected progeny-derived carcasses (Figure 1C). These potentially new P-element insertions set an upper limit to the false-positive rate. For protected ovaries at 18°C, the 34 potentially new P-element insertions can be explained by the false-positive rate. In contrast, invaded ovaries gained 527 new insertions (Figure 1C). P-elements prefer to target ~100 genomic loci (Spradling et al., 2011). Of the new P-element insertion sites in invaded ovaries, a large fraction, 19%, corresponds to these hotspots, as expected.
Supporting the finding that P-elements indeed mobilize in invaded ovaries at 18°C, P-element total RNA was 3-fold greater in these ovaries than protected controls, as measured by quantitative RT-PCR (qRT-PCR; Figure S1B), Strand-specific paired-end RNA sequencing (RNA-Seq; Figure S1C), and single-molecule RNA fluorescent in situ hybridization (RNA-FISH; Figure S1D). Since the splicing of the last intron is essential to generate the mRNA isoform encoding the active transposase, we used isoform-specific qRT-PCR to measure the abundance of the active P-element mRNA isoform. Invaded ovaries produced 31-fold more spliced mRNA than protected controls at 18°C (P-value = 0.0001; Figure S1E). In animal germ cells, piRNAs silence transposons to maintain animal fertility (Aravin et al., 2006; Aravin et al., 2001; Girard et al., 2006; Grivna et al., 2006; Lau et al., 2006; Saito et al., 2006; Vagin et al., 2006; Weick and Miska, 2014). Consistently, we found that P-element-derived piRNAs were ~8-fold less abundant in ovaries from 2–4 day-old invaded progeny reared at 18°C compared to the age-matched protected progeny (Figure 1D). Together, our data suggest that at 18°C, P-elements mobilize in invaded ovaries, but the intensity of transposon invasion is not high enough to interfere with fly oogenesis, and the hosts maintain seemingly normal fertility.
Vigorous invasion leads to the rebirth of oogenesis
In contrast to 18°C, P-element invasion at 25°C leads to rudimentary ovaries and host sterility (Figure 1B), indicating that P-elements are much more active at higher temperature. By designing a high sensitivity assay that can monitor P-element mobilization with single-cell resolution, we validated this hypothesis and established that temperature-switching serves as a powerful tool to adjust the intensity of P-element invasion (see below). Accordingly, we designed a temperature-shift scheme to investigate how the hosts respond to vigorous transposon invasion in the adult ovary, in which the entire oogenesis process can be precisely characterized. Under this scheme, the hosts were reared at 18°C until the adult stage to ensure normal ovarian development, then newly eclosed adult flies were shifted to 25°C to induce vigorous invasion (Figure 2A). The temperature switch itself appears to have no effect on oogenesis: the control protected progeny maintained normal ovary morphology and fertility for 15 days after the temperature-shift (Figure 2B and 2C). In contrast, shifting invaded progeny to 25°C resulted in fertility defects after 4 days. These flies had small ovaries and laid few eggs, few of which hatched (Figure 2B and 2C). Intriguingly, these defects resolved ~9 days after the temperature-shift: invaded progeny showed normal ovary morphology, egg laying, and hatch rates indistinguishable from age-matched, protected control flies (Figure 2B and 2C).
Figure 2. Vigorous invasion leads to rebirth of fly oogenesis.
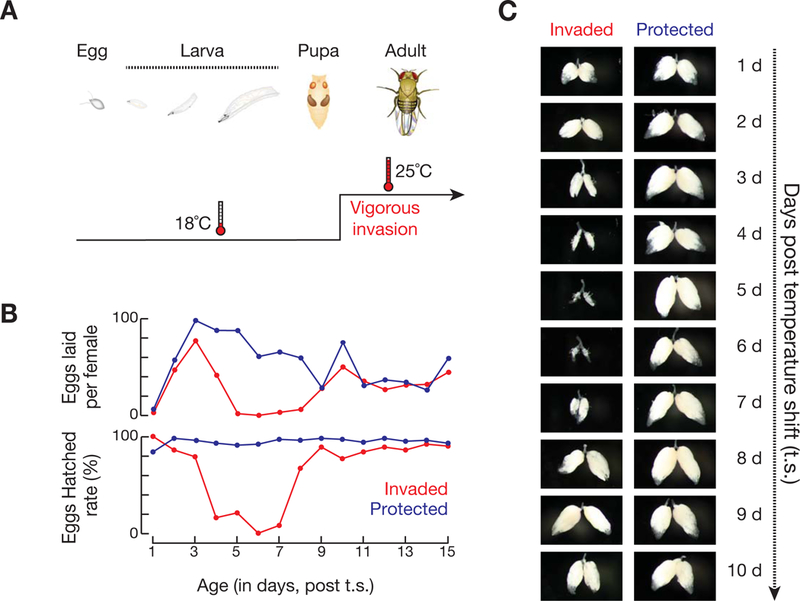
(A) Schematic diagram of the temperature-shift design.
(B) Fertility of invaded and protected female flies after the temperature-shift/vigorous P-element invasion.
(C) Ovarian morphology per day after vigorous P-element invasion.
Invading transposons are silenced in the recovered ovaries
To determine whether the recovered ovaries achieve transposon silencing, we measured P-element activity at both transcriptional and transposition levels. While P-element transcripts were initially abundant in ovaries prior to recovery, the abundance of transcripts in recovered ovaries dropped to similarly low levels as protected progeny (Figure 3A and S2A). Sequencing ovarian DNA further confirmed that P-elements are shackled in the genome of recovered ovaries (Figure S2B and S2C). With ~37-fold genome coverage, the number of P-element insertion events significantly decreased from 629 prior to recovery to 281 after recovery on average (P-value = 0.0018; Figure S2B and S2C). In fly ovaries, piRNAs silence transposons by coating them with the repressive histone mark, H3 lysine 9 trimethylation (H3K9me3) (Huang et al., 2013; Le Thomas et al., 2013; Rozhkov et al., 2013; Sienski et al., 2012). We found that this repressive mark doubled on P-elements when they were silenced in recovered ovaries, compared with younger siblings before recovery (Figure 3B). Accordingly, the amount of P-element-silencing piRNAs dramatically increased in recovered flies (Figure 3C). Except for P-element, the piRNA level for other transposon families stayed the same after recovery (Figure S2D), suggesting that the invasion-recovery process only affects the production of P-element-silencing piRNAs. Overall, these data indicate that adult flies have robust ability to rapidly silence invading transposons upon vigorous invasion.
Figure 3. P-elements are silenced in the recovered ovaries.
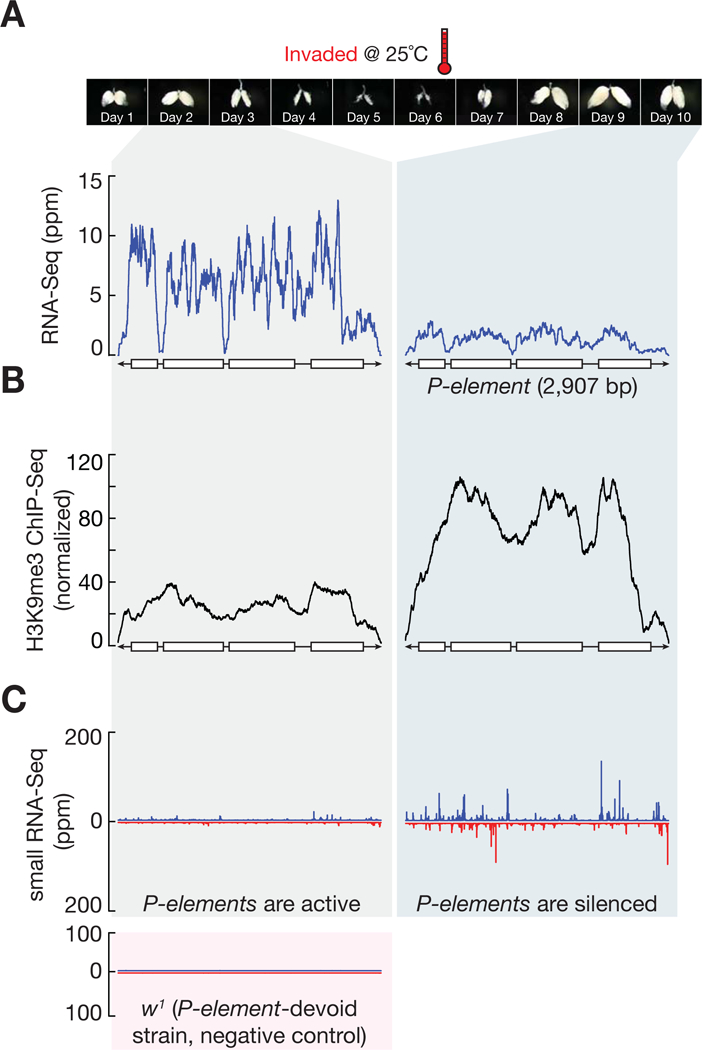
(A) RNA-Seq profiles for P-element from invaded progeny.
(B) H3K9me3 occupancy on P-element, as measured by H3K9me3 ChIP-Seq.
(C) Small RNA-Seq assay to quantify the production of P-element piRNAs. Blue, sense piRNAs. Red, antisense. Ovary morphology pictures are re-used from Figure 2C to serve as time scale.
See also Figure S2.
De novo piRNA production necessitates the expression of transcripts that contain P-element sequences from piRNA clusters, the genomic loci that transcribe piRNA precursors (Brennecke et al., 2007; Khurana et al., 2011). Our genomic sequencing data showed that the P-element-containing strain indeed has P-elements in two major clusters: cluster 40 on the left arm of the third chromosome and cluster 3 on chromosome 4 (Table S1). Alternatively, P-element could hop into piRNA clusters and thereby initiate piRNA production and oogenesis. This model predicts that the germ cells formed during the recovery process would carry these events and therefore the new landing sites detected in recovered ovaries should be enriched in piRNA clusters. However, we only found 7 sites in piRNA clusters from the total 844 transposition events that P-element made in the recovered ovaries, disfavoring the “jumping-in-cluster” model. Hence, consistent as previously reported (Khurana et al., 2011), paternally inherited cluster 3 and 40 likely are the major sources for P-element-silencing piRNA production.
Germline stem cells endogenize the invading transposons
To explore which ovarian cell type is responsible for endogenization, we thoroughly examined oogenesis during the entire invasion-recovery process using immunofluorescence to detect the germ cell marker protein Vasa and Hu-li tai shao (Lasko and Ashburner, 1988; Xie and Spradling, 2000). The 1B1 antibody against Hu-li tai shao labels the spectrosome, a round cytoplasmic structure in germline stem cells and undifferentiated cystoblasts. However, it labels the branched fusome in dividing cysts, and outlines the cell membrane in egg chambers (Lasko and Ashburner, 1988; Xie and Spradling, 2000).
One day after the shift to 25°C, 72% of germaria (n = 78) from invaded ovaries contained early-stage germ cells followed by a “gap” region devoid of germ cells (Figure 4A). The pattern from 1B1 labeling showed that these cells were either germline stem cells or cystoblasts, and occasionally retained differentiated cysts (Figure 4A). Besides blocking differentiation, it appears that selective germ cell death also contributes to the formation of the “Vasa-negative zone”, as we detected a wave of germ cell apoptosis in germaria upon P-element invasion (Figure S3). Examination of ovarioles at 2 and 3 days after the temperature-shift showed that egg chambers continued to develop even in ovarioles with arrested germaria (Figure 4A and S4), explaining why the invaded progeny remain fertile for three days after the temperature shift. When the resulting oocytes are laid, no new egg chambers are produced to sustain the assembly line of egg-making. Thus, by 4 days after the temperature-shift, the ovaries become small and fertility declines precipitously (Figure 2B and 2C). Concurrently, the arrested undifferentiated cells rapidly responded to the challenge from the intensive invasion by resuming development and reinitiating oogenesis on day four, as evidenced by the branched structure of 1B1 labeling and the presence of new egg chambers by day 6 (Figure 4A). The restoration of oogenesis ultimately reestablished normal ovary structure and female fertility (Figure 2B and 2C). Based on these findings, we conclude that transposon adaptation occurs in the germline stem cells, and this would ensure permanent P-element silencing in all daughter cells. During the recovery process, since the first emerging egg chamber appeared to come from the accumulated cystoblast, our data also indicate that the undifferentiated cystoblasts maintain similar transposon adaptation potential as germline stem cells.
Figure 4. Germline stem cells silence the invading P-elements via the activation of Chk2/p53.
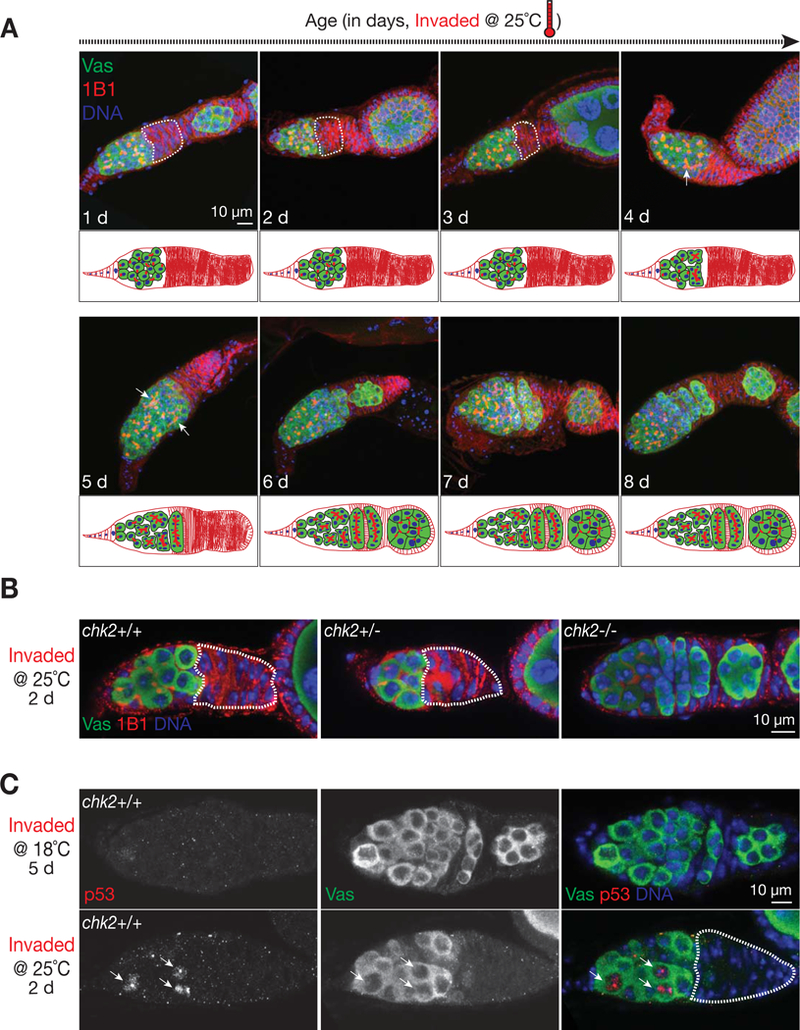
(A) Germarial structure from invaded progeny after vigorous P-element invasion. The germ cells are labeled by Vasa protein (green). The germaria are co-stained with 1B1 (red), an antibody that targets Hu-li tai shao protein that specifies germ cell stages in germarium and outlines cell membrane. Upon intensive invasion, the early stage germ cells are arrested, as evidenced by a “gap” region (circled by dash line) that contains no Vasa positive cells. Note that germ cells in the germarium display “dot” shaped structure from 1B1 staining, indicating that they are undifferentiated germ cells. Oogenesis reinitiates at day 4 or 5, as suggested by forming the “branch” structure from 1B1 staining (pointed by arrow).
(B) Germarial structure from invaded progeny that are either wild type (left), heterozygous (middle), or homozygous (right) for the chk2 gene. Mutating chk2 gene completely rescues the arrest phenotype, as evidenced by the germarium containing continuous germ cells and normal formation of cysts at different stages.
(C) p53 staining to detect the activation of DNA damage response. p53 signals (arrows) are readily detectable in the germarium from 25°C incubation.
Knowing it is the undifferentiated cell that responds to temperature-shift/vigorous invasion prompted us to precisely quantify P-element activity in these cells at different temperatures. To monitor P-element mobilization with single-cell resolution at different temperatures, we designed a GFP::Vasa mobilization assay (Figure 5). Briefly, the mobilization, most likely excision, of the P-element vector based GFP::Vasa transgene in invaded progeny would lead to disappearance or reduction of the GFP signal. Since the excised transgene may still insert back into genome and let the cell regain GFP signal, our current assay may have underestimated the overall mobilization frequency. To apply this assay, we first confirmed that the fly strain that carries this transgene indeed serves as a P-element-devoid strain (Figure 5A). Compared to 18°C, shifting invaded progeny to 25°C appears to increase P-element activity by 7 to 11-fold in undifferentiated germ cells (Figure 5C and 5D). The percentage of germaria that displayed GFP-negative germ cells increased from 8.1% at 18°C (n = 37) to 56% at 25°C (n = 99). On average, the number of marked P-element mobilized cells escalated from 0.1 per germarium at 18°C to 1.1 at 25°C (Figure 5D). Our data therefore suggest that P-element mobilizes at a compromised rate at 18°C, and higher temperature stimulates its activity by at least 7-fold, likely caused by the increased enzymatic activity of the P-element transposase (Kaufman and Rio, 1992; Robertson et al., 1988).
Figure 5. The P-element mobilization assay to quantify the effect of temperature on P-element activity.
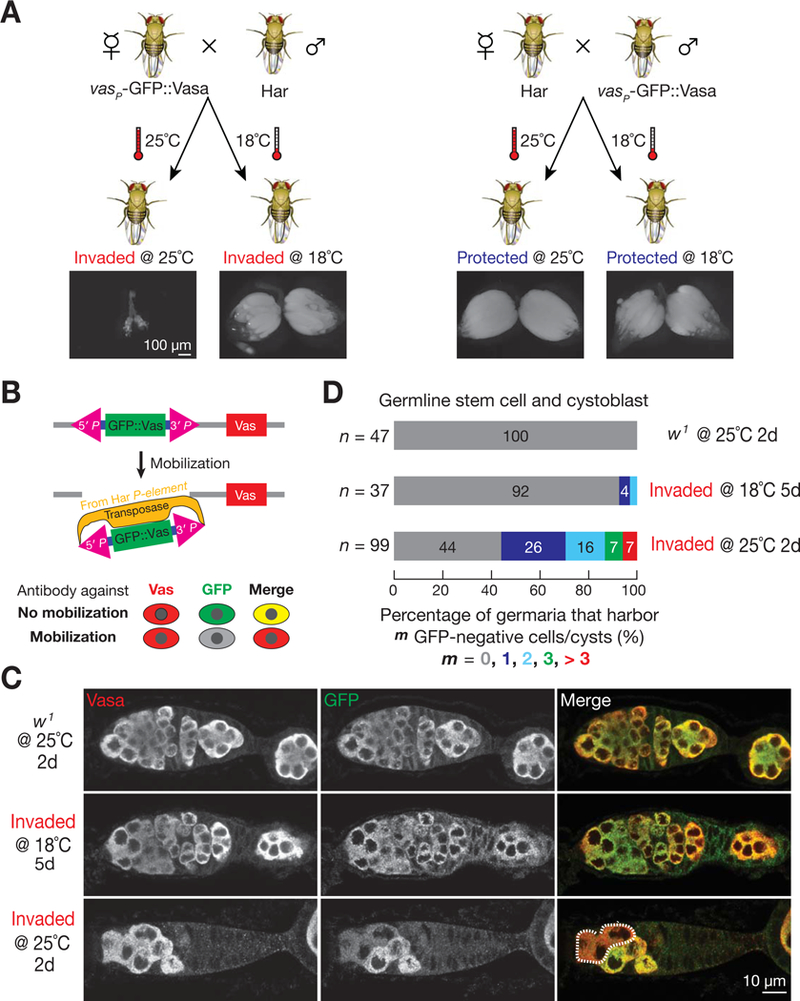
(A) The GFP::Vasa reporter fly serves as a P-element-devoid strain to set up the invading cross.
(B) Strategy for the P-element mobilization assay, used in panel (C) and (D). P-element mobilization, most likely excision, leads to disappearance of GFP signal.
(C) Representative images for the mobilization assay. Germarium is co-stained by Vasa (red) and GFP (green) antibodies. P-element transposition produces GFP negative, but Vasa positive germ cells. Germ cells that have P-element mobilization events are circled by dash line. w1 flies, which do not contain P-element transposases, serve as negative controls.
(D) Quantification of P-element mobilization assay.
Chk2 activation leads to germ cell differentiation arrest upon intensive invasion
What mediates the arrest of undifferentiated cells? P-element mobilization creates DNA double-strand breaks at both the site abandoned by the element and the new site of insertion (Beall and Rio, 1997; Johnson-Schlitz and Engels, 1993), therefore potentially triggering activation of the DNA damage checkpoint and leading to oogenesis arrest in germarium. Similar with previous findings (Rangan et al., 2011), either mutating the key kinase gene in DNA damage signaling–chk2, or only depleting its protein product in germ cells by cell-type-specific RNAi produced normal looking germaria, which are filled with germline stem cells, cystoblasts, differentiated cysts, and newly formed egg chambers (Figure 4B and S5). We further tested the potential role of p53, the downstream factor of the DNA damage checkpoint pathway, in mediating the differentiation arrest of stem cells. The robust p53 signal was readily detected in cells that are under differentiation arrest (Fig. 4C). Consistently, depleting p53 in germ cells by RNAi released stem cells from arrest in invaded progeny (Figure S6A). These results indicate that upon intensive transposon invasion, the hosts shut down oogenesis by activating Chk2/p53-mediated DNA damage signaling in germline stem cells, providing a time window to repair DNA lesions and subdue invading elements. Different from P-element invasion, it appears that mutating the piRNA pathway largely activates Chk2 in the developing oocytes (Chen et al., 2007; Klattenhoff et al., 2007), likely resulting from the mobilization of retrotransposons, but not DNA transposons, in oocytes (Wang et al., 2018).
As suggested by previous studies, p53 might also have a function in maintaining germline stem cells (Bakhrat et al., 2010; Ma et al., 2017; Tasnim and Kelleher, 2018; Wylie et al., 2014). We next quantified the number of germline stem cells in both invaded and control animals. While depleting p53 had no effect on stem cell maintenance in protected control flies, the number of germline stem cells significantly dropped in invaded flies upon p53 inactivation (Figure S6B, P-value = 0.00001). Our data thus suggest that besides restricting differentiation, p53 activity is also required for germline stem cell maintenance upon P-element invasion.
Chk2-mediated arrest is required for transposon endogenization
Either mutating chk2 in the whole animal or depleting it only in germ cells prevented invaded progeny from fertility recovery (Figure 6A and S7A). Without Chk2, invaded flies gained 884 new P-element insertions on average, which is significantly 3-fold greater than wild-type recovered flies at the same age (P-value = 0.0006; Figure S2B). Consistently, instead of being silenced at the transcriptional level in wild type recovered ovaries (Figure 3A), P-elements still actively transcribe mRNAs in the absence of Chk2-mediated adaptive response (Figure 6B). Therefore, the DNA damage checkpoint mediated arrest is essential for oogenesis recovery and P-element endogenization.
Figure 6. Chk2-mediated arrest response tames invading transposons at 25°C.
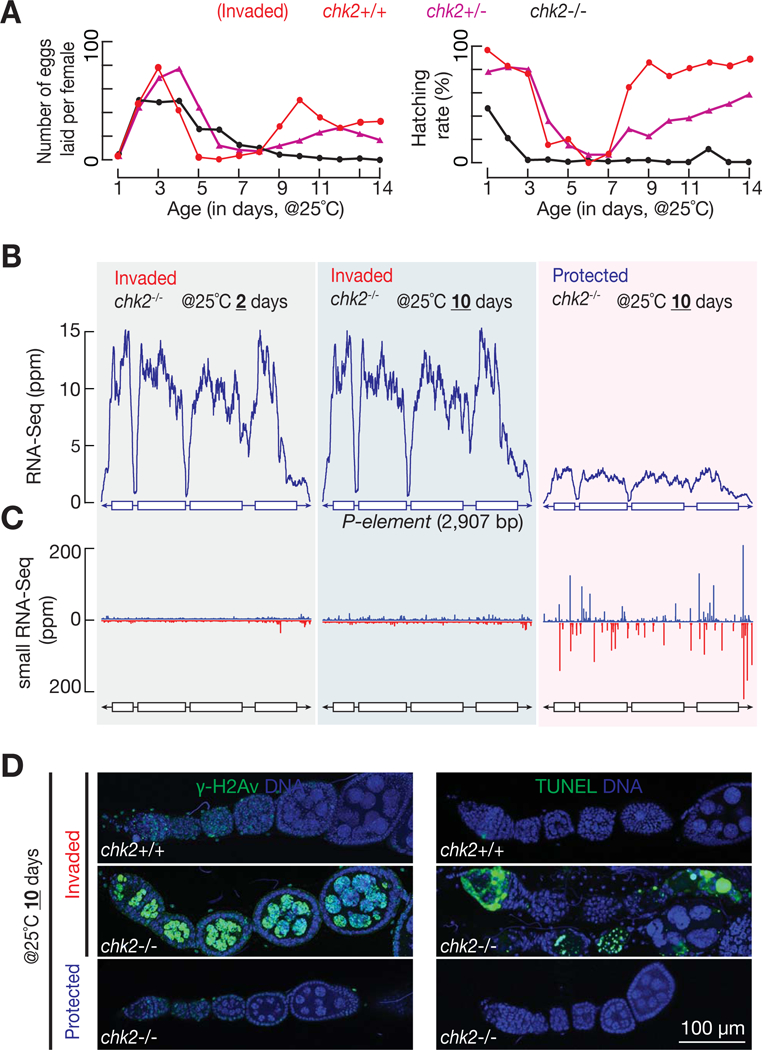
(A) Fertility of invaded progeny that are in either chk2 heterozygous or homozygous background. Fertility of wild-type progeny (from Figure 2B) is reused for comparison.
(B) RNA-Seq profiles for P-element from invaded progeny with chk2 null mutation. Unlike wild-type flies (Figure 3A), chk2 mutants are unable to silence invading transposons. Note that Chk2 is not required for P-element silencing in protected progeny.
(C) Small RNA-Seq assay to quantify the production of P-element piRNAs. Blue, sense piRNAs. Red, antisense. Without Chk2-mediated arrest response, piRNA production stays at a low level from 10 days old flies at 25°C, comparing with abundant piRNA production from age-matched wild-type flies (Figure 3C).
(D) Left panel: γ-H2Av staining to probe the amount of DNA damage. Right panel: TUNEL staining displays dying cells.
See also Figure S7.
We next asked whether Chk2 is still essential for fertility and transposon silencing when the animals are not facing transposon challenge. Neither mutating chk2 in whole animals nor depleting it only in germ cells gave any noticeable defects in protected progeny (Figure S7A and S7B). Meanwhile, assaying the level of P-element transcripts and its mobilization activity indicates that loss of Chk2 has no effect on P-element suppression in protected offspring (Figure 6B and S2B). Therefore, our results demonstrate that Chk2 is nonessential for transposon silencing when the animals are not facing intensive transposon invasions.
Chk2-mediated arrest promotes P-element-silencing piRNA production
Given the essential role of piRNAs in transposon silencing, we next sought to examine whether Chk2-mediated arrest response is required for the production of P-element-silencing piRNAs. It appears that Chk2 is not required for initiating piRNA production, as at day 2 after the temperature-shift, chk2 mutants gave the same low amount of piRNAs that target P-element as wild-type flies (sequencing piRNAs from the P-element-devoid w1 strain served as a negative control to verify this low abundance of piRNAs are bona fide P-element piRNAs; Figure 3C and 6C). However, in contrast to abundant P-element-silencing piRNAs being detected in wild-type recovered progeny (10 days after the temperature-shift), the piRNAs from age-matched chk2 mutants remained at a low level (Figure 3C and 6C). Thus, our data indicate that the Chk2-mediated developmental arrest provides a critical time window to propel piRNA amplification.
To explain this observation, we propose that, by concentrating newly generated P-element-silencing piRNAs in a small number of cells, halting differentiation prevents them from being diluted (Figure 7A). This would allow the accumulated piRNAs to rapidly reach a certain level that is required to initiate Ping-Pong amplification, which cleaves P-element transcripts and establishes Piwi-mediated transcriptional silencing (Brennecke et al., 2007; Gunawardane et al., 2007; Han et al., 2015; Mohn et al., 2015). With the invading P-elements being permanently silenced and the DNA lesions being repaired, ovarian stem cells reinitiate differentiation to recover oogenesis (Figure 7A). Supporting this model, removing just one copy of aub and ago3 (aub+/−; ago3+/−), which encode two piRNA pathway core machinery proteins that mediate Ping-Pong cycles, led to recovery failure (Figure 7B and 7C). These results suggest that rapidly initiating the piRNA amplification cycle with sufficient amount of Ping-Pong partner proteins, which is likely promoted by the arrest, is essential for transposon endogenization.
Figure 7. Role of Chk2 in mediating piRNA amplification and transposon endogenization in germline stem cells.
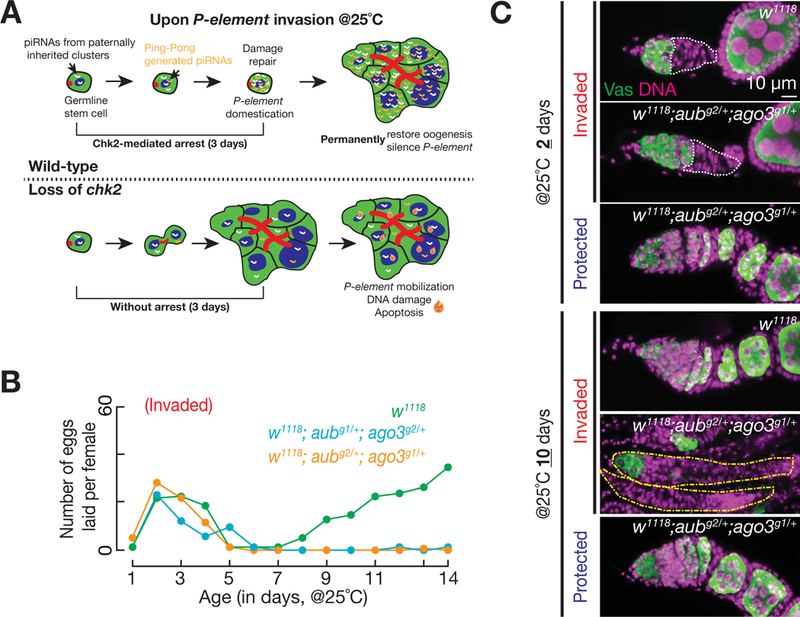
(A) A model explains the role of Chk2 in mediating piRNA amplification. Chk2-mediated arrest restricts the newly produced piRNAs, which are most likely generated from paternally inherited piRNA clusters, within a small volume. This can quickly increase the piRNA concentration to reach a level that initiates Ping-Pong amplification, which cleaves P-element transcripts and initiates transcriptional silencing via Piwi.
(B) Fertility of invaded females, which are the heterozygotes of piRNA pathway factors, after experiencing the P-element invasion. All mutant alleles were generated from the same genetic background as the w1118 controls.
(C) Germarial structure of the heterozygotes of piRNA pathway factors after experiencing the P-element invasion. Germaria from the heterozygotes of piRNA pathway factors contain nearly no germ cells (occasionally a few) at 10 days, suggesting that germ cells were unable to recover from P-element invasion. Note that yellow dotted lines are used to outline germaria.
Through promoting piRNA amplification, the Chk2-mediated adaptive response ensures that all daughter cells in recovered ovaries are protected from transposon mobilization. While chk2 mutants showed strong signals of γ-H2Av (a phosphorylated H2A variant in Drosophila that coats DNA break sites) and cell death at all oogenesis stages (Figure 6D), we did not detect any signals for DNA damage or cell death in wild-type recovered ovaries. The intensive occurrence of apoptosis in chk2 mutants at 10 days after the temperature-shift suggests that loss of Chk2 does not suppress cell death, but rather delays its occurrence. Intriguingly, 42% of egg chambers (n= 138) from these damaged ovaries had an abnormal number of germ cells, and 25% appeared to be full of the stem-cell-like undifferentiated cells (Figure S7C), which reminisces the stem cell tumor phenotype (McKearin and Spradling, 1990). Altogether, our data highlight the essential function of Chk2 in promoting piRNA production and transposon silencing upon vigorous invasion. Without the Chk2-mediated arrest response, the hosts accumulate unrepaired DNA breaks and suffer with inappropriate germ cell proliferation and differentiation, which finally leads to animal sterility.
DISCUSSION
Considered as “selfish DNA sequences”, transposons have heavily accumulated in the genome of nearly all organisms during evolution (Levin and Moran, 2011). Although capable of fueling genomic divergence, the transposon invasion process is disruptive to host cells, and often severely impacts host fertility or even survival (Slotkin and Martienssen, 2007). Therefore, taming invading transposons is an essential and endless task for the host organism. In this study, by using P-element invasion as a model, we first established temperature-shifting as a powerful tool to adjust the intensity of transposon invasion. By investigating the response from the Drosophila adult ovaries, in which we can measure P-element activity and germ cell development in detail, we uncovered a robust transposon-endogenizing mechanism from the germline stem cells. Centered on the key DNA damage checkpoint component Chk2, this robust adaptive response renders hosts the ability to permanently silence invading transposons within just four days.
P-element activity in ovarian cells
Our GFP::Vasa mobilization assay shows that P-element actively hops in germline stem cells. Does P-element also mobilize in other ovarian cells? Since nurse cells are polyploid and the developing oocytes are transcriptionally inactive, our current assay could not faithfully monitor P-element mobilization in them. However, previous study shows that nurse cells express the protein PSI (Labourier et al., 2002), which can block intron removal of P-element transcripts and lead to the production of inactive transposases (Ignjatovic et al., 2005; Labourier et al., 2002; Siebel et al., 1995; Siebel et al., 1994). Therefore, it is unlikely that P-elements mobilize within developing egg chambers. As a type of DNA transposon, which employs the cut-and-paste mechanism for transposition, P-elements cannot directly increase their copy number through mobilization (Spradling et al., 2011). Instead, the propagation is likely achieved via homologous repair from the sister DNA strand during S-phase of the cell cycle (Spradling et al., 2011). Hence, to amplify themselves during Drosophila oogenesis, perhaps P-elements evolved to preferentially mobilize in the dividing germline stem cells, but not in the developing oocytes, which are under cell cycle arrest.
Chk2-mediated transposon adaptation in germline stem cells
By investigating adult oogenesis of Drosophila, we uncovered the Chk2-mediated adaptive response from germline stem cells upon P-element transposon invasion. Interestingly, it appears that arrested germ cells are not equally capable of taming transposons, and Chk2 activation promotes adaptation by eliminating the cells with lower competency. Several lines of evidence support the occurrence of selective cell elimination. First, we detected a significant increase of cell death once P-elements became hyperactive after the temperature-shift. Second, although we occasionally observed GFP negative egg chambers directly connected to germaria at 25°C from the GFP::Vasa mobilization assay, we did not detect any GFP negative cells in later stage egg chambers at any time points. This suggests that the germ cells that maintained high P-element activity, and presumably less competent to adapt, were eliminated at early stages of oogenesis. Third, the number of new P-element insertion events declined to 44% in recovered ovaries after adaptation. This dramatic decline indicates that only the stem cells that had lower transposition rates survived from the selection. Therefore, it is tempting to speculate that not all germ cells are created equal, and that in addition to germarial arrest, the Chk2-mediated DNA break checkpoint also has a role in selecting the survivors from P-element invasion and promoting adaptation.
In the survived ovarian stem cells, Chk2-mediated oogenesis arrest provides a critical time window to propel piRNA generation from the paternally inherited clusters, initiating the amplification cycles for piRNA biogenesis. With at least two piRNA clusters containing P-element sequences in the paternally inherited genome, invaded progeny are capable of generating P-element-silencing piRNAs de novo. Although it is still unclear when the clusters become active during pre-adult development, it has been shown that the primordial germ cells in larval ovaries can already initiate de novo piRNA production (Marie et al., 2017). Consistently, we detected low levels of piRNAs corresponding to P-element before adaptation. However, it appears that the amount of piRNAs produced at this stage is too scarce to silence invading P-elements. Their activation results in sterility and triggers the Chk2-dependent acute adaptive response from germline stem cells. Subsequently, the Chk2-mediated arrest blocks differentiation, which would allow the newly produced P-element-silencing piRNAs to quickly reach a concentration sufficient for Ping-Pong amplification. Finally, these newly produced piRNAs silence transposons at the post transcriptional level and also initiate transcriptional silencing (Brennecke et al., 2007; Gunawardane et al., 2007; Han et al., 2015; Mohn et al., 2015).
Besides promoting piRNA production, the arrest period also allows germ cells to repair DNA lesions before reinitiating oogenesis, thereby preventing the proliferation of cells with DNA damage and defective differentiation. Having the ability to repair damage and endogenize invading transposons in germline stem cells ensures permanent restoration of robust oogenesis and protection of all daughter cells from transposon activation.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhao Zhang (zzhang@carnegiescience.edu)
Experimental Model
Fly stock and husbandry
Flies were raised at 25°C on a standard corn-meal agar diet. A piggybac insertion line harboring nanos-GAL4 (Bl# 32180) is obtained from the Bloomington Drosophila Stock Center (Indiana University, IN, USA). Other fly lines used in this study were listed in the Key Resource Table. For the temperature-shift experiment, crosses were set up at 18°C, and newly eclosed female progeny were shifted to 25 °C. Flies were transferred into a new vial supplemented with yeast paste every 2–3 days until a desired time point. The Gal4 allele and mnkp6 allele were backcrossed into w1 and Harwich strain for at least 7 generations.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-hts | Developmental Studies Hybridoma Bank (DSHB) |
1B1 |
| Mouse monoclonal anti-γH2Av | Developmental Studies Hybridoma Bank (DSHB) |
UNC93–5.2.1 |
| Mouse monoclonal anti-P53 | Developmental Studies Hybridoma Bank (DSHB) |
25F4 |
| Rabbit polyclonal anti-H3K9me3 | Active motif | 39161 |
| Rabbit polyclonal anti-Vasa | Lasko et al., Genes & Dev., 1990 | |
| Chicken polyclonal anti-GFP | Molecular Probes | A10262 |
| Rabbit monoclonal anti-pSmad3 | Abcam | Ab52903 |
| Alexa 488 Donkey anti-Mouse | Molecular Probes | A21202 |
| Alexa 488 Donkey anti-Rabbit | Molecular Probes | A21206 |
| Alexa 488 Goat anti-Chicken | Molecular Probes | A11039 |
| Alexa 594 Donkey anti-Mouse | Molecular Probes | A21203 |
| Alexa 594 Goat anti-Rabbit | Molecular Probes | A11037 |
| Alexa 660 Goat anti-Mouse | Molecular Probes | A21054 |
| Critical Commercial Assays | ||
| DeadEnd Fluorometric TUNEL System | Promega | G32350 |
| LysoTracker® Red DND-99 | Molecular Probes | L7528 |
| Deposited Data | ||
| Raw sequencing reads | This paper | SRP159373 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: w1 | Khurana et al., Cell, 2011 | N/A |
| D. melanogaster: Har | Khurana et al., Cell, 2011 | N/A |
| D. melanogaster: w*;mnkp6 | Klattenhoff et al., Dev. Cell, 2007 | N/A |
| D. melanogaster: w*; p[VASp-GFP::Vasa] | Johnstone et al., Development, 2004 | N/A |
|
D. melanogaster: w*; PBac[w+mW.hs= GreenEye.nosGAL4]Dmel6 |
Bloomington Stock Center (Indiana University, IN, USA) |
32180 |
|
D. melanogaster: y1 sc* v1; P[y+t7.7 v+t1.8= TRiP.HMS01573]attP2 (chk1 RNAi) |
Bloomington Stock Center (Indiana University, IN, USA) |
36685 |
|
D. melanogaster: y1 sc* v1; P[y+t7.7 v+t1.8= TRiP.GL00020]attP2/TM3, Sb1 (chk2 RNAi) |
Bloomington Stock Center (Indiana University, IN, USA) |
35152 |
|
D. melanogaster: y1 sc* v1; P[TRiP.GL00094]attP2 (white RNAi) |
Bloomington Stock Center (Indiana University, IN, USA) |
35573 |
|
D. melanogaster: y1 v1; P{y[+t7.7] v[+t1.8]= TRiP.GL01220}attP40 (p53 RNAi) |
Bloomington Stock Center (Indiana University, IN, USA) |
41638 |
| D. melanogaster: w1118 | Vienna Drosophila Research Center (Vienna, Austria) |
313595 |
|
D. melanogaster: w1118; aubg1/CyO; ago3g2/TM6b, Tb, Hu |
Vienna Drosophila Research Center (Vienna, Austria) |
313593 |
|
D. melanogaster: w1118; aubg2/CyO; ago3g1/TM6b, Tb, Hu |
Vienna Drosophila Research Center (Vienna, Austria) |
313594 |
| Oligonucleotides | ||
| P-element_qPCR_F 5’-TGAGTGCTCGCAACCTTATG-3’ |
This paper | N/A |
| P-element_qPCR_R 5’-TTTGAAATGGGAGCCTTTTG-3’ |
This paper | N/A |
| P-element_SPLICED_F 5’-ATAATAGCCAGGAATACAGAAA-3’ |
This paper | N/A |
| P-element_SPLICED_R 5’-TCATCGACAGGCTCATCATC-3’ |
This paper | N/A |
| chk2_F 5’-TACGAGATTACGGGGCTACG-3’ |
This paper | N/A |
| chk2_R 5’-CGTACAATCGACCCCAGACT-3’ |
This paper | N/A |
| Software and Algorithms | ||
| piPipes | Han et al., Bioinformatics, 2015 | N/A |
| TEMP | Zhuang et al., Nucleic Acids Res, 2007 | N/A |
| R | The R foundation | N/A |
| Other | ||
Methods Details
Antibody staining
Briefly, ovaries were dissected in 1X PBS (pH 7.8) and fixed by 4% paraformaldehyde (PFA) in PBS for 15 minutes at room temperature, followed by washing with PBS containing 0.1% TritonX-100 (PBST) three times for 10 minutes each. Ovaries then were incubated with blocking solution containing 2% bovine serum albumin (BSA) and 2% normal goat serum (NGS) in PBS for 1 hour at room temperature. The samples were incubated with desired primary antibodies in the blocking solution at 4°C overnight, followed by washing with PBST three times for 10 minutes each. Secondary antibodies in the blocking solution were added for 2 hours at room temperature. After three washes (10 minutes each) with PBST, the samples were mounted with VECTASHIELD antifade mounting medium (VECTOR LABORATORIES INC MS, 101098–042) and examined under SP5 microscope (Leica). Primary antibodies used in this study were: anti-Hu-li tai shao (1B1 antibody, 1:100), anti-GFP (1:300), anti-γH2Av (1:500), anti-P53 (1:500), anti-pSmad3 (1:100), and anti-Vasa (1:5,000). All of secondary antibodies in this study were used at 1:500 and chromatin was visualized by DAPI.
TUNEL and LysoTracker assay
For TUNEL assay, ovaries were dissected in PBS (pH 7.8), fixed by 4% paraformaldehyde and washed with PBST three times for 10 minutes each at room temperature, followed by incubation with equilibration buffer (200 mM potassium cacodylate (pH 6.6), 25 mM Tris-HCl (pH 6.6), 0.2 mM DTT, 0.25 mg/ml BSA, and 2.5 mM cobalt chloride) for 10 minutes at room temperature. Then samples were incubated with the equilibration buffer containing Recombinant Terminal Deoxynucleotidyl Transferase (rTdT), and fluorescein-12-dUTP for three hours at 37 °C. After three washing steps with PBST for 10 minutes, the samples were mounted. For LysoTracker assay, ovaries were dissected in PBS (pH 7.8) and incubated with 50 μM of Lysotracker in PBS for 5 minutes at room temperature. Samples were washed with PBS three times for 5 minutes, followed by incubation with 4% paraformaldehyde for 10 minutes at room temperature. After fixation, samples washed with PBST, and mounted. All samples were examined under a fluorescent microscope, ApoTome (ZEISS).
Quantitative real time PCR
Total RNA was extracted from dissected ovaries with mirVana™ miRNA isolation kit (Invitrogen, AM1561) according to manufacturer’s instructions. Isolated total RNA was treated with Turbo DNase (Invitrogen, AM2238) at 37 °C for 30 minutes to get rid of genomic DNA contamination, followed by purification with RNA clean and Concentrator 5 (Zymo Research, R1015) according to manufacturer’s instruction. 1–2 μg of total RNA was used to generate cDNA with the iScript™ Reverse Transcription Supermix (Bio-Rad, 170–8840). Quantitative PCR was set up using SsoFast™ EvaGreen® Supermix (Bio-Rad, 1725201), and performed by CFX96 Touch Real-Time PCR Detection System (Bio-Rad, 1841100). Each experiment was performed in three biological replicates with three technical triplicates. Relative RNA levels were normalized to rp49 levels and calculated by the 2−∆∆CT method. For the regular PCR experiment, a targeted region of P-element was amplified (35 cycle) by a primer set that detects both unspliced and spliced form. Twenty five cycles were used to amplify chk2 amplicon. Regular PCR results were visualized on 2 % of agarose gel. Sequences of the primers are available in the table S2.
RNA-FISH
Stellaris RNA FISH probe set was purchased from LGC Bioresearch Techlologies, and the probe sequences were listed in Table S2. Five ovaries were dissected in cold PBS, and fixed for 20 minutes in PBS with 4% formaldehyde. Ovaries were washed once in PBST and twice with PBS, and then immersed in 70% (v/v) ethanol for at least two hours at 4°C. Then ovaries were washed once with 1 ml Wash buffer A (LGC Biosearch Tech, Cat# SMF-WA1–60) for 5 minutes, and then add 50 µl Hybridization Buffer (LGC Biosearch Tech, Cat# SMF-HB1–10) containing probe set (125 nM) to hybridize at 37 °C for overnight. Ovaries were washed twice in Wash buffer A for 30 minutes at 37 °C and then once with Wash Buffer B (LGC Biosearch Tech, Cat# SMF-WB1–20) for 5 minutes at room temperature. Mounting samples with 20 μl Vectashield Mounting Medium.
High throughput sequencing library preparation and data analysis
RNA-Seq libraries were prepared according to the published protocol (Zhang et al., 2012). Three µg total RNA was first subjected to rRNA depletion by Ribo-Zero rRNA Removal Kit (illumina, Cat# MRZH11124). After DNase treatment, the RNA was first fragmented, then converted into cDNA by using random primers. dUTP was incorporated into the second strand when converting cDNA:RNA hybrid into dsDNA, and the dsDNA was subjected to end-repair, A tailing, Y-shape adapter ligation, UDG treatment to remove the second strand, and final PCR amplification. piPipes was used to analyze sequencing results.
For genomic DNA library, 150 ng DNA was fragmented into 300–500 bp by Biorupter. The fragmented DNA was subject to end-repair, A tailing, Y-shape adapter ligation, and PCR amplification to add barcode. TEMP was used to detect P-element insertions in all samples.
For small RNA libraries, 18–30 nt small RNAs were first purified from 20 μg total RNA. After 2S RNA depletion, 3’ adapter and 5’ adapter were ligated to small RNAs sequentially. After reverse transcription, the libraries were amplified by PCR and barcodes were added. piPipes was used to analyze sequencing results.
To prepare ChIP-Seq libraries, 50 ovaries were dissected and fixed by formaldehyde. Then, the ovaries were sonicated to break the chromatin into 200–400 bp. For each library, 5 µl H3K9me3 antibodies were used for immunoprecipitation. The purified DNA was subjected to end-repair, A tailing, Y-shape adapter ligation, and PCR amplification to add barcode. The sequencing results were also analyzed by piPipes. Detailed protocol for all library preparation can be obtained upon request. Table S3 summaries all of the libraries made in this study.
QUANTIFICATION AND STATISTICAL ANALYSIS
Student’s T test was carried out using either Microsoft Excel or R to calculate significance. P values are indicated on plots. Results are expressed as mean ± standard error (SD). The number of animals examined are indicated in figure legends.
Supplementary Material
Table S1. Potential P-element insertion in Harwich, Related to Figure 1
Table S2. P-element FISH probes, Related to Figure S1
Table S3. Summary of deep sequencing libraries, Related to STAR Methods
ACKNOWLEDGMENTS
We thank Paul Lasko, Julius Brennecke, William Theurkauf, and Ruth Lehman for providing antibodies and fly alleles. We thank Allan Spradling, Steve DeLuca, John Urban and other Spradling lab members, as well as Bortvin lab for critical comments during project development and manuscript preparation. We also thank Marnie Halpern, Phillip Zamore, William Theurkauf, and Alex Bortvin for reading manuscript. This work was supported by the grant from the National Institutes of Health to Z.Z. (DP5 OD021355) and Carnegie Endowment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
DATA AND SOFTWARE AVAILABILITY
All sequencing data generated from this study are available at NCBI: SRP159373.
REFERENCES
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. (2006). A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, and Gvozdev VA (2001). Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Current biology : CB 11, 1017–1027. [DOI] [PubMed] [Google Scholar]
- Bakhrat A, Pritchett T, Peretz G, McCall K, and Abdu U (2010). Drosophila Chk2 and p53 proteins induce stage-specific cell death independently during oogenesis. Apoptosis 15, 1425–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall EL, and Rio DC (1997). Drosophila P-element transposase is a novel site-specific endonuclease. Genes & development 11, 2137–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, and Hannon GJ (2007). Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, and Hannon GJ (2008). An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 322, 1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pane A, and Schupbach T (2007). Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Current biology : CB 17, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels WR (1997). Invasions of P elements. Genetics 145, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels WR, and Preston CR (1979). Hybrid dysgenesis in Drosophila melanogaster: the biology of female and male sterility. Genetics 92, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, and Carmell MA (2006). A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202. [DOI] [PubMed] [Google Scholar]
- Grivna ST, Beyret E, Wang Z, and Lin H (2006). A novel class of small RNAs in mouse spermatogenic cells. Genes & development 20, 1709–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, and Siomi MC (2007). A slicer-mediated mechanism for repeat-associated siRNA 5’ end formation in Drosophila. Science 315, 1587–1590. [DOI] [PubMed] [Google Scholar]
- Han BW, Wang W, Li C, Weng Z, and Zamore PD (2015). Noncoding RNA. piRNA-guided transposon cleavage initiates Zucchini-dependent, phased piRNA production. Science 348, 817–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, and Lin H (2013). A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24, 502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignjatovic T, Yang JC, Butler J, Neuhaus D, and Nagai K (2005). Structural basis of the interaction between P-element somatic inhibitor and U1–70k essential for the alternative splicing of P-element transposase. Journal of molecular biology 351, 52–65. [DOI] [PubMed] [Google Scholar]
- Johnson-Schlitz DM, and Engels WR (1993). P-element-induced interallelic gene conversion of insertions and deletions in Drosophila melanogaster. Molecular and cellular biology 13, 7006–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman PD, and Rio DC (1992). P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell 69, 27–39. [DOI] [PubMed] [Google Scholar]
- Khurana JS, Wang J, Xu J, Koppetsch BS, Thomson TC, Nowosielska A, Li C, Zamore PD, Weng Z, and Theurkauf WE (2011). Adaptation to P element transposon invasion in Drosophila melanogaster. Cell 147, 1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Kidwell JF, and Sved JA (1977). Hybrid Dysgenesis in DROSOPHILA MELANOGASTER: A Syndrome of Aberrant Traits Including Mutation, Sterility and Male Recombination. Genetics 86, 813–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, and Novy JB (1979). Hybrid Dysgenesis in DROSOPHILA MELANOGASTER: Sterility Resulting from Gonadal Dysgenesis in the P-M System. Genetics 92, 1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RC, Aggarwal SK, and Aggarwal U (1968). The development of the female Drosophila reproductive system. J Morphol 124, 143–166. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Bratu DP, McGinnis-Schultz N, Koppetsch BS, Cook HA, and Theurkauf WE (2007). Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Dev Cell 12, 45–55. [DOI] [PubMed] [Google Scholar]
- Labourier E, Blanchette M, Feiger JW, Adams MD, and Rio DC (2002). The KH-type RNA-binding protein PSI is required for Drosophila viability, male fertility, and cellular mRNA processing. Genes & development 16, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko PF, and Ashburner M (1988). The product of the Drosophila gene vasa is very similar to eukaryotic initiation factor-4A. Nature 335, 611–617. [DOI] [PubMed] [Google Scholar]
- Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, and Kingston RE (2006). Characterization of the piRNA complex from rat testes. Science 313, 363–367. [DOI] [PubMed] [Google Scholar]
- Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, and Toth KF (2013). Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes & development 27, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, and Moran JV (2011). Dynamic interactions between transposable elements and their hosts. Nature reviews. Genetics 12, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Zhu X, Han Y, Story B, Do T, Song X, Wang S, Zhang Y, Blanchette M, Gogol M, et al. (2017). Aubergine Controls Germline Stem Cell Self-Renewal and Progeny Differentiation via Distinct Mechanisms. Dev Cell 41, 157–169 e155. [DOI] [PubMed] [Google Scholar]
- Marie PP, Ronsseray S, and Boivin A (2017). From Embryo to Adult: piRNA-Mediated Silencing throughout Germline Development in Drosophila. G3 7, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKearin DM, and Spradling AC (1990). bag-of-marbles: a Drosophila gene required to initiate both male and female gametogenesis. Genes & development 4, 2242–2251. [DOI] [PubMed] [Google Scholar]
- Mohn F, Handler D, and Brennecke J (2015). Noncoding RNA. piRNA-guided slicing specifies transcripts for Zucchini-dependent, phased piRNA biogenesis. Science 348, 812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill RJ, O’Neill MJ, and Graves JA (1998). Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393, 68–72. [DOI] [PubMed] [Google Scholar]
- Rangan P, Malone CD, Navarro C, Newbold SP, Hayes PS, Sachidanandam R, Hannon GJ, and Lehmann R (2011). piRNA production requires heterochromatin formation in Drosophila. Current biology : CB 21, 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, Benz WK, and Engels WR (1988). A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, and Hannon GJ (2013). Multiple roles for Piwi in silencing Drosophila transposons. Genes & development 27, 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Nishida KM, Mori T, Kawamura Y, Miyoshi K, Nagami T, Siomi H, and Siomi MC (2006). Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes & development 20, 2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebel CW, Admon A, and Rio DC (1995). Soma-specific expression and cloning of PSI, a negative regulator of P element pre-mRNA splicing. Genes & development 9, 269–283. [DOI] [PubMed] [Google Scholar]
- Siebel CW, Kanaar R, and Rio DC (1994). Regulation of tissue-specific P-element pre-mRNA splicing requires the RNA-binding protein PSI. Genes & development 8, 1713–1725. [DOI] [PubMed] [Google Scholar]
- Sienski G, Donertas D, and Brennecke J (2012). Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151, 964–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, and Martienssen R (2007). Transposable elements and the epigenetic regulation of the genome. Nature reviews. Genetics 8, 272–285. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Bellen HJ, and Hoskins RA (2011). Drosophila P elements preferentially transpose to replication origins. Proceedings of the National Academy of Sciences of the United States of America 108, 15948–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton RE, Meers J, and Young PR (2006). Retroviral invasion of the koala genome. Nature 442, 79–81. [DOI] [PubMed] [Google Scholar]
- Tasnim S, and Kelleher ES (2018). p53 is required for female germline stem cell maintenance in P-element hybrid dysgenesis. Developmental biology 434, 215–220. [DOI] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, and Zamore PD (2006). A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313, 320–324. [DOI] [PubMed] [Google Scholar]
- Wang L, Dou K, Moon S, Tan FJ, and Zhang ZZ (2018). Hijacking Oogenesis Enables Massive Propagation of LINE and Retroviral Transposons. Cell [DOI] [PMC free article] [PubMed]
- Weick EM, and Miska EA (2014). piRNAs: from biogenesis to function. Development 141, 3458–3471. [DOI] [PubMed] [Google Scholar]
- Wylie A, Lu WJ, D’Brot A, Buszczak M, and Abrams JM (2014). p53 activity is selectively licensed in the Drosophila stem cell compartment. eLife 3, e01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, and Spradling AC (2000). A Niche Maintaining Germ Line Stem Cells in the Drosophila Ovary. Science 290, 328–330. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Theurkauf WE, Weng Z, and Zamore PD (2012). Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly(A) selection. Silence 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Wang J, Theurkauf W, and Weng Z (2014). TEMP: a computational method for analyzing transposable element polymorphism in populations. Nucleic acids research 42, 6826–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Potential P-element insertion in Harwich, Related to Figure 1
Table S2. P-element FISH probes, Related to Figure S1
Table S3. Summary of deep sequencing libraries, Related to STAR Methods