

Summary:
ATP-competitive kinase inhibitors often bind several kinases due to the high conservation of the ATP binding pocket. Through clustering analysis of a large kinome profiling dataset we found a cluster of eight promiscuous kinases that on average bind more than five times more kinase inhibitors than the other 398 kinases in the dataset. To understand the structural basis of promiscuous inhibitor binding, we determined the co-crystal structure of the receptor tyrosine kinase DDR1 with the type I inhibitors dasatinib and VX-680. Surprisingly, we find that DDR1 binds these type I inhibitors in an inactive conformation typically reserved for type II inhibitors. Our computational and biochemical studies show that DDR1 is unusually stable in this inactive conformation, giving a mechanistic explanation for inhibitor promiscuity. This phenotypic clustering analysis provides a strategy to obtain functional insights not available by sequence comparison alone.
Graphical Abstract
eTOC Blurb
Hanson et al identify a group of kinases that bind a surprising number of inhibitors and reveal the molecular basis for this promiscuity. Functional clustering of proteins identifies groups of co-inhibited kinases. The findings have implications for the development of specific kinase inhibitors and drug resistance.
Introduction
Protein kinases represent one of the largest enzyme families in the human genome and act as signaling mediators in a variety of cellular processes (Manning et al., 2002). Because many diseases are associated with aberrant protein kinase activity, targeted kinase inhibitors are clinically highly successful, such as imatinib in cancer therapy (Druker et al., 2006; Fabbro, 2015; Wu et al., 2016).
ATP-competitive kinase inhibitors can be classified by the conformation of the highly conserved Asp-Phe-Gly (DFG) motif of the kinase upon inhibitor binding (Zhao et al., 2014). Type I inhibitors bind to the active, DFG-Asp-in, conformation. Type II inhibitors bind an inactive conformation in which the aspartate of the DFG motif faces away from the active site into the bulk solvent (DFG-Asp-out) (Wodicka et al., 2010). While both type I and type II kinase inhibitors have been clinically successful, specific kinase inhibition remains challenging due to the high conservation of the ATP binding pocket (Zhang et al., 2009). For example, the type I inhibitor dasatinib and the type II inhibitor imatinib bind 86 and 19 kinases out of 317, respectively (Karaman et al., 2008). While low inhibitor selectivity seems to be clinically tolerable for treating certain types of leukemia, inhibition of off-target kinases often limits the application of kinase inhibitors against solid tumors (Cohen et al., 2017; Eckstein et al., 2014; Lee and Wang, 2009).
For this reason, the specificity relationship between inhibitor and kinase is typically viewed from the perspective of the inhibitor (i.e., which kinases does a single inhibitor target?). Instead, examining this relationship from the perspective of the kinase (i.e., which inhibitors does an individual kinase bind?) can lead to a kinome-wide understanding of inhibitor binding behavior (Anastassiadis et al., 2011; Huang et al., 2010). To identify relationships among kinases as determined by the similarity of their inhibition phenotype – the profile of kinase inhibitors for which they have measurable affinity – we perform hierarchical clustering on a previously published dataset assessing the inhibition of 406 kinase constructs by 645 inhibitors (Drewry et al., 2017). We identify two groups of kinases with strikingly different promiscuity towards kinase inhibitors. The group of promiscuous kinases consists of eight Ser/Thr and Tyr kinases including established clinical targets (PDGFRA/B, KIT, and CSF1R), as well as kinases that are not prominent clinical targets (DDR1, DDR2, YSK4, and MEK5) (Wu et al., 2015). Importantly, kinases that are the target of many drug development programs such as EGFR, Abl, BRAF, and IGF1R are not part of this promiscuous group.
To determine the structural basis for promiscuity towards kinase inhibitors, we solved the co-crystal structure of DDR1 in complex with two type I inhibitors: the aurora kinase inhibitor, VX-680, and the pan-tyrosine kinase inhibitor, dasatinib (Harrington et al., 2004; Lombardo et al., 2004). DDR1 is a receptor tyrosine kinase that binds to the extracellular matrix and is characterized by low kinase activity and slow activation kinetics. Surprisingly, our structures show that DDR1 binds both type I inhibitors in the DFG-Asp-out conformation, which is the binding conformation typically reserved for type II inhibitors. This suggests that DDR1 is stable in the DFG-Asp-out inactive conformation. This in itself is unusual since the first structures of kinases in the DFGAsp-out conformation were considered to be induced by the high affinity type II inhibitors (Nagar et al., 2002; Schindler et al., 2000). Here, we show that the DFG-Aspout conformation is not only stable in DDR1, but facilitates promiscuous inhibitor binding. Moreover, we find within the subset of promiscuous kinases a conserved salt bridge that stabilizes the DFG-Asp-out conformation. Disruption of this salt bridge shifts the population of DDR1 towards the active DFG-Asp-in conformation and increases the specific kinase activity tenfold. The study provides an example of how large functional datasets can be used to group proteins by functional phenotype instead of sequence homology to elucidate a common mechanism. While here we define a single phenotypically different subgroup of kinases and its underlying mechanism, we expect that further analyses of this type will reveal additional phenotype-based kinase subgroups and mechanisms.
Results
Phenotypic clustering identifies a group of unusually promiscuous kinases
Several screens of kinase inhibitor collections against large panels of kinases have been conducted in an effort to characterize the selectivity of kinase inhibitors, and to determine which fraction of the kinome can be inhibited with existing inhibitors (Drewry et al., 2017; Fabian et al., 2005; Karaman et al., 2008; Klaeger et al., 2017). Hierarchical clustering has been performed on these datasets to identify patterns among ligands (Paricharak et al., 2013). Here, we perform hierarchical cluster analysis on a recently published dataset of 645 small molecule inhibitors and 406 human kinases (392 wild-type kinases and 14 variants) (Drewry et al., 2017) to identify the relationships among kinases based on their binding phenotype to inhibitors. We obtain a tree in which eight kinases form a cluster distant from all other kinases (Figure 1A and Figure S1). When we assessed for each kinase the number of ligands that inhibit KinoBead binding by more than 90% at 1 mM inhibitor, the group of eight kinases includes the most promiscuous kinases in this panel (Figure 1B). We find that there is a wide spread of promiscuity ranging from extremely discriminant kinases (five kinases are inhibited by only one inhibitor each) to promiscuous kinases that are inhibited by more than 100 inhibitors. Strikingly, the group of eight promiscuous kinases binds a mean of 98.6 inhibitors (standard deviation (SD) = 31.9) per kinase whereas the group of 398 discriminant kinases binds a mean of 17.3 (SD = 14.0) inhibitors (Wilcoxon p-value < 0.0001) (Figure 1C).
Figure 1: Hierarchical clustering of the human kinome by inhibition phenotype reveals a group of highly promiscuous kinases.

A) Hierarchical clustering analysis of the PKIS2 dataset assessing KinoBead binding inhibition for 406 kinases by 645 ligands at 1 μM concentration. Individual kinases are shown as dots and colored by the number of inhibitors capable of displacing more than 90% of kinase from covalently-tethered pan-kinase inhibitors (green being most inhibitors, red being fewest inhibitors). The promiscuous branch is circled in green. B) The most basal branch of this dendrogram is a single branch of eight promiscuous kinases that bind a mean number of 98.6 ± 31.9 ligands (green) while the other 398 in the panel bind 17.3 ± 14.0 ligands (red) (mean ± SD). C) Number of kinases that are inhibited by given number of inhibitors to more than 90%. D) Superposition of the promiscuity of kinases onto the kinase phylogenetic tree. Circle diameter is proportional to kinase promiscuity for emphasis, and colors are as in C. See also Figures S1 and S2.
The eight promiscuous kinases include PDGFRA, PDGFRB, KIT, CSF1R, DDR1, DDR2, MEK5, and YSK4 and are inhibited by 75, 145, 126, 75, 104, 50, 121 and 97 compounds respectively (Figure 1B). One question is whether the number of bound inhibitors reflects the inhibitor development effort targeted against this kinase rather than promiscuity. The group of eight kinases includes both clinical targets (PDGFRA, PGFDRB, KIT, and CSF1R) and kinases of minor clinical interest (DDR1, DDR2, MEK5 and YSK4) (Wu et al., 2015). This indicates that these eight kinases are not inhibited by many compounds simply because they are the most prominent drug targets. Overlaying kinase promiscuity on the kinome phylogenetic tree clearly shows that our eight most promiscuous kinases do not share most recent common ancestry (Figure 1D).
Next, we set out to determine the structural and energetic basis for promiscuity towards kinase inhibitors. We focused on DDR1 since it is a promiscuous kinase in various inhibitor screens and binds to the type II inhibitors imatinib and nilotinib, as well as the type I inhibitor VX-680 (Day et al., 2008; Karaman et al., 2008; Manley et al., 2010; Rix et al., 2010). DDR1 is a receptor tyrosine kinase that binds extracellular collagen and exhibits unusually slow activation kinetics and low kinase activity. DDR1 acts as a sensor for anchorage to the extracellular matrix which is important for cellular adhesion, migration and invasion. In order to elucidate its role in disease, chemical biology efforts have resulted in DDR1 specific inhibitors (Canning et al., 2014; Deng et al., 2013; Kothiwale et al., 2015; Murray et al., 2015; Richters et al., 2014). These studies however have not addressed why DDR1 is so promiscuous. Here, we set out to understand the structural basis for promiscuity in DDR1.
DDR1 binds both type I and type II inhibitors in the inactive conformation.
First, we wanted to determine if DDR1 binds type I and type II kinase inhibitors in similar conformations as less promiscuous kinases. We therefore co-crystallized DDR1 with type I inhibitors since previously published structures of DDR1 were only in complex with type II inhibitors (Canning et al., 2014; Deng et al., 2013; Murray et al., 2015). We expected that DDR1 would bind the Aurora kinase inhibitor VX-680 and the pan-tyrosine kinase inhibitor dasatinib in the DFG-Asp-in conformation observed with other kinases (Tokarski et al., 2006; Young et al., 2006). To our surprise, we found that DDR1 adopts in both complexes the inactive DFG-Asp-out conformation (DDR1 residues Asp747-Phe748-Gly749) typically observed in kinases binding type II inhibitors (Figure 2A, B).
Figure 2: DDR1 binds type I inhibitors in the DFG-Asp-out conformation.

A) Co-crystal structure of DDR1·VX-680 [PDB-entry: 6BRJ] with activation loop (blue), phosphate binding P-loop (red), helix αC (orange). B) The co-crystal structure of DDR1·dasatinib [PDB-entry: 6BSD] with regulatory elements colored as in A. C) Comparison of type I (top, VX-680/dasatinib) and type II (bottom, imatinib/ponatinib) inhibitors binding to Abl (green, right column) and DDR1 kinase (blue, left column). Only the protein surrounding the DFG-motif is shown for clarity. D) Comparison of Φ dihedral angle for Asp747 in DDR1 (blue) and Abl (green) bound to the type I and type II inhibitors in panel C. See also Figure S3.
Since DDR1 binds VX-680 in the DFG-Asp-out conformation, we were curious to see if the inhibitor interacted differently with DDR1 than with Abl and Aurora kinases. The interactions between VX-680 and DDR1 were similar to interactions seen with Abl and Aurora kinases, however, the hydrophobic cyclopropyl group of VX-680 was shielded by side chains of Phe748 and Met750, and a unique salt-bridge formed between Asp671 and Arg752 (Figure 2A). In the equivalent structures of Aurora and Abl kinases bound to VX-680, this hydrophobic moiety is shielded by the phosphate-binding loop (P-loop) folding over the active site (Adrián et al., 2006; Consortium et al., 2008; Elkins et al., 2012; Salah et al., 2011).
Similar to our DDR1[circle4]VX-680 structure, dasatinib also binds DDR1 in the DFGAsp-out conformation with similar interactions between the drug and DDR1 as seen in structures of Abl kinase bound to dasatinib (Figure 2B, 2C) (Shah et al., 2004). The DFG motif in DDR1[circle4]dasatinib is rotated in comparison to DDR1[circle4]VX-680, suggesting the DFG motif may be in an intermediate state. Furthermore, due to the lack of electron density, we were unable to build in residues following the DFG motif: Met750, Ser751, and Arg752. Some electron density is present where the side chain of Arg752 would fall, and when aligned to the activation loop of DDR1[circle4]VX-680, suggests that the salt bridge between Asp671 and Arg752 would be intact.
When bound to type II inhibitors (imatinib and ponatinib), the conformation of the DFG motif of DDR1 resembles the DFG-Asp-out conformation in Abl. However, the DFG-Asp-out conformation in DDR1[circle4]VX-680 and DDR1[circle4]dasatinib differs from the conformations of Abl bound to either type I or type II inhibitors (Figure 2C, D). Similar to other structures of kinases in the DFG-Asp-Out conformation, such as Abl kinase bound to imatinib, the salt-bridge between the catalytic lysine and the αC-helix is intact in DDR1. Both type I and type II inhibitors form similar interactions with DDR1 as they do in other kinases, including in particular the hinge region of the kinase (the loop connecting the N- and C-lobe) (Zhang et al., 2009). DDR1 is able to accommodate the larger type II kinase inhibitors by allowing inhibitors to bind in a hydrophobic pocket at the back of the active site that is located under the aC-helix and only accessible in the DFG-Asp-out inactive conformation (Canning et al., 2014; Murray et al., 2015; Zhang et al., 2009). However, in the case of type I inhibitors, DDR1 binds them in the DFG-Aspout conformation and can adjust the position of the DFG motif to accommodate them (Figure 2C, D). This curious behavior suggests that the inactive conformation of DDR1 is relatively stable, and that accessing the active DFG-Asp-in conformation may be energetically unfavorable for DDR1, which also helps explain the low cellular kinase activity (Leitinger, 2014).
DDR1 is stable in the DFG-Asp-out conformation.
To determine the stability of the DFG-Asp-out conformation in DDR1, we performed massively distributed molecular dynamics (MD) simulations starting from available crystal structures of DDR1, all of which are in the DFG-Asp-out conformation. Simulations were initiated from the VX-680 bound structure reported here, as well as previously published structures of DDR1 in complex with ponatinib and imatinib (PDB IDs 3ZOS and 4BKJ, respectively) (Canning et al., 2014). In these initial ~450 μs aggregate simulations of DDR1 protonated at Asp747, we observed four independent transitions from the DFG-Asp-out to the DFG-Asp-in conformation. (Figure 3A).
Figure 3: Wild-type DDR1 is stable in the DFG-Asp-out conformation and mutations destabilize this inactive conformation.

A) Snapshots from one of the trajectories seen to flip from DFG-Asp-out to DFG-Asp-in are superimposed on the final free energy landscape for WT DDR1 are highlighted in yellow. HMM macrostates representing DFG-in (red) and DFG-out (purple) states are shown, and transparency is proportional to the membership of each k-means cluster center to the macrostate. B) The Asp671-Arg752 salt bridge, Asp671-Tyr755 hydrogen bond, and Asp729-Tyr759 interaction thought to stabilize the inactive DFG-Asp-out conformation. C) Free energy landscapes for WT, D671N, Y755A, and Y759A simulations superimposed onto the first two (slowest) TICs of the WT simulations. Dotted line corresponds to the linear separation between states seen in C. D) Free energy difference between DFG-out and DFG-in states calculated from the free energy landscapes in panel C. Positive values indicate stabilization in the DFG-out conformation; negative values indicate stabilization in the DFG-in conformation. Error bars indicate 95% confidence intervals from a Bayesian MSM with 1000 samples. E) Kinase activity assays of DDR1. F) Binding affinity of DDR1 wt and mutant proteins for the type II inhibitor imatinib. Binding affinity for imatinib is reported as inhibitory constant Ki for competition with a general kinase inhibitor. See also Figure S4 and Table S1.
To collect a larger number of DFG transitions, we initiated new simulations from snapshots of these four simulations in which the transition was observed. The resulting free energy landscape for DDR1 WT clearly shows a preference of ~1 kT for the DFGAsp-out conformation over the DFG-Asp-in conformation (Figure 3A,C).
Mutations destabilize the DFG-Asp-out conformation and increase kinase activity.
Next, we wanted to understand the biophysical basis for the stability of the DFG-Aspout conformation. We noticed in the structure of DDR1[circle4]VX-680 that a network of salt bridges and hydrogen bonds in DDR1 around Asp671, Arg752, Tyr755, and Tyr759 were likely stabilizing the DFG-Asp-out conformation (Figure 3B). We therefore performed MD simulations to determine the free energy landscape for three DDR1 constructs in which the stabilizing interactions were disrupted by mutation (D671N, Y755A, or Y759A) (Figure 3C). We find that all mutants are more stable in the DFG-Asp-in conformation by about ~0.5 kT (Figure 3D).
We hypothesized that these mutations disrupt the inactive (DFG-Asp-out) conformation and increase kinase activity. Indeed, we found that the DDR1 D671N mutant was 9.3-fold more active in biochemical kinase assays compared to wild-type DDR1 (activity increased from 24 fmol sec−1 to 224 fmol sec−1) (Figure 3E). This increase in activity suggests that the Asp671-Arg752 salt bridge is critical for the stabilization of the inactive conformation and destabilizing it allows the kinase to more readily adopt the active conformation. The Y755A and Y759A mutants also showed an increase in activity compared to wild-type DDR1 (7.5-fold and 7.8-fold increase, respectively), implicating them in the stabilization of the inactive DDR1 conformation (Figure 3E). Interestingly, the fold change in kinase activation upon mutation is consistent with the ~5-fold change in DFG-Asp-in conformation predicted by the simulations (Figure 3D).
If the mutations shift the population of DDR1 from the inactive DFG-Asp-out to the active DFG-Asp-in conformation, we would predict that the mutant proteins bind less tightly to type II inhibitors such as imatinib that require the DFG-Asp-out conformation for binding. Consistent with this model, we find that the tested mutants bind imatinib 3–4-fold less tightly (Figure 3F).
Discussion
In this study we grouped protein kinases by their inhibition phenotype towards a set of 645 kinase inhibitors. We identified eight promiscuous kinases that bind on average five times more inhibitors than the other kinases and we selected DDR1 as a model kinase in which to explore the potential mechanism of this promiscuous behavior.
The sequence homology based clustering of the human kinome by Manning et al., has been extremely useful to organize kinases into families by sequence relationship (Manning et al., 2002). Since kinases are such prominent drug targets, we expect that clustering of the kinases by their inhibition phenotype could allow medicinal chemists to quickly visualize what kinases and signaling cascades are often co-inhibited. This classification could reveal interactions relevant to polypharmacology or negative side effects.
Importantly, promiscuity is not only a feature of kinase but may also affected by the specificity of inhibitors tested to identify promiscuous kinases. The inhibitors studied here were chosen to be selective probes and their selectivity scores are superior to those of the average clinically approved kinase inhibitor. However, when we repeat this clustering analysis with independent inhibitor sets including clinically approved drugs, we identify essentially the same promiscuous kinases (Figure S2B).
Similarly, clusters of kinases can be analyzed for their shared preference towards certain inhibitor scaffolds. For example, we asked which chemical scaffolds preferentially inhibit the group of eight promiscuous kinases. We found that 40 ligands bound significantly more tightly to the promiscuous kinases than the rest. These inhibitors were enriched for scaffolds containing: 2,4-dianilinopyrimidine, 4-anilinoquinoline, 2-aminobenzimidazole, 6-phenoxy-imidazopyridazine, and 4-anilino or phenoxy pyrrolopyrimidine. Further analysis of these and other ligands that target certain clades of kinases, clustered by phenotype and not genotype, will improve our understanding of targeted kinase inhibitor design.
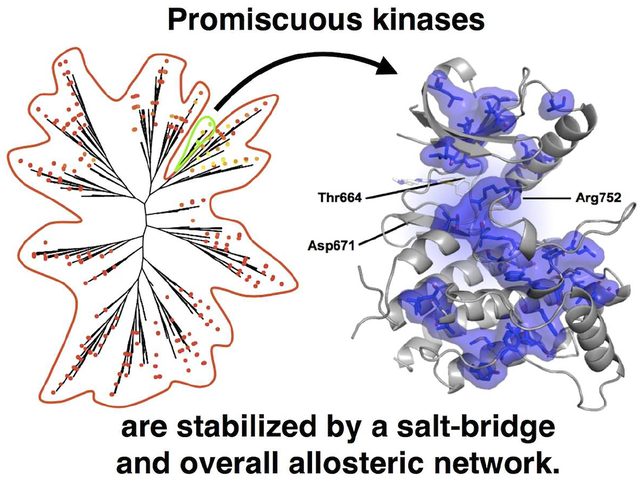
Since the DFG-Asp-out conformation appears to facilitate the promiscuity of DDR1, we wanted to understand whether other promiscuous kinases are also stable in the DFGAsp-out conformation. Therefore, we compared the kinase domain sequences of the eight promiscuous kinases to the rest of the human kinome using the Two Sample Logo (TSL) server (Manning et al., 2002; Vacic et al., 2006). This analysis revealed that residues stabilizing the DFG-Asp-out conformation in DDR1 (Asp671 and Arg752) are enriched among the promiscuous tyrosine kinases (Figure 4A and Figure S5) predicting that this salt bridge could stabilize the DFG-Asp-out conformation also in the other promiscuous kinases. In fact, we found that 25 of the 27 available crystal structures of promiscuous kinases in the PDB are in the DFG-Asp-out conformation and form this salt bridge (Figure 4B and Supplemental Table 2) (Illig et al., 2011; Mol et al., 2004; Murray et al., 2015; Walter et al., 2007; Wang et al., 2016). This includes the apo structures of CSF1R, KIT, and PGDFRA showing that the DFG-Asp-out conformation is energetically accessible in the absence of ligands (Liang et al., 2016; Mol et al., 2004; Walter et al., 2007). It will be interesting to determine the structural basis for the two promiscuous non tyrosine kinases MEK5 and YSK4. They both lack the equivalent to the Asp671-Arg752 (DDR1 numbering) salt bridge and currently no structure is available for these kinases.
Figure 4: Structural hallmarks of DDR1 and other promiscuous kinases.

A) Sequence alignment of the eight promiscuous kinases around Thr664 (the ‘gatekeeper’ mutant), Asp671, and Arg752 (DDR1 numbering). Dark blue highlights residues enriched in the promiscuous set by 50% or more and cyan indicates residues enriched by 10–50% according to Two Sequence Logo comparison (using p-value cutoff 0.01) of the promiscuous kinases to the other 483 kinases analyzed. Bottom: All residues enriched in our promiscuous set by 50% or more are shown as blue surface on the DDR1-VX680 structure. B) Potential salt bridges between residues equivalent to DDR1 Asp671 and Arg752 are shown for representative structures: CSF1R (PDB IDs: 3KRL and 2IOV) in pale cyan, c-Kit (PDB IDs: IT45 and IT46) in pink, DDR1 (PDB IDs: 5FDP and 5BVO) in orange, and PDGFRA (PDB ID: 5GRN) in teal. C) The structures of VX-680 bound to DDR1 (top) and Abl (bottom, PDB entry 2F4J) illustrate the hydrophobic shield formed by the activation loop (blue) and the P-loop (red), respectively. See also Figure S5.
While we identify DDR1 here as a promiscuous kinase, DDR1 can still be inhibited with selective inhibitors. The salt bridge formed between the activation loop and the C-lobe of DDR1 forms a hydrophobic cage that facilitate the binding of many chemically different inhibitors. However, this unique hydrophobic cage has been exploited by highly selective inhibitors for DDR1, such as DDR1-IN-1 (Canning et al., 2014). Interestingly, DDR1-IN-1 bypasses interactions with the threonine gatekeeper residue in DDR1 and instead forms a hydrogen bond with the αD-helix in the C-lobe of DDR1. This interaction is only possible due to anchoring of the activation loop to the C-lobe by the Asp671-Arg752 salt bridge in the DFG-Asp-Out conformation adapted by DDR1.
In addition to providing insights about promiscuous kinases in general, the results presented here suggest a molecular mechanism for the unusually low kinase activity of DDR1 (Leitinger, 2014). Among promiscuous kinases, Tyr755 is only present in DDR1 and DDR2. This tyrosine is part of DDR1/2’s YxxxYY motif, which is an autoinhibitory motif conserved between DDR1/2 and the insulin receptor kinase (IRK) family (Artim et al., 2012; Leitinger, 2014). This autoinhibition is released upon tyrosine phosphorylation at this motif. Unlike the promiscuous tyrosine kinases, the IRK family kinases do not contain the Asp-Arg salt bridge. Consequently, they are catalytically more active, but less promiscuous than DDR1. The presence of both the Tyr755 and the Asp-Arg salt bridge explains why, despite other sequence similarities, DDR1 is less active than both other promiscuous kinases and the kinases of the IRK family (Wei et al., 1995).
Consistent with previous simulations on the DFG-Asp-out/in interconversion of Abl kinase we only observe the DFG flip with protonated Asp747 (Shan et al., 2009). We showed previously that the pKa for the DFG-Asp in Abl is elevated at 6.5. Further ‘constant pH’ simulations where protonation events can happen freely would be required to predict the pKa of Asp747 in DDR1 and the absolute populations of DFG-in/out more precisely (Radak et al., 2017). Importantly, our simulations predict the change in DFG populations (5-fold), in good agreement with the 7–10 fold increase in biochemical kinase activity.
Interestingly, we observed that during the DFG-Asp-out/in interconversion in the DDR1 simulations the aC-helix remained in the ‘in’ conformation. This is different from other kinases, e.g. Abl, in which the aC helix transiently moves out of the way for the DFG-flip to occur (Shan et al., 2009). We speculate that differences in the mechanism of the DFG-flip could lead to differences in the kinetics of the DFG-flip. For Abl kinase, the transition of the protonated DFG aspartate through a hydrophobic pocket accessible upon aC helix movement can become the rate limiting step for the DFG-flip (Shan et al., 2009). Since the DFG-flip in DDR1 appears to occur without movement of the aC helix, this might indicate the absence of this rate limiting step.
To examine the relationship of the relative stability of the DFG-Asp-out conformation to the promiscuous property of DDR1 and other kinases, we compared the conformations of DDR1·VX-680 and Abl·VX-680. As stated earlier, Abl binds VX-680 in the active conformation, where the activation loop extends outwards, but the P-loop of the kinase folds over to shield the inhibitor from the solvent. In DDR1, the kinase is in the inactive conformation, and instead of the P-loop the activation loop provides the hydrophobic contact VX-680 needs to bind (Figure 4C). Similarly, in structures of KIT, DDR1, and CSF1R bound to imatinib, the activation loop provides the hydrophobic shield that the kinked P-loop of Abl provides.
To test our hypothesis that the relative stability of the DFG-Asp-out conformation is related to promiscuity, we looked at data available in the PKIS2 set for a kinase that was tested in a state stabilized in DFG-Asp-in and the DFG-Asp-out conformation. We found data for activation loop phosphorylated and non-phosphorylated Abl kinase. Phosphorylated Abl is stabilized in the DFG-Asp-in active conformation (Hari et al., 2013). The number of compounds Abl binds with high affinity increases in the nonphosphorylated state, in which the DFG-Asp-out conformation is favored (Figure S2C). This implies that also for Abl kinase, the relative stability of the DFG-Asp-out conformation correlates with promiscuity.
How could disease-related mutations affect the stability of the DFG-Asp-out conformation and the ability of kinases to bind ligands with high affinity? The Two Sample Logo analysis identified residues across the entire kinase domain that are specific to the group of promiscuous kinases. These residues could impact the overall stability of the DFG-Asp-out conformation, and in turn, impact the ability of the kinase to bind ligands (Figure 4A). We found that seven of them correspond to sites of clinical mutations that confer imatinib resistance in Abl (Azam et al., 2003) (Figure S5B). Two of these residues (Abl resistance mutants M370T/I and M491I) are distant from the imatinib binding site and the mechanism of their resistance is unclear. Our model suggests that mutations at these sites destabilize the DFG-Asp-out conformation and thereby weaken the affinity for imatinib. In addition, the D681N/Y/G mutations in PDGFRA (Asp671 of the salt bridge in DDR1) are activating mutations which confer resistance to imatinib and sunitinib (COSMIC Study IDs: COSU419, COSU375)(Zehir et al., 2017). This is consistent with our previous finding that a distributed network of residues stabilizes the DFG-Asp-out conformation in kinases (Seeliger et al., 2007). Mutating these residues reduces the stability of the DFG-Asp-out conformation, conferring resistance to ligands like imatinib that favor this conformation. Similarly, when we compare the sequences of DDR1 and the less promiscuous DDR2, we find that no amino acids within 5 Å of the inhibitors differ between DDR1 and DDR2. This indicates that in fact differences in secondary shell or even more remote residues underlie the difference in promiscuity between DDR1 and DDR2 (Figure S5 C,D,E).
Our data indicate that the ability of these promiscuous kinases to bind chemically diverse inhibitors is defined by the hydrophobic pocket formed by the activation loop, which is only accessible in the DFG-Asp-out conformation. Inhibitors do not artificially induce the DFG-Asp-out conformation as was once surmised, but it is a stable, accessible conformation of kinases. Analysis of the available apo-structures of the promiscuous kinases PDGFRA, c-Kit, and CSF1R and our MD analysis of DDR1 show that the DFGAsp-out inactive conformation is the preferred conformation of this unusual set of kinases (Supplementary Table 2). This analysis of a large chemical genomic dataset identified a phenotypically distinct class of medically important signaling enzymes. Further analysis revealed a shared structural mechanism underlying this characteristic that would not have been obtained by sequence comparison alone. We speculate that future analysis of similar datasets will yield further insight into the function, regulation, and druggability of enzymes.
Significance
In this study we have analyzed a publically available data set of protein kinase inhibitors. Rather than analyzing the specificity of kinase inhibitors as is typically done to identify which off-target kinases an inhibitor will affect, we analyzed how kinases differ in their ability to bind a set of fairly specific inhibitors. We were able to group the 400 kinases into families with similar inhibitor binding properties. To our surprise a group of 8 kinase was significantly more promiscuous than the rest. We found that the stability of the DFGAsp-out inactive conformation underlies the promiscuity of these kinases. As more large scale functional data sets become available, we expect that grouping of proteins by functional conservation (e.g. with respect to inhibitor binding) will complement the insight from grouping by sequence conservation to reveal how conserved structural features underlie the function of proteins. In the case of kinases, such studies may aid the development of better therapeutics (e.g. by identifying clusters of commonly co-inhibited kinases) and the understanding the mechanism of resistance mutations.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, John Chodera (john.chodera@choderalab.org) and Markus A. Seeliger (markus.seeliger@stonybrook.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sf9 cells were grown in Sf-900-II medium supplemented with 5% fetal bovine serum and 1x antibiotic/antimitotic solution. Cells were grown according to the Bac-to-Bac protocol from Invitrogen.
METHOD DETAILS
Hierarchical clustering of a large kinase inhibitor set:
Pairwise distances were computed among the 645 inhibitors and 406 kinases to generate the Published Kinase Inhibitor Set 2 (PKIS2) in (Drewry et al., 2017). Inhibition refers here to competitive binding of the inhibitor of interest to the kinase and thereby preventing binding of the kinase to immobilized generic, ATP-competitive kinase inhibitors using the commercial DiscoverX KINOMEscan service.
Distance calculations were computed as Euclidean distances and performed using the dist function in R (version 3.4.0). We performed hierarchical clustering analysis of this dataset using the ‘complete linkage’ method (hclust), in which the similarity of two clusters of kinases is calculated as the similarity of the affinity fingerprint of their most dissimilar members. The resulting dendrogram, in which a cluster of eight kinases is most distant from all other clusters on the tree, i.e., forms the basal cluster when rooted to the midpoint of the tree (Figure 1 and Figure S1). To test the robustness of this result, a consensus tree of 1000 bootstrapped datasets was generated. In this tree, DDR1 and other promiscuous kinases separated into two clades, and not one, distant from other kinases (with YSK4, MEK5, KIT, and PDGFRB in one and RAF1, BRAF, DDR1, DDR2, p38α, and p38β in the other, see Figure S2A for details). Overall, this bootstrap result supports the general trend in which we found our promiscuous kinases distinct from the majority of kinases. We also found that results using the UPGMA or ‘average’ method, while varying in detail, also followed this trend (Figure S1C).
In addition to construction of this affinity-based tree, we quantified promiscuity by counting the number of inhibitors that bind a single kinase with 90% or greater inhibition. The more inhibitors inhibit a given kinase, the more promiscuous is the kinase. A cutoff of either 90% inhibition or 75% inhibition results in similar patterns (Figure S1B), though throughout this paper we have used the 90% cutoff. Mapping these promiscuity values onto the kinome phylogenetic tree was performed using the kinhub.org KinMap tool (Eid et al., 2017). To define ligand chemotypes that are more predominant in the promiscuous kinases, ligands were sorted by those with p values < 0.05 when comparing the mean and SD inhibition within promiscuous kinases to the mean and SD of all other kinases using the Bonferroni-corrected Student’s t-test.
Cloning and constructs:
The construct of human DDR1 kinase domain (residues 526–876, DDR1a numbering, UniProtKB accession ID: Q08345–2) was amplified from a pDNR-Dual mammalian expression vector containing full length human DDR1 isoform A (residues 1–876; purchased from the ASU Gene Repository) and subcloned into the pFastBac HTb vector (Invitrogen) using BamHI and XhoI restriction sites located after the His6-tag and TEV protease cleavage site. Individual mutations were introduced into pFastBac HTb DDR1 KD (D671N, Y755A, and Y759A) by site directed mutagenesis and verified by DNA sequencing. Cloning and baculovirus generation for the kinase domain of DDR1a utilized the Bac-to-Bac baculovirus expression system (Invitrogen).
Protein expression and purification:
His6-tagged DDR1 kinase domain (KD) was expressed in Spodoptera frugiperda (Sf9) insect cells (RRID: CVCL_0549) and purified following the Bac-to-Bac system protocol from Invitrogen. Sf9 cells were grown in Sf-900-II medium supplemented with 5% fetal bovine serum and 1x antibiotic/antimitotic solution. A 1 L culture of Sf9 cells at 0.8 × 106 cells/mL was infected with 30 mL of P3 virus and incubated for 72 hours. Infected cells were centrifuged for 5 minutes at 3000 g at 4°C using the SLC-6000 rotor. Cells were resuspended in lysis buffer (50 mM Tris pH 8.5, 5 mM β-mercaptoehanol, 100 mM NaCl, 1 mM PMSF, and 1% NP-40) in a ratio of 5 mL lysis buffer per 1 g of cells. Lysates were centrifuged at 20,000 g for 60 minutes at 4°C. The resulting supernatant was loaded onto 1 mL of NiNTA resin (Qiagen) per 2 L of insect cells on a gravity column in the cold room (4°C). The resin was washed with 10 column volumes of low salt wash buffer (20 mM Tris pH 8.5, 20 mM imidazole, 500 mM NaCl, 5 mM β-mercaptoehanol, and 10% glycerol) and 10 column volumes of high salt wash buffer (20 mM Tris pH 8.5, 1 M NaCl, 5 mM β-mercaptoehanol and 10% glycerol) followed by 2 column volumes of low salt wash buffer. The protein was eluted and fractionated with 20 mM Tris pH 8.5, 125 mM NaCl, 200 mM Imidazole, 10% glycerol and 5 mM β-mercaptoehanol. The His6-tag of the protein was then cleaved by TEV protease digest overnight at 4°C. The cleaved protein was further purified by size exclusion chromatography (GE 16/60 Superdex 200) in 20 mM Tris pH 8.5, 125 mM NaCl, 20 mM imidazole, 10% glycerol and 5 mM β-mercaptoehanol. The typical yields for DDR1 were approximately 2 mg of protein per liter of Sf9 cells. Protein identity, purity, and verification of no post-translational state was confirmed by LC-MS/MS (Figure S3). No post-translational modifications were detected. For storage, protein was frozen in liquid N2 and stored at −80°C.
DDR1 crystallization:
DDR1 KD was complexed with dasatinib at a concentration of 10 mg/mL protein and 423 μM inhibitor, in buffer containing 20 mM Tris pH 8.5, 125 mM NaCl, 5 mM β-mercaptoehanol and 10% glycerol. DDR1·dasatinib crystals were grown using the hanging drop vapor diffusion method and micro seeding in a mother liquor of 18% PEG 3350 and 0.1 M Bis-Tris pH 5.5. Micro seeds were generated from DDR1·dasatinib crystals grown previously in a mother liquor of 22% PEG 3350, and 0.1 M Bis-Tris pH 5.5. Crystals were cryoprotected in mother liquor plus 20% ethylene glycol and stored in liquid nitrogen.
DDR1·VX-680 complex was formed at 10 mg/mL protein and 423 μM VX-680 in an identical buffer to the DDR1·dasatinib complex. The hanging drop vapor diffusion method and DDR1·dasatinib micro seeds were used to grow crystals in a mother liquor of 18% PEG 3350, 0.05 M NH4I and 0.1 M Bis-Tris pH 5.5. DDR1a·VX-680 crystals were cryoprotected in mother liquor plus 20% glycerol, and stored in liquid nitrogen.
X-ray diffraction data and processing:
X-ray diffraction data were collected at the National Synchrotron Light Source at Brookhaven National Laboratories beamline x29. Data for all protein-drug complexes were collected at 100 K and 1.075 Å wavelength.
DDR1·VX-680 and DDR1·dasatinib crystals diffracted to a resolution of 2.2 Å and 2.6 Å respectively (Table 1). Data for both complexes were processed in space group P212121 using HKL2000 (Otwinowski and Minor, 1997). The structures were solved by molecular replacement using the kinase domain of DDR1 bound to imatinib (PDB entry 4BKJ; residues 599–913) with the αC-helix (residues 660–680), activation loop (residues 775–814), and ligand removed as a search model in Phaser (McCoy et al., 2005). The models were built in Coot (Emsley and Cowtan, 2004) and refined in Phenix (Adams et al., 2002).
Table 1.
Data collection and refinement statistics (molecular replacement)
| DDR1•Dasatinib PDB: 6BSD |
DDR1•VX-680 PDB: 6BRJ |
|
|---|---|---|
| Crystal Parameters | ||
| Space group | P212121 | P212121 |
| a, b, c (Å) | 61.7, 72.3, 74.7 | 61.5, 75.4, 77.2 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Data Collection Statistics | ||
| Wavelength (Å) | 1.075 | 1.075 |
| Resolution (Å) | 47.6–2.61 (2.64–2.61) | 48.1–2.23 (2.26–2.22) |
| Rmerge | 0.174 (1.00) | 0.115 (0.558) |
| I / σI | 13.3 (2.25) | 24.4 (2.97) |
| Completeness (%) | 99.8 (98.0) | 99.3 (93.04) |
| Redundancy | 13.2 (8.0) | 12.2 (4.5) |
| Refinement Statistics | ||
| Protein atoms | 2218 | 2183 |
| B factors (Å2) | 42.2 | 37.3 |
| Ligand/water | 33/61 | 33/104 |
| B factors (Å2) | 41.6/38.5 | 39.3/42.1 |
| Rwork/Rfree | 0.1781/0.2552 | 0.1849/0.2225 |
| RMSD | ||
| Bond lengths (Å) | 0.009 | 0.014 |
| Bond angles (°) | 1.16 | 1.04 |
| Ramachandran Plot (% residues) | ||
| Most Favored | 265 (98.2) | 264 (98.5) |
| Additional Allowed | 5 (1.85) | 4 (1.49) |
| Disallowed Regions | 0 | 0 |
Highest-resolution shell is shown in parentheses.
RMSD, root mean square deviation
DDR1 kinase activity assays:
In vitro kinase activity for DDR1 KD was measured by substrate peptide phosphorylation using [γ−32P] ATP in a phosphocellulose paper binding assay (Casnellie, 1991). Reactions included 20 mM Tris pH 7.4, 10 mM MgCl2, 400 μM ATP, 200 μM Axltide (KKSRGDYMTMQIG), 0.5 μM DDR1 KD and 50–100 cpm/pmol [γ−32P] ATP. Reaction mixtures were incubated for 60 minutes at 30°C and stopped by the addition of trichloroacetic acid (TCA) and centrifuged to separate the precipitated kinase from the soluble substrate peptide. Supernatant was blotted onto phosphocellulose paper and washed three times with phosphoric acid. The phosphocellulose paper was dried and radioactivity was quantified by scintillation counting.
DDR1 drug binding assay:
Drug binding assays were performed using the Invitrogen LanthaScreen Eu kinase binding assay according to manufacturer’s instructions. First we determined the dissociation constant of DDR1 wt and mutant proteins to the Alexa Fluor-647-labeled tracer 178 molecule that binds to the ATP binding site of DDR1. 5 uL of DDR1 at 15 nM concentration was mixed with 5 uL of Eu-labeled anti-His6 antibody at 6 nM concentration and 5 uL of tracer at concentration ranging from 1500 nM to 0 nM in assay buffer (50 mM Hepes pH7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% Brij-35) and 3% DMSO. A series of control experiments contained 30 μM dasatinib in the assay buffer. Samples were mixed in a white 384-well plate and incubated in the dark at room temperature for 60 min. Time-resolved fluorescence energy transfer was measured in a Molecular devices SpectraMax M5 Microplate reader using the following settings: Excitation 332 nm, Emission 620/665 nm, Emission cutoff 550 nm, TRF integration delay 50 ms, Integration time 400 μs, 100 flashes per read, calibration on. The TR-FRET ratio at 665 and 620 nm was calculated for each concentration of tracer. The TR/FRET ratio for the dasatinib control was subtracted from the TR FRET ratio of the dasatinib-free samples at the same tracer concentration. The tracer binding isotherm was fit to a quadratic binding equation to yield the dissociation constant KDtracer of the tracer to each of the kinase constructs tested.
Binding of imatinib to DDR1 proteins was measured by competition with tracer 178. 5 μL of DDR1 at 15 nM and 6 nM Eu-labeled anti-His body in assay buffer was mixed with 5 μL of tracer 178 at 30 nM in assay buffer and 5 μL of imatinib (30 μM −0.004 μM) in assay buffer with 3% DMSO. Samples were mixed in a white 384-well microtiter plate and incubated in the dark for 60 min at room temperature. TR FRET ratios were determined as described above. The TR FRET signal at different imatinib concentrations was fit to the following equation using Kaleidagraph:
where m1 is the minimum TR FRET value, m2 is the maximum TR FRET value and m3 is the concentration at which the amplitude of the TRFRET signal decreases by 50% (IC50imatinib).
Using the Cheng-Prusoff relationship, the IC50imatinib values are converted into inhibitory constants Kimatinibi (Cheng and Prusoff, 1973):
where [tracer] is the concentration of tracer 178 in the binding competition reaction and KDtracer is the dissociation constant of tracer for the specific kinase or mutant kinase tested.
Molecular dynamics simulation of DDR1 wild-type:
Simulations of WT DDR1 kinase domain (residues 567–875 according to human DDR1a numbering) were initiated from the structure of DDR1 in complex with VX-680 (this study, PDB ID 6BRJ), as well as complexes with imatinib and ponatinib (PDB IDs 4BKJ and 3ZOS, respectively) (Canning et al., 2014). In all cases, the ligand was removed and only the kinase domain was simulated. Separate sets of simulations were performed with the Asp747 in the DFG motif either protonated or deprotonated, producing a total of six initial structures. Previous simulations of Abl kinase showed that protonation of the Asp in the DFG motif enhances the likelihood of seeing the interconversion between DFG-Asp-in and DFG-Asp-out, the so-called DFG flip (Shan et al., 2009).
Simulations were run using OpenMM 6.3 derived core (core21 v0.0.18) on Folding@home after a short equilibration period (consisting of 100 ps implicit solvent simulation using the OBC GBSA implicit solvent model (Onufriev et al., 2004) followed by 100 ps explicit solvent simulation under isothermal-isobaric (NPT) conditions) (Eastman et al., 2017, Shirts and Pande, 2000, Eastman et al., 2013). Simulations were run in a box of 16,046 explicit TIP3P water molecules (Jorgensen et al., 1983) using the Amber99SB-ILDN force field (Lindorff-Larsen et al., 2010), at 300 K, using the Leapfrog Langevin integrator with a timestep of 2 fs, and a collision rate of 1 / ps. A molecular-scaling Monte Carlo barostat was used with default update interval of 25 steps and pressure of 1 atm. Particle Mesh Ewald (PME) with a cutoff of 9 Å and default parameters was used for long-range electrostatics. For each of the six WT starting conditions, 50 simulations of ~3 μs each were run totaling ~150 μs, resulting in ~0.9 ms in aggregate simulation time for all starting structures.
Molecular dynamics of DDR1 mutants and wild-type started along the DFG flip:
To enhance sampling of the DFG flip, new simulations were adaptively seeded along the DFG flip observed from the unbiased simulations above. From this original set of WT DDR1simulations, four of the Asp747-protonated simulations interconverted from the DFG-Asp-out conformation to the DFG-Asp-in conformation. From each of these four simulations, nine snapshots were chosen at even intervals along the coordinate originally used to identify trajectories: the distance between the CZ atom of the Phe748 of the DFG motif and CA atom of the Gly716 in the αE-helix, seen to be either comparable or preferable to the Asp747 phi dihedral as an order parameter. This resulted in 36 structures from which new simulations were initiated for eight chemically distinct systems: WT, D671N, Y755A, and Y759A, each with Asp747 either protonated or deprotonated. Simulations from the original three starting structures (DDR1·VX-680, 4BKJ, and 3ZOS) were also started for all eight of these systems. Additionally all eight systems were started from the DDR1·dasatinib structure, with the unresolved loop in the N-lobe of this structure modeled in from the DDR1·VX-680 structure. To ensure uniform preparation for all 320 (8 systems × 40 structures) of these new simulations, Ensembler was used to automate the process (Parton et al., 2016). Ensembler uses Modeller to model in mutants where needed (Fiser and Sali, 2003). The overall simulation protocol was the same as described above for the WT simulations and 20 simulations of 1 μs each were generated by Folding@home for each of 320 starting conformations, totaling 6.5 ms in aggregate for simulations restarted along the DFG flip, more than previously published for any kinase.
Molecular simulation analysis using Markov models:
Free energy landscapes of DDR1 simulations were built using the PyEMMA Python library (Scherer et al., 2015). Trajectories were first featurized according to 25 features of interest that were either previously used to investigate the DFG-Asp-in vs. DFG-Asp-out conformation (Mobitz, 2015, Meng et al., 2015, Hu et al., 2015) or were of particular interest to this system, e.g. the Asp671-Arg752 distance (Supplemental Table 1). The time-lagged independent component analysis (TICA) projection was then calculated from these coordinates to find the most kinetically relevant linear combination (Perez-Hernandez et al, 2013), and cumulative sum of the eigenvalues began to flatten despite the small number coordinates used (Figure S4A). Commute mapping and a lag time of 25 ns were used. K-means clustering of the TICA coordinates was used to generate a 300 microstate model and corresponding transition matrix, which was then used to determine the parameters of a two-state Bayesian hidden Markov Model (HMM) (Noé et al., 2013). The two-state HMM separated the DFG-Asp-in and DFG-Asp-out states for the WT simulation (Figure S4D), and the largest time-scale separation consistently supported a two-state model (Figure S4C), though these two states were different between WT and mutants (Figure S4E). Markov state models (MSM) (Prinz et al., 2011) were then built for all the mutant variants within the TICA space of the WT-protonated simulations. A lag time of 50 ns was used for constructing the MSM. Note that for the WT simulations, all simulations, including initial simulations and re-seeded simulations were analysed, though the nature of the MSM analysis ought to correct for any error introduced by this additional data. Chapman-Kolmogorov tests of the WT HMM and the MSMs of all mutants indicated that these models were valid representations of our real data (Figure S4B). To compare the relative free energies of the states, a line was found using the scikit learn C-support vector classification algorithm with C=1.0 and gamma=0.7 that separates the two HMM states in our WT simulations (Figure S4D). The ratio between the populations of the two states was used to estimate the free energy difference between the states.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean values ± standard error of the mean (s.e.).
DATA AND SOFTWARE AVAILABILITY
The structures of DDR1·VX-680 and DDR1·dasatinib were deposited in the PDB under ID codes 6BRJ and 6BSD respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| VX-680 (MK-0457, Tozasertib) | Selleck Chemicals | Cat # S1048 |
| Dasatinib | Cayman Chemical | Cat#11498 |
| Imatinib | Novartis | |
| Axltide | Sigma Aldrich | Cat # 12–516 |
| [Y-32P] ATP | Perkin Elmer | Cat # NEG002A |
| Trichloroacetic acid | Sigma Aldrich | Cat # T4885–500G |
| TEV Protease | Lab generated | N/A |
| Tris-HCl | Sigma Aldrich | Cat # T3253–5KG |
| PMSF | EMD Millipore | Cat # 52332–25GM |
| NP-40 | Fluka | Cat # 74385 |
| β-mercaptoehanol | Sigma Aldrich | Cat # M6250 |
| Sodium Chloride | Fisher Scientific | Cat # BP358–10 |
| NiNTA resin | Qiagen | Cat # 30230 |
| Imidazole | Alfa Aesar | Cat # A10221–2500g |
| Glycerol | Fisher Scientific | Cat # G37–20 |
| PEG 3350 | Sigma Aldrich | Cat # P3015–500G |
| Bis-Tris | Sigma Aldrich | Cat # B9754–100G |
| Ethylene glycol | Sigma Aldrich | Cat # 102466 |
| Ammonium Iodide | Sigma Aldrich | Cat # 03101–100G |
| Kinase tracer 178 | Invitrogen | Cat # PV5593 |
| LanthaScreen Eu-anti-His Antibody | Invitrogen | Cat # PV5596 |
| EGTA | Acros Organics | Cat # 428570100 |
| Brij-35 | Acros Organics | Cat # 329581000 |
| MgCl2 | Sigma Aldrich | Cat # M9272 |
| HEPES | Spectrum | Cat # H1084 |
| Critical Commercial Assays | ||
| Experimental Models: Cell Lines | ||
| Sf9 | ATCC | Cat # CRL-1711 CVCL_0549 |
| Software and Algorithms | ||
| HKL-2000 | HKL Research, Inc |
http://www.hkl-xray.com/hkl-2000 SCR_015547 |
| CCP4 - Phaser | University of Cambridge; Cambridge; United Kingdom |
https://www.phenix-online.org/documentation/reference/phaser.html SCR_014219 |
| Phenix Crystallography Suite | Lawrence Berkeley National Laboratory, University of California at Berkeley; California; USA |
https://www.phenix-online.org/ SCR_014224 |
| Coot | MRC Laboratory of Molecular Biology |
http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ SCR_014222 |
| Kaleidagraph | Synergy software | |
| Other | ||
| Phosphocellulose paper | Milipore | IPVH 00010 |
| Fluorimeter | Horriba Scientific | Fluoromax 4 |
| Microplatereader | Molecular Devices | SpectraMax M5 |
| Deposited Data | ||
| Structure of DDR1•VX680 | This study | PDB 6BRJ |
| Structure of DDR1•Dasatinib | This study | PDB 6BSD |
| Molecular simulations for DDR1 | This study | Open Science Framework at https://osf.io/4r8x2/. |
| Dendrogram presented in Figure 1A | This study | GitHub at https://github.com/choderalab/DDR1_and_kinase_promiscuity_materials |
| Kinase inhibition data | (Drewry et al., 2017) | |
| Structure of Abl•VX680 | https://www.rcsb.org/ | PDB 2F4J |
| Structure of Abl•Dasatinib | https://www.rcsb.org/ | PDB 2GQG |
| Structure of DDR1•Imatinib | https://www.rcsb.org/ | PDB 4BKJ |
| Structure of CSF1R | https://www.rcsb.org/ | PDB 3KRL |
| Structure of CSF1R | https://www.rcsb.org/ | PDB 2IOV |
| Structure of c-Kit | https://www.rcsb.org/ | PDB IT45 |
| Structure of c-Kit | https://www.rcsb.org/ | PDB IT46 |
| Structure of DDR1 | https://www.rcsb.org/ | PDB 5FDP |
| Structure of DDR1 | https://www.rcsb.org/ | PDB 5BVO |
| Structure of PDGFRA | https://www.rcsb.org/ | PDB5GRN |
| Structure of DDR1•ponatinib | https://www.rcsb.org/ | PDB 3ZOS |
| Structure of Abl•ponatinib | https://www.rcsb.org/ | PDB 3OXZ |
| Structure of Abl•imatinib | https://www.rcsb.org/ | PDB 2HYY |
Highlights.
Clustering of kinome by inhibition phenotype identifies promiscuous kinases.
Promiscuous tyrosine kinases are stable in the DFG-Asp-out inactive conformation.
Residues define promiscuous kinases and stabilize inactive conformation.
Acknowledgements
We acknowledge support for this work by NIH R35 GM119437 (M.A.S), R01 GM108904 (J.S.R), P30 CA008748, R01 GM1215 (J.D.C), R01 CA58530 (W.T.M), SKI and Cycle for Survival (J.D.C). We acknowledge the generosity of donated CPU time of Folding@home donors and statistical consulting by the Stony Brook University Biostatistical Consulting Core. X-ray diffraction data were collected at the National Synchrotron Light Source at Brookhaven National Laboratories beamline x29.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
J.D.C. is a member of the S.A.B. of Schrödinger LLC.
Data generated in this study are available at https://github.com/choderalab/DDR1_and_kinase_promiscuity_materials https://osf.io/4r8x2/ and upon request to the corresponding authors, John D. Chodera (john.chodera@choderalab.org) and Markus A. Seeliger (markus.seeliger@stonybrook.edu)
References
- Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, et al. (2006). Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nature Chemical Biology 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, and Peterson JR (2011). Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature Biotechnology 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artim SC, Mendrola JM, and Lemmon MA (2012). Assessing the range of kinase autoinhibition mechanisms in the insulin receptor family. The Biochemical journal 448, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Latek RR, and Daley GQ (2003). Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843. [DOI] [PubMed] [Google Scholar]
- Canning P, Tan L, Chu K, Lee SW, Gray NS, and Bullock AN (2014). Structural Mechanisms Determining Inhibition of the Collagen Receptor DDR1 by Selective and Multi-Targeted Type II Kinase Inhibitors. Journal of molecular biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, and Prusoff WH (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- Cohen NA, Kim TS, and DeMatteo RP (2017). Principles of Kinase Inhibitor Therapy for Solid Tumors. Ann Surg 265, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium SG, Consortium CSG, Consortium NSG, Gräslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, et al. (2008). Protein production and purification. Nature Methods 5, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E, Waters B, Spiegel K, Alnadaf T, Manley PW, Buchdunger E, Walker C, and Jarai G (2008). Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. European journal of pharmacology 599, 44–53. [DOI] [PubMed] [Google Scholar]
- Deng X, Elkins JM, Zhang J, Yang Q, Erazo T, Gomez N, Choi HG, Wang J, Dzamko N, Lee J-D, et al. (2013). Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones. Journal of Biotechnology 70, 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewry DH, Wells CI, Andrews DM, Angell R, Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen M, et al. (2017). Progress towards a public chemogenomic set for protein kinases and a call for contributions. Plos One 12, e0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, amp, apos, Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MWN, Silver RT, et al. (2006). Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. The New England journal of medicine 355, 2408–2417. [DOI] [PubMed] [Google Scholar]
- Eckstein N, Röper L, Haas B, Potthast H, Hermes U, Unkrig C, Naumann-Winter F, and Enzmann H (2014). Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. Journal of Experimental & Clinical Cancer Research 33, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Santaguida S, Musacchio A, and Knapp S (2012). Crystal structure of human aurora B in complex with INCENP and VX-680. J Med Chem 55, 7841–7848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro D (2015). 25 years of small molecular weight kinase inhibitors: potentials and limitations. Mol Pharmacol 87, 766–775. [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. (2005). A small molecule-kinase interaction map for clinical kinase inhibitors. Nature Biotechnology 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Hari SB, Perera BG, Ranjitkar P, Seeliger MA, and Maly DJ (2013). Conformation-selective inhibitors reveal differences in the activation and phosphate-binding loops of the tyrosine kinases Abl and Src. Acs Chem Biol 8, 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, et al. (2004). VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 10, 262–267. [DOI] [PubMed] [Google Scholar]
- Huang D, Zhou T, Lafleur K, Nevado C, and Caflisch A (2010). Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics (Oxford, England) 26, 198–204. [DOI] [PubMed] [Google Scholar]
- Illig CR, Manthey CL, Wall MJ, Meegalla SK, Chen J, Wilson KJ, Ballentine SK, Desjarlais RL, Schubert C, Crysler CS, et al. (2011). Optimization of a potent class of arylamide colony-stimulating factor-1 receptor inhibitors leading to anti-inflammatory clinical candidate 4-cyano-N-[2-(1-cyclohexen-1-yl)-4-[1-[(dimethylamino)acetyl]-4-piperidinyl]pheny l]-1H-imidazole-2-carboxamide (JNJ-28312141). J Med Chem 54, 7860–7883. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. (2008). A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnology 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C, et al. (2017). The target landscape of clinical kinase drugs. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothiwale S, Borza CM, Lowe EW, Pozzi A, and Meiler J (2015). Discoidin domain receptor 1 (DDR1) kinase as target for structure-based drug discovery. Drug discovery today 20, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, and Wang JYJ (2009). Exploiting the promiscuity of imatinib. Journal of biology 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger B (2014). Discoidin Domain Receptor Functions in Physiological and Pathological Conditions. In (Elsevier; ), pp. 39–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Yan X-E, Yin Y, and Yun C-H (2016). Structural and biochemical studies of the PDGFRA kinase domain. Biochemical and Biophysical Research Communications 477, 667–672. [DOI] [PubMed] [Google Scholar]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, et al. (2004). Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 47, 6658–6661. [DOI] [PubMed] [Google Scholar]
- Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, Mestan J, Trappe J, Wartmann M, and Fabbro D (2010). Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochimica et biophysica acta 1804, 445–453. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science (New York, NY) 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, and Wilson KP (2004). Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem 279, 31655–31663. [DOI] [PubMed] [Google Scholar]
- Murray CW, Berdini V, Buck IM, Carr ME, Cleasby A, Coyle JE, Curry JE, Day JE, Day PJ, Hearn K, et al. (2015). Fragment-Based Discovery of Potent and Selective DDR1/2 Inhibitors. ACS Med Chem Lett 6, 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, and Kuriyan J (2002). Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer research 62, 4236–4243. [PubMed] [Google Scholar]
- Paricharak S, Klenka T, Augustin M, Patel UA, and Bender A (2013). Are phylogenetic trees suitable for chemogenomics analyses of bioactivity data sets: the importance of shared active compounds and choosing a suitable data embedding method, as exemplified on Kinases. Journal of cheminformatics 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak BK, Chipot C, Suh D, Jo S, Jiang W, Phillips JC, Schulten K, and Roux B (2017). Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. Journal of chemical theory and computation 13, 5933–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters A, Nguyen HD, Phan T, Simard JR, Grütter C, Engel J, and Rauh D (2014). Identification of Type II and III DDR2 Inhibitors. Journal of medicinal chemistry 57, 4252–4262. [DOI] [PubMed] [Google Scholar]
- Rix U, Remsing Rix LL, Terker AS, Fernbach NV, Hantschel O, Planyavsky M, Breitwieser FP, Herrmann H, Colinge J, Bennett KL, et al. (2010). A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, UK 24, 44–50. [DOI] [PubMed] [Google Scholar]
- Salah E, Ugochukwu E, Barr AJ, von Delft F, Knapp S, and Elkins JM (2011). Crystal structures of ABL-related gene (ABL2) in complex with imatinib, tozasertib (VX-680), and a type I inhibitor of the triazole carbothioamide class. J Med Chem 54, 2359–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, and Kuriyan J (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science (New York, NY) 289, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, and Kuriyan J (2007). c-Src Binds to the Cancer Drug Imatinib with an Inactive Abl/c-Kit Conformation and a Distributed Thermodynamic Penalty. Structure 15, 299–311. [DOI] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, and Sawyers CL (2004). Overriding Imatinib Resistance with a Novel ABL Kinase Inhibitor. Science 305, 399–401. [DOI] [PubMed] [Google Scholar]
- Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MØ, Dror RO, Kuriyan J, and Shaw DE (2009). A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proceedings of the National Academy of Sciences of the United States of America 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, et al. (2006). The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res 66, 5790–5797. [DOI] [PubMed] [Google Scholar]
- Vacic V, Iakoucheva LM, and Radivojac P (2006). Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 22, 1536–1537. [DOI] [PubMed] [Google Scholar]
- Walter M, Lucet IS, Patel O, Broughton SE, Bamert R, Williams NK, Fantino E, Wilks AF, and Rossjohn J (2007). The 2.7 A crystal structure of the autoinhibited human c-Fms kinase domain. J Mol Biol 367, 839–847. [DOI] [PubMed] [Google Scholar]
- Wang Z, Bian H, Bartual SG, Du W, Luo J, Zhao H, Zhang S, Mo C, Zhou Y, Xu Y, et al. (2016). Structure-Based Design of Tetrahydroisoquinoline-7-carboxamides as Selective Discoidin Domain Receptor 1 (DDR1) Inhibitors. J Med Chem 59, 5911–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Hubbard SR, Hendrickson WA, and Ellis L (1995). Expression, Characterization, and Crystallization of the Catalytic Core of the Human Insulin Receptor Protein-tyrosine Kinase Domain. Journal of Biological Chemistry 270, 8122–8130. [DOI] [PubMed] [Google Scholar]
- Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S, Hua XH, Ford JM, Armstrong RC, Zarrinkar PP, et al. (2010). Activation State-Dependent Binding of Small Molecule Kinase Inhibitors: Structural Insights from Biochemistry. Chemistry & Biology 17, 1241–1249. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, and Clausen MH (2015). FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 36, 422–439. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, and Clausen MH (2016). Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today 21, 5–10. [DOI] [PubMed] [Google Scholar]
- Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs WH, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, et al. (2006). Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer research 66, 1007–1014. [DOI] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, and Gray NS (2009). Targeting cancer with small molecule kinase inhibitors. Nature Reviews Cancer 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, and Gray NS (2014). Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? Acs Chem Biol 9, 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures of DDR1·VX-680 and DDR1·dasatinib were deposited in the PDB under ID codes 6BRJ and 6BSD respectively.