

SUMMARY
Ectopic expression of combinations of transcription factors (TF) can drive direct lineage conversion, thereby reprogramming a somatic cell’s identity. To determine the molecular mechanisms by which Gata4, Mef2c, and Tbx5 (GMT) induce conversion from a cardiac fibroblast toward an induced cardiomyocyte, we performed comprehensive transcriptomic, DNA-occupancy, and epigenomic interrogation throughout the reprogramming process. Integration of these data sets identified new TFs involved in cardiac reprogramming and revealed context-specific roles for GMT, including the ability of Mef2c and Tbx5 to independently promote chromatin remodeling at previously inaccessible sites. We also find evidence for cooperative facilitation and refinement of each TF’s binding profile in a combinatorial setting. A reporter assay employing newly defined regulatory elements confirmed binding of a single TF can be sufficient for activation, suggesting co-binding events do not necessarily reflect synergy. These results shed light on fundamental mechanisms by which TFs direct lineage conversion.
Keywords: Reprogramming, cardiomyocyte, cardiac fibroblast, transcription factor, ATAC-seq, single cell RNA-seq, ChIP-seq
Graphical Abstract
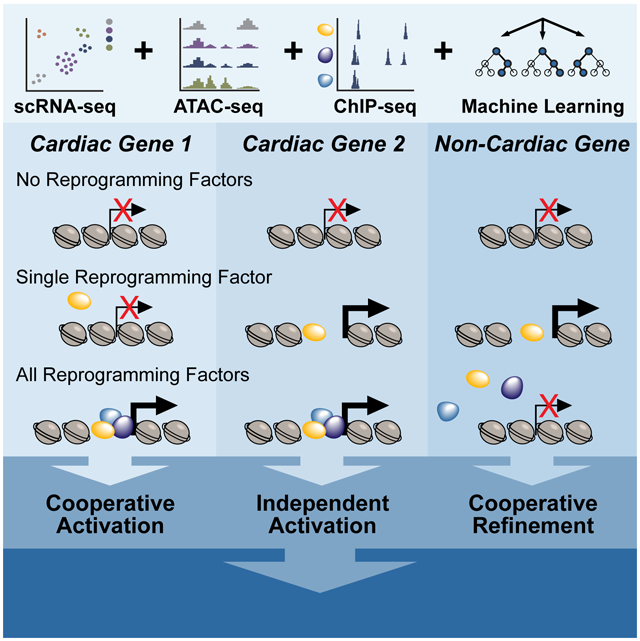
eTOC
Srivastava and colleagues integrate multiple (epi)genomic assays to dissect the mechanisms by which transcription factors function independently and combinatorially to initiate cell fate transitions. Heterogeneous binding relationships between Gata4, Mef2c, and Tbx5 highlight the context-specific mechanisms that dictate cardiac reprogramming.
INTRODUCTION
Somatic cellular identity is established by complex gene regulatory networks during embryonic development. Knowledge of these networks has been exploited to devise combinations of transcription factors that facilitate direct reprogramming of somatic cells from one lineage to another without progression through an intermediate pluripotent state (Feng et al., 2008; Ieda et al., 2010; Rackham et al., 2016; Wernig et al., 2008). However, the precise mechanisms by which various combinations lead to cellular specificity is only beginning to be understood (Treutlein et al., 2016; Wapinski et al., 2017).
Direct reprogramming of cardiac fibroblasts to cardiomyocytes has been achieved in a variety of ways, including ectopic expression of cardiac-enriched transcription factors (Fu et al., 2013; Ieda et al., 2010; Nam et al., 2013; Qian et al., 2012; Song et al., 2012). Ectopic expression of Gata4, Mef2c, and Tbx5 (GMT) is sufficient to alter the fibroblast epigenome and promote expression of genes associated with cardiomyocytes while simultaneously repressing the fibroblast gene program (Ieda et al., 2010; Liu et al., 2017; Zhou et al., 2016). Perturbation of epigenetic remodelers and ectopic expression of additional transcription factors such as Hand2 and Myocd, or microRNAs, were found to influence reprogramming as well, with addition of Hand2 resulting in a greater portion of pacemaker-like cells (Addis et al., 2013; Christoforou et al., 2013; Jayawardena et al., 2015; Nam et al., 2014; Protze et al., 2012; Zhou et al., 2016). Concomitant chemical inhibition of TGFβ and Wnt signaling resulted in improved reprogramming both in vitro and in vivo (Ifkovits et al., 2014; Mohamed et al., 2017). Introduction of reprogramming factors directly into the heart after damage by a gene therapy approach resulted in significantly improved cardiac function, suggesting potential therapeutic benefits of in vivo cardiac reprogramming for regenerative medicine (Jayawardena et al., 2015; Qian et al., 2012; Song et al., 2012).
Cardiac reprogramming factors, like other reprogramming factors, are typically tissue-enriched rather than tissue-specific, yet somehow still induce a unique fate switch. Gata4, Mef2c, and Tbx5 each have essential functions in a wide range of tissues during embryonic development, and their deletion individually leads to embryonic lethality and gross malformation of various organ systems, including the developing cardiovascular system (Bruneau et al., 2001; Lin et al., 1997; Molkentin et al., 1997). Gata4 interacts with Nkx2-5 and Tbx5 to promote cardiovascular development, but it also cooperates with Foxa2 during endoderm development to promote expression of a transcriptional network required for foregut development (Holtzinger and Evans, 2005). Similarly, beyond cardiogenesis Tbx5 is also required for limb development (Agarwal et al., 2003; Ahn et al., 2002), while Mef2c is also essential for neural development (Li et al., 2008; Shalizi et al., 2006). Thus, transcription factor combinations such as GMT likely provide specificity to genome-wide epigenomic remodeling, but how they achieve tissue-specific transcriptional regulation remains unknown.
Gata4 is a zinc finger transcription factor capable of interacting with heterochromatin, but its ability to interact with regions containing DNA methylation is limited (Cirillo et al., 2002; Oda et al., 2013). Little is known regarding the ability of Tbx5 and Mef2c to bind to compact chromatin. One screen identified TBX5 as a factor capable of inducing DNA demethylation when expressed ectopically (Suzuki et al., 2017). While the function of Mef2c in this regard remains unclear, a study of the closely related factor Mef2d in photoreceptor cells found that it requires additional co-factors to access regions that do not encode strong consensus Mef-response motifs (Andzelm et al., 2015). Which of these factors, if any, functions to open closed chromatin in the context of direct reprogramming, and whether they require combinatorial interaction to do so, remains unknown.
Here, we investigated the genome-wide consequences of Gata4, Mef2c, and Tbx5 expression, alone and in combination, in cardiac fibroblasts as the cells underwent reprogramming towards a cardiomyocyte-like state. By combining single cell RNA-sequencing, ChIP-seq of GMT, and ATAC-seq analyses, we found that epigenomic and transcriptional changes occurred rapidly within the first 24-48 hours of reprogramming. Cells that adopted a trajectory toward the cardiac fate could largely be predicted by virtue of early gene expression changes and reprogramming factor expression. A machine learning model of gene expression changes as a function of transcription factor binding motifs in dynamic open chromatin regions identified new candidate factors involved in reprogramming. Although GMT are each capable of promoting chromatin remodeling when expressed individually, we found that accessibility changes during direct reprogramming were primarily associated with Mef2c and Tbx5 binding only. Cooperative activity between Gata4, Tbx5, and Mef2c was evident as combinatorial expression resulted in refinement and facilitation of DNA binding by these factors compared to single factor expression, and combinatorial binding correlated with opening of chromatin particularly at cardiac loci.
RESULTS
Cardiac Reprogramming Occurs Rapidly and at Variable Rates
To determine the discrete temporal transcriptional response to reprogramming with GMT in the context of TGFβ and Wnt inhibition, we performed single cell RNA-sequencing during cardiac reprogramming of Thy1 positive (Thy1+) cells, largely representing fibroblasts, isolated from neonatal mouse hearts that encode an αMHC-GFP reporter activated during reprogramming (Ieda et al., 2010) (Figure 1A). We collected and analyzed 29,718 cells representing five time points after transduction with retroviruses encoding Gata4, Mef2c, and Tbx5 (days −1, 1,2, 3, 7). We additionally collected cells sorted at day 14 using the aforementioned reporter.
Figure 1. Single Cell Expression Analysis of Direct Cardiac Reprogramming.

(A) Schematic of reprogramming system and main reprogramming milestones.
(B) UMAP visualization of the cell clustering (0-13).
(C) UMAP visualization of cells in (B) colored by collection time (days).
(D) Stacked bar plot indicating the relative contribution of cells from each time point (as shown in C) to each cluster (as shown in B).
(E) Heatmap showing expression of top 20 differentially expressed genes for each cluster. Red indicates higher expression; blue lower. Clusters (top bar, colors as in (B)) are ordered according to dendrogram in (D). Two representative marker genes per cluster are labelled at right.
(F) Pseudotime trajectory of cells from clusters 0-5, 7, and 10. Cell color is based on cluster color in (B).
(G) Expression [log10(UMI+0.1)] of branch marker genes (Tnni3, Mmp3, Ccnbl, and Slc1a6) visualized in pseudotime trajectory plots from (F). See also Figure S1 and Table S1.
Transcript information from all samples was aggregated and 14 distinct transcriptional signatures were identified (Figure 1B, Table S1) (Becht et al., 2018; Butler et al., 2018). Excluding the clusters that exclusively represent day −1 (clusters 6 and 11), all clusters included cells collected at each time point, highlighting the limited technical variability between our timepoints and the heterogeneous response to GMT (Figure 1C, D).
To understand the biological significance of the 6 main groups of cells identified through hierarchical clustering of our populations, we identified representative gene signatures for each cluster (Figure 1E). 7,395 genes were differentially expressed (p < 0.01, average log fold change > 0.3) across the time course (Table S1). Three populations represented non-fibroblast cell types present in the starting population: epicardium-derived (clusters 11 and 12, Lrrn4 and Mgp), endocardium (cluster 13, Emcn and Egfl7), and macrophages (clusters 8 and 9, Lyz2 and Spp1) (Figure 1D-E, S1B) (Cavallero et al., 2015; Xiao et al., 2018). Notably, the expression of genes such as Lrrn4 and Emcn exclusively within endocardium and epicardium cells throughout our time course contradicts a previous report that suggested these genes are expressed at early stages in reprogramming cells but are subsequently repressed by GMT expression (Figure 1C-D) (Liu et al., 2017). Instead, our analysis suggests these distinct cell types persist in the population in low numbers and likely were not detected in the previous study due to a limitation in the number of cells captured.
Four signatures putatively represented various stages or outcomes of cardiac reprogramming. The initial stages (early iCMs) were identified by activation of genes such as Nid2 and Tnnt2 and incomplete repression of fibroblast-associated genes such as Lmcd1 and Ptgs2 (clusters 0 and 5; Figure 1E, S1B). This signature was present within 48 hours of GMT transduction, in agreement with previous reports (Liu et al., 2017; Sauls et al., 2018). The late stages (late iCMs) were marked by expression of cardiomyocyte-related genes such as Tnni3 and Myl7, and downregulation of the fibroblast genes such as Ptgs2 (cluster 2; Figure 1E). Unexpectedly, this cluster contained cells collected from days 3 (5% of day 3 cells) and 7 (26% of day 7 cells), as well as reporter-positive cells collected on day 14 (“+14r”; Figure 1C-D), suggesting a reprogrammed state can be acquired rapidly.
The two additional signatures appear to contain alternative reprogramming outcomes. The first exhibits expression of various genes associated with the cell cycle such as Cdk1 and Ccnbl (clusters 4 and 10; Figure 1E). The second activates genes that become more robustly expressed in cluster 7, such as Mmp3 and Figf (Figure 1E, S1B). Cluster 7 is similar to clusters 11 and 12, which are found in the starting population, suggesting cluster 7 represents cells that do not acquire a cardiac fate nor enter the cell cycle (Figure 1D-E). Gene ontology (GO) analysis of genes activated in cluster 7 (compared to cluster 11, n=488 genes, average log fold change > 0.3, p < 1e-10) revealed biological processes associated with vasculature and blood vessel development (p = 1.76e-13 and 5.32e-13, respectively). These cells uniquely activate genes associated with vascular developmental processes such as Epas1, Figf, and Sox9 (Figure 1E, Table S1) (Achen et al., 1998; Lincoln et al., 2007; Tian et al., 1997). They also continue to express genes associated with the starting fibroblast state, such as Dcn and Tbx20 (Table S1).
To better understand the associations between identified clusters and establish a transcriptional trajectory of the reprogramming process, we next ordered clusters in pseudotime using Monocle (Cao et al., 2019). Clusters containing fibroblasts (cluster 6) and non-fibroblast cell types identified in the starting population (clusters 8, 9, 11, 12, and 13) were eliminated as they do not reflect the outcome of reprogramming. This analysis revealed a tree structure with distinct branches indicating three possible outcomes that originate from Slc1a6 positive cells (Figure 1F): one characterized by progressive activation of Mmp3, another by activation of cardiac genes such as Tnni3, and the third by markers of cell cycle progression (e.g. Ccnb1) (Figure 1G). The inhibitory effect imposed by proliferation during cardiac reprogramming is consistent with the prior observation that continued proliferation prevents fibroblast reprogramming to a pluripotent state (Xu et al., 2013). Collectively, these data suggest that a reprogramming trajectory can be acquired within 48 hours of GMT transduction but that reprogramming progresses at variable rates in individual cells.
Reprogramming Trajectory is Entered Quickly and Driven by GMT Transduction
To understand how ectopic GMT expression may dictate the observed transcriptional trajectories, we assayed expression of Gata4, Mef2c, or Tbx5 in each cell by generating 5’ single cell RNA-sequencing data for 2,593 cells collected on day 1 of reprogramming. This approach circumvented the limitation of our initial analysis where the individual ectopic retroviral plasmids could not be distinguished because they each encode the same 3’ polyadenylation sequence. After eliminating the myeloid lineage, we identified 12 clusters within this population, confirming the prompt rate in which cells alter their transcriptional landscape in our system (Figure 2A). A pseudotime analysis again identified three main branches in the main trajectory as well as a separate group of clusters (clusters 6, 7, 11, and 12) that were unlinked from the main trajectory (Figure 2B).
Figure 2. Cardiac Reprogramming Trajectory Is Entered Rapidly.

(A) UMAP visualization of cells collected at day +1 and colored by clusters.
(B) Pseudotime trajectories of all cells in (A). Color indicates progression in pseudotime space. Grey indicates a disjointed trajectory.
(C) Dot plot showing expression [log10(UMI+0.1)] and cell percentage of top marker genes from each of the 12 clusters in (A) based on specificity calculated by Moran’s I test. Background color indicates branches from (B).
(D) Expression [log10(UMI+0.1)] of Gata4, Mef2c, and Tbx5, overlaid on the trajectory plot from (B).
(E,F) Violin plots depicting normalized UMI levels for (E) Gata4, Mef2c, and Tbx5, and (F) Pdlim3, Tbx20, and Tnnt2 in clusters 1, 4, and 8, color-coded as in (A). Stars indicate negative binomial adjusted p-values.
(G) Expression [log 10(UMI+0.1)] of branch marker genes, overlaid on the trajectory plot from (B). See also Figure S2 and Table S2.
The three branches identified in the main trajectory represent gene signatures analogous to those presented in Figure 1 based on a differential expression analysis: cells that are reprogramming (A, 33%), those that are likely proliferating (B, 22%) and fibroblast-like cells (C, 29%) (Figure 2C, Table S2). Evaluation of GMT expression revealed their collective expression in branch A (Figure 2D), which contains cells that have activated markers of a cardiomyocyte fate (e.g. Pdlim3 and Smpx; Figure 2C-E) and downregulated genes associated with a fibroblast identity (e.g. Postn and Tbx20; Figure 2C, Table S2). Gata4 is expressed in cardiac fibroblasts and is therefore detected throughout the population; however, it is increased 1.6-fold in cluster 1 (branch A) compared to cluster 2 (branch C). GMT is also expressed in cluster 10, which lies within branch B and expresses Smpx and Pdlim3 at increased levels, suggesting expression of GMT can initiate reprogramming even if the cells enter a proliferative state (Figure 2C-D). While this type of reprogramming may not produce more advanced iCMs, it suggests proliferation does not prevent the initial stages of reprogramming (Liu et al., 2017). In contrast to branches A and B, the fibroblast-like cells that populate branch C exhibited only baseline levels of all three factors (Figure 2D). This analysis indicates that branch C represents fibroblasts that do not express ectopic GMT, rather than representing a newly acquired state driven by transduction with one or two factors. Therefore, unlike observations made during direct neural reprogramming, we do not detect the emergence of an alternative cell type in our experiments (Treutlein et al., 2016).
GMT expression was also detected within the unlinked trajectory in clusters 6 and 11 (Figure 2D). These clusters contain epicardial cells expressing Ddx4, Lrrn4, and Msln (Figure 2C, S2A, Table S2). Cluster 6 additionally upregulated early markers of the iCM trajectory such as Pdlim3 and Smpx, suggesting that, although unlinked from the main trajectory, this cell type may be capable of acquiring a cardiomyocyte-like gene expression signature upon transduction with GMT (Figure 2C, Table S2). A cell cycle-related phenomenon was observed in the epicardial cells similar to what we observed in branch B, as epicardial cells in cluster 11 entered the cell cycle and activated expression of Pdlim3 and Smpx (Figure 2C, S2B, Table S2).
To identify variables that may dictate progress in the reprogramming trajectory, we next compared the gene expression profiles of clusters 1 and 4 as they represent early, yet distinct iCM reprogramming states. Examination of GMT expression levels found a statistically significant difference in Gata4 (p-value = 9.58e-45) and Tbx5 (p-value = 2.08e-15) between clusters 1 and 4, but not Mef2c (Figure 2E, S2B). Cluster 1 exhibited stronger upregulation of early markers of reprogramming (e.g. Cd24a, Smpx, and Tnnt2) and downregulation of fibroblast-associated genes (e.g. Postn, Sdpr, and Tbx20) (Figure 2G, Table S2). Therefore, while robust expression of Mef2c is required, this variation suggests lower levels of Gata4 and/or Tbx5 may limit the rate of reprogramming but nonetheless allow initiation of the process. While cluster 8 is most similar to clusters 1 and 4, it has not robustly activated markers of reprogramming but it has downregulated markers of the starting fibroblasts (Figure 2C, E-G). There was a significant difference in Mef2c expression between clusters 4 and 8 (p-value = 1.17e-08; Figure 2E), further supporting the necessity of robust expression of this gene.
Chromatin Remodeling Occurs within 72 Hours of GMT Expression
To identify the dynamics in chromatin accessibility underlying the aforementioned transcriptional changes, we performed ATAC-seq on αMHC-GFP+ cells collected at five time points during reprogramming (days 2, 3, 7, 14, and 21), and compared regions of accessible chromatin to those detected in the starting fibroblast population. This analysis identified 100,691 total dynamic regions, which included a rapid gain of accessibility by day 2 of reprogramming at the early reprogramming marker gene Slc6a6 and cardiac Tnnt2 loci (Figure 3A, S3A-B, Table S3). Principal component analysis of the genome-wide chromatin accessibility data showed extensive chromatin remodeling by day 2, in agreement with the transcriptional dynamics presented in Figure 1 (Figure S3C).
Figure 3. Cardiac Reprogramming Initiates Rapid and Distinct Patterns of Chromatin Accessibility Changes.

(A) Gain of ATAC-seq (normalized to sample read depth of condition with highest number of mapped read pairs) signal in iCMs (purple) harvested at indicated days of GMT-induced reprogramming compared to fibroblasts (black) near the early reprogramming marker gene Slc6a6 (left) and cardiomyocyte gene locus Tnnt2 (right).
(B) Hierarchical clustering of tag density over fibroblasts at 10,000 regions with most differentially accessible chromatin status in αMHC-GFP+ iCMs harvested at indicated days of reprogramming.
(C) Medoid plots representative of overall trends, showing ATAC-seq tag density over time at dynamic regions from each cluster in (B).
(D) Tables listing transcription factor families with motifs significantly enriched (p ≤ 1e-19) within dynamic regions from each cluster compared to all stably accessible (non-dynamic) regions. P-values listed are from the top ranked motif from each transcription factor family.
(E) Bar charts showing top three ranked biological process terms enriched in dynamic regions from each cluster compared to all stably accessible (non-dynamic) regions. See also Figure S3, Table S3, and Table S4.
To uncover factors that direct the most robust changes in chromatin accessibility, we performed hierarchical clustering on the 10,000 most differentially accessible regions identified during our time course and found eight primary patterns (Figure 3B). Approximately half of the most dynamic regions (n=4,480) identified in our time course lost accessibility during transdifferentiation, while the other half gained accessibility (n=5,520). Regardless of chromatin remodeling dynamics, the vast majority of changes occurred distal from transcriptional start sites, with dynamic regions underrepresented in promoter proximal regions (p = 2.2e-16; Figure S3D). The majority of regions that lost accessibility exhibited this change within 3 days of GMT induction (clusters A1-4; Figure 3B, C). Motif enrichment analysis identified the TEAD family as most associated with loss of chromatin accessibility (Figure 3D, Table S3), specifically motifs for TEA transcription factors Tead1 and Tead4, which are both expressed throughout reprogramming (Table S4, Figure S3E).
In contrast to the similar dynamics observed in clusters that exhibited the strongest loss of accessibility, there were multiple distinct patterns associated with gain of accessibility. Cluster A5 demonstrated a gain in chromatin accessibility by day 2, but then exhibited a return towards the fibroblast accessibility state at later time points, suggesting that accessible chromatin at those sites was not stabilized (n=385; Figure 3B, C). This cluster showed limited enrichment of transcription factor sequence motifs, which may have prevented stable GMT binding similar to findings reported for Mef2c, Gata4, and FOXA2 where a lack of motif leads to transient sampling rather than stable binding (Figure 3D, Table S3) (Andzelm et al., 2015; Donaghey et al., 2018). Cluster A6 demonstrated an initial trend similar to cluster A5; however, the extent of accessibility loss at later time points was reduced (n=1,352; Figure 3B-C). Clusters A7 and A8 represent the majority of regions associated with a gain in accessibility, and the maximum gain in these regions was observed after day 3 (n=2,471 and n=1,212, respectively; Figure 3B-C). Regions in these clusters maintained higher levels of accessibility over the time course, compared to clusters A5 and A6, and also contained significant enrichment of multiple motif families (Figure 3D, Table S3). Thus, regions that transition from closed to open during cardiac reprogramming have a number of distinct patterns over time, including several that are only transiently open, and each is associated with different sequence motifs.
To assess the potential functional roles of each cluster, we annotated regions exhibiting changes in chromatin accessibility using GREAT (Figure 3E) (McLean et al., 2010). Regions that exhibited loss of accessibility were associated with the inflammatory response (cluster A2) and monocytes (cluster A3), supporting a previous report that demonstrates reprogramming is promoted by repression of inflammatory signaling pathways (Zhou et al., 2017). Clusters A7 and A8, which gain and maintain accessibility, were associated with cardiovascular terms such as cardiac and striated muscle development. Regions that did not maintain accessibility in cluster A5 were also associated with cardiac function (Figure 3E). It remains possible that while Tbx5 may transiently sample those sites during reprogramming, it requires developmentally regulated binding partners such as Eomes, or others, to initiate and/or stabilize the interactions with DNA that are not robustly detected in our system (McLane et al., 2013).
Computational Modeling Reveals Additional Factors Involved in Cardiac Reprogramming
In an effort to discern additional transcription factors that direct the initial stages of reprogramming, we devised a multivariate machine learning approach to predict which transcription factor sequence motifs are most associated with transcriptional changes that occur during the first 2 days of reprogramming (Figure 4A). We found a stronger correlation between sequence motif content of dynamic chromatin regions 2-500kb from the TSS (Pearson correlation = 0.37 between observed versus predicted fold-change, p-value < 0.05, t-test for correlation coefficient) as compared to within 2kb of the TSS (Pearson correlation = 0.22, p-value < 0.05, t-test for correlation coefficient), supporting a previous report that suggested chromatin dynamics proximal to the TSS were poor predictors of gene expression dynamics (Pliner et al., 2018). This model predicted 48 motifs significantly associated with transcriptional changes that occur during the fibroblast to day 2 time frame (Figure 4B). A lower correlation was detected when incorporating only the motifs of GMT that are within accessible chromatin at day 2 (correlation = 0.19), suggesting additional transcription factors are indeed involved in early transcriptional dynamics associated with reprogramming.
Figure 4. Model of Transcription Factor Association with Changing Rate of Gene Expression over Time.

(A) Schematic representation of input features for the multivariate machine-learning approach used to predict transcription factors influencing gene expression changes during reprogramming.
(B) Bar chart displaying significant motifs identified in (A). Motif family is listed in parentheses.
(C) Heatmap of clustered gene-association signatures for identified motifs. Rows represent transcription factor motifs; columns are genes associated with these motifs.
(D) Total predicted synergistic interactions for each candidate transcription factor with at least one additional candidate interactor. Black bars indicate candidates selected for knockdown experiments.
(E) Reprogramming outcomes following knockdown of candidate transcription factors at day 2 of reprogramming, relative to control knockdown. Values indicated are the mean of three replicates. Paired t-test uncorrected p-value < 0.05 indicated by “*”. See also Table S4 and Table S5.
We ranked the identified motifs based on a net importance score (Figure 4B). A positive net importance score suggests an increase in transcription while a negative score suggests a repressive influence; a score close to zero indicates a mixed influence. This analysis found that the Tbx5 motif (T-box) was most associated with gene expression changes (Figure 4B). Notably, while Mef2 motifs (MADS) were also among the top ranked set of putative early regulators, Gata motifs were absent from this list, suggesting that Tbx5 and Mef2c are more influential than Gata4 in regulating gene expression changes during the early stages of reprogramming. The lack of Gata4 motif detection in this prediction combined with ATAC-sequencing results suggests that its link to gene expression changes is weak. Motifs for Smads2/3/4 were also predicted to influence gene expression, supported by previous work that showed TGFβ inhibition positively influences reprogramming outcome (Ifkovits et al., 2014; Mohamed et al., 2017).
To reveal if the identified transcription factors target similar or distinct gene sets, we next performed hierarchical clustering to discover groups of motifs with similar relationships to gene expression changes (Figure 4C). Motifs clustered into two groups, one of which contained many motifs associated with cardiomyocyte development such as Mef2 and Tbx family motifs, as well as Sox, Fox, and SMAD motifs (Figure 4C, lower). The Tbx5 motif was most closely linked to changes at regions that also encode Tgif1 motifs, a TGFβ-induced transcriptional homeodomain-containing repressor (Figure 4C, bold) (Wotton et al., 1999). However, dynamics associated with the Mef2 motifs were not closely linked to any other motifs, suggesting Mef2c may function independently in the reprogramming context (Figure 4C, bold). Next we used a linear model to determine motif pairs whose influence on gene expression could not be explained by either of the motifs alone. Ranking transcription factor motifs by the total number of predicted interactions revealed strong enrichment for the motifs Tcfcp2l1, Sox4, and Hif1a, providing evidence that their role in reprogramming involves cooperative interactions with additional factors (Figure 4D, Table S5). These results further support a model in which multiple transcription factors jointly regulate gene expression dynamics during reprogramming.
To leverage the predictions made by the TF interaction model, we next examined the effect of shRNA-induced knockdown of selected factors on reprogramming efficiency at day 2. Of the 18 genes tested, 5 exhibited a statistically significant reduction in reprogramming efficiency (Sp1, Foxo1, Tcfp2l1, Tgif1, and Foxo1) while 3 improved reprogramming (Hifla, Prdml, and Smad3) (Figure 4E). Future studies of the remaining candidates may reveal additional factors that can enhance or serve as barriers of cardiac reprogramming, and their mechanisms of action.
Mef2c and Tbx5 Binding Is Associated with Changes in Chromatin Accessibility
Given that GMT drive cardiac reprogramming, we next performed ChIP-seq at day 2 of reprogramming to assess Gata4, Mef2c, and Tbx5 occupancy to dissect direct versus indirect consequences of their binding to DNA when introduced in combination. Unlike the ATAC-seq presented in Figure 3, this experiment was performed in immortalized neonatal cardiac fibroblasts to obtain sufficient numbers of cells (Figure S4A); therefore, we created a new, matched ATAC-seq dataset for integration with the ChIP-seq data. This analysis identified 5,100, 6,904, or 5,307 peaks for Gata4, Mef2c, or Tbx5, respectively (Figure 5A-B, S4B). The majority of Gata4 and Mef2c peaks were located further than 2 kilobases (kb) from the nearest transcription start site (TSS), while 45% of Tbx5 peaks were within 2kb of a TSS (Figure S4C).
Figure 5. Chromatin Accessibility Dynamics Associated with Gata4, Mef2c, and Tbx5 Occupancy during Reprogramming.

(A) ChIP-seq profiles display GMT binding at day 2 of reprogramming near Nid2 (left panel) and Actn2 (right panel). ChIP-seq track height is normalized to sample read depth of condition with highest number of reads. ATAC-seq track height is normalized to sample read depth of condition with highest number of mapped read pairs.
(B) Heatmap displays hierarchical clustering of ChIP-seq peaks. Grey scale displays average tag density across replicates, normalized to input, with black indicating an increase in tag density in sample over input. At right, co-clustered changes in accessibility.
(C) Tables display transcription factor families with motifs significantly enriched within GMT-bound regions from each cluster, compared to stably accessible (non-dynamic) regions. P-values listed are from the top ranked motif within each transcription factor family. See also Table S6.)
(D) Bar plot displays the percentages of accessible chromatin regions (ATAC-seq peaks) from each cluster (A1-A8 in Figure 3B), bound by Gata4, Mef2c, or Tbx5. See also Figure S4 and Table S6.
To reveal relationships between reprogramming factor binding and chromatin accessibility, we performed hierarchical clustering on the merged region set (n=14,138) bound by Gata4, Mef2c, and/or Tbx5 during reprogramming, including changes in chromatin accessibility, and identified eight primary patterns (Figure 5B, S4B). The regions in two groups (clusters B1 and B3, n=2,630) were generally bound by all three factors. However, these clusters exhibited disparate chromatin dynamics. Cluster B1 contained regions that were accessible in the starting population, and their accessibility increased slightly by day 2 (Figure 5B). In contrast, regions within cluster B3 were inaccessible in the starting fibroblasts, and their accessibility increased (6.33-fold mean increase by day 2) (Figure 5B).
Two clusters contain regions bound by Mef2c alone (clusters B2 and B4; n=1,968 and n=2,240, respectively), each displaying opposing trends in chromatin accessibility (Figure 5B). While regions in cluster B2 lost accessibility on average, regions in cluster 4 experienced a 2.55-fold mean increase (Table S6). While the trend identified in cluster B4 suggests the machinery Mef2c requires to promote chromatin remodeling is active during reprogramming, we did not observe significant chromatin remodeling at regions bound by Tbx5 alone (cluster B7, n= 2,370) nor Gata4 alone (cluster B5, n=1,419) (Figure 5B). Cluster B7 exhibited little change in accessibility during reprogramming (0.64-fold mean change), while regions bound by Gata4 and Tbx5 together (cluster B6; n=2,129), exhibited a 3.84-fold mean increase in accessibility, suggesting synergistic binding of Gata4 and Tbx5 has a positive impact on chromatin remodeling at those regions (Figure 5B).
We next identified potential non-GMT cofactors within these regions by searching for known motifs enriched within ChIP-seq peaks and summarized them based on TF family (Figure 5C, Table S5). As expected, top-ranked motifs correspond to families of reprogramming transcription factors; however, additional motif families were also significantly enriched, including those that bind bZip, Homeobox, and Forkhead proteins (Figure 5C, Table S5). These families include transcription factors such as Atf1/2/3/7, Fosl2, Jun, and Bach2 (bZip); Tgif1/2 and Meis1 (H-box); and Foxm1 (Forkhead), all of which are expressed during reprogramming (Figure S3E). This result suggests that combinatorial binding at dynamic chromatin is important beyond GMT.
Finally, we analyzed the binding of Gata4, Mef2c, and Tbx5 at regions that exhibited the most dynamic chromatin accessibility changes during reprogramming (regions from Figure 5B-C). We detected no enrichment of GMT binding at day 2 in regions that lose accessibility during reprogramming (clusters A1-4), while we detected statistically significant enrichment of GMT binding at day 2 in almost all regions that gained accessibility during reprogramming (clusters A6, A7, and A8; Figure 5D). Cluster A5 is the only ATAC-seq cluster that gained accessibility at day 2 of reprogramming without significant enrichment of binding by reprogramming factors, providing a potential explanation for why this cluster exhibits only a transient accessibility gain (Figure 5D, 3B-C). Taken together, these data suggest that chromatin accessibility dynamics directed by binding of GMT are context-specific.
Transcription Factor DNA Occupancy Defines Chromatin Accessibility Trends
To understand how GMT binding, individually and in combination, is related to changes in chromatin accessibility, we next performed ChIP-seq and ATAC-seq at day 2 on immortalized neonatal cardiac fibroblasts with single factors (SF) as well as pairs of factors ectopically expressed (double factor, DF) (Figure 6A). Overall, each individual factor’s binding pattern differed from that detected during reprogramming with all factors (AF) (Figure 6B). A large shift occurred for Gata4, whose binding became more similar to that of Tbx5 during AF reprogramming, supporting a previously reported cooperative binding relationship between these two transcription factors in developing mouse and human cardiomyocytes (Ang et al., 2016; Luna-Zurita et al., 2016; Maitra et al., 2009). Mef2c exhibited a decidedly distinct binding pattern compared to Tbx5 and Gata4, but its binding was altered by the addition of Gata4 and Tbx5 (Figure 6B).
Figure 6. Chromatin Accessibility Dynamics Associated with Gata4, Mef2c, and Tbx5 Occupancy Following Independent and Combinatorial Expression.

(A) Profiles display ChIP-seq signal for GMT in single factor (SF) and all factor (AF) conditions, and ATAC-seq signal in SF, double factor (DF), and AF conditions, near the early reprogramming marker gene locus, Ehd4. Grey rectangle highlights region bound by Gata4, Mef2c, and Tbx5 in AF condition, without binding by any of these factors in SF conditions. Blue rectangles highlight regions bound primarily by Mef2c when ectopically expressed alone, but bound by Gata4, Mef2c, and Tbx5 when all factors are expressed. Profiles represent read density from merged biological replicates normalized to read depth.
(B) Principal component analysis of all ChIP-seq replicates.
(C) Heatmap displays hierarchical clustering of ChIP-seq peaks, with grey scale displaying average tag density across replicates, normalized to input (left) and color scale displaying changes in accessibility compared to fibroblasts (right).
(D) Violin plots show distribution of distances from ChIP-seq peaks in each cluster to nearest TSS of differentially expressed genes in cells from “Early iCM”, “Late iCM”, and untransduced fibroblast single cell RNA-sequencing clusters. See also Figure S5 and Table S7.
Hierarchical clustering of the merged region set when AF or an SF were detected, together with changes in chromatin accessibility, resulted in 8 distinct clusters (Figure 6C). Most clusters were driven by binding of a single factor (Figure 6C). Clusters C2 and C4-C6 represent clusters bound in single factor conditions that are refined by the addition of all factors (Figure 6C, S5A). Binding of a single factor in C2 and C5 was associated with a concomitant increase in chromatin accessibility. The single binding events refined by the addition of the other reprogramming factors coincide with overrepresentation of the single factor’s sequence motif (Figure S5B, Table S6). Regions in C5 exhibited an increase in chromatin accessibility only when Gata4 was present alone, which may suggest these regions act as regulatory elements in cell types for which Gata4 is involved but Tbx5 and Mef2c are not, such as at the TSS of the endothelial gene Lecam2 (Figure S5A). Similarly, Mef2c was sufficient to induce an increase in chromatin accessibility in a subset of regions within C2 that was abrogated by the addition of Gata4 and Tbx5 (mean fold changes, M=1.75, MG=1.24, MT=1.12, AF=0.84) (Figure 6C, Table S7).
Mef2c and Tbx5 SF binding events were also associated with a loss of chromatin accessibility (C4 and C6), suggesting their ability to interact with DNA and alter chromatin accessibility is context-dependent. C8 confirms the divergent response to Tbx5 binding as a subset of these regions were also bound by Gata4, and were associated with minimal changes in chromatin accessibility (Figure 6C). We did not identify a cluster in which Gata4 was independently capable of stably increasing chromatin accessibility, suggesting its function in the reprogramming context is downstream of epigenomic remodeling.
We noted unique trends in clusters C3 and C7. They are dominated by regions that exhibit binding of all three factors in the AF reprogramming condition, but limited binding in the SF conditions (Figure 6C). While Mef2c binding was sufficient to promote a 3.09-fold increase in chromatin accessibility in cluster C3, Tbx5 comparably directed a 3.08-fold increase in cluster C7, representing the greatest average increases in chromatin accessibility among regions included in this analysis (Figure 6C, Table S7). This suggests that chromatin changes induced by Mef2c and Tbx5 create a chromatin landscape amenable to the binding of the additional reprogramming factors, adding another layer of regulatory complexity.
We next ascertained the relationship between these clusters and the transcriptional signatures identified by single cell RNA-sequencing (Figure 1). To that end, we defined genes that represent early iCMs, late iCMs and untransduced fibroblasts (p < 0.0001; Table S1) and calculated the distance from the TSS to the closest dynamic region. Regions whose chromatin dynamics were largely associated with Mef2c binding (clusters C2 and C3) were significantly associated with genes marking “Early iCM” populations (p < 0.001; Figure 6D, left). Clusters C3 and C7 were associated with “Late iCM” genes and an increase in occupancy by all three reprogramming factors during reprogramming (p < 1e-10; Figure 6D, middle). This association between C3 and C7 with gene expression suggests that the “Late iCM” trajectory results from cooperative binding by all three reprogramming factors while Mef2c is associated with the initial gene expression changes that define early iCMs.
To identify chromatin dynamics associated with untransduced cells that do not reprogram, we next evaluated the distance between observed chromatin dynamics and genes that represent this trajectory. Indeed, we found that regions in cluster C5 that were bound by Gata4 only in the Gata4 SF condition were significantly closer to genes that represent the untransduced fibroblasts (p < 0.001; Figure 6D, right). The lack of chromatin accessibility changes in the DF conditions in C5 indicate that neither Mef2c (MG) nor Tbx5 (GT) bind to or prevent Gata4 from binding to or altering chromatin accessibility at these regions, supporting our conclusion that these cells represent an untransduced population that expresses and is regulated by endogenous levels of Gata4 (Figure 6A, C). Cumulatively, these data reveal the complexity of the mechanisms through which transcription factors influence each other, both enabling and refining one another’s ability to bind DNA and affect accessibility changes.
Individual Factors Activate Transcription of Reprogramming Genes
To understand the extent to which GMT synergy leads to gene expression changes during reprogramming, we next identified genomic regions bound by GMT that are proximal to differentially expressed genes. Ldb3 and Ptrf represent two genes whose expression increases in early iCMs by day 1 compared to the starting population and are bound by reprogramming factors; Ptrf is bound by Mef2c and Tbx5 in the AF context, while Ldb3 is bound by all three reprogramming factors in the AF context (Figure 7A, B). We predicted putative enhancer elements that may be responsive to GMT guided by the ChIP-seq and ATAC-seq data and designed reporter constructs with these regions to identify which factor(s) are sufficient to induce gene expression. Neonatal cardiac fibroblasts were concurrently transduced with these reporters as well as GMT, and reporter expression was assessed using FACS.
Figure 7. Individual Reprogramming Factors Activate Transcription at GMT-bound Regulatory Regions of Early Reprogramming Genes.

(A) Violin plots depicting normalized UMI levels for early iCM marker genes Ldb3 and Ptrf in fibroblasts and iCMs at day 1 of reprogramming. Fold change (fc) listed above plot.
(B) Read density profiles display ChIP-seq and ATAC-seq signal near Ldb3 and Ptrf loci in the setting of single factor (SF), double factor (DF) or all factor (AF) conditions, normalized to read depth. Peak calls indicated above ChIP-seq profiles in gray. Blue rectangles highlight putative regulatory regions investigated by reporter assays in panels (C, D).
(C) Ldb3-mKate and (D) Ptrf-mKate reporter activation at day 1 of reprogramming with single, double, and all factor (AF) infections. Values displayed are means of four replicates. Error bars indicate standard deviation. T-test uncorrected p-value thresholds as indicated.
Despite evidence of binding by multiple factors at these sites during reprogramming, ectopic expression of a single factor was sufficient to induce reporter gene expression from both of these constructs. Mef2c, but neither Gata4 nor Tbx5, was sufficient to induce robust Ldb3-driven reporter expression, consistent with Mef2c’s binding to the endogenous Ldb3 locus independently, with subsequent binding of Tbx5 and Gata4 (Figure 7B, C). Conversely, while both Mef2c and Tbx5 were independently capable of binding to the endogenous Ptrf locus, only Tbx5 was sufficient to activate expression of the Ptrf-driven reporter when introduced alone (Figure 7B, D). While Mef2c had no discernable effect on reporter expression, the addition of Gata4 limited the ability of Tbx5 to induce reporter expression resulting in a mean decrease of 61% (uncorrected p-value = 0.006), demonstrating an example of the coregulatory refinement suggested by ChIP- and ATAC-seq data (Figure 5). Although both the Ldb3 and Ptrf genes are expressed in early iCMs, our results demonstrate that the role of each reprogramming factor is not limited to synergistic activation, but rather differs between these two loci, indicating context-specific effects in regulatory element usage for each transcription factor.
DISCUSSION
Here, we interrogated the transcriptional and epigenomic dynamics underlying direct cardiac reprogramming in an in vitro mouse cardiac fibroblast system, revealing numerous insights into the mechanisms associated with the cell fate transition from a fibroblast toward a cardiomyocyte. Epigenomic and transcriptional changes occurred broadly within the first 72 hours, and cells destined to reprogram could largely be predicted by virtue of early gene expression dynamics and reprogramming factor expression. Single cell assays addressed longstanding questions regarding heterogeneity and response to combinations of reprogramming factors, clarifying existing interpretation of bulk transcriptome data sets. A machine learning approach revealed clusters of co-located transcription factor motifs within dynamically changing chromatin regions associated with coordinate gene expression changes, pointing to additional factors that may promote or inhibit reprogramming. Integration of GMT DNA occupancy with genome-wide chromatin accessibility and single cell RNA-sequencing in the setting of individual or combinations of transcription factors revealed an interdependency of their binding patterns and suggests possible mechanisms through which they facilitate successful reprogramming.
Despite similarities to rapid transcriptional and chromatin remodeling seen in other systems, our results highlight differences between direct cardiac reprogramming and other reprogramming types. For example, during the transition from fibroblast to neuron induced using a combination of Ascl1, Brn2, and Mytl (Treutlein et al., 2016), an alternative fate characteristic of skeletal muscle was observed. In contrast, for cells that were successfully transduced with GMT, no major alternative fates, compared to starting cell types, were observed. Alternatives may be limited in the cardiac setting, because, as we show here, GMT binding is refined when they are expressed combinatorially, perhaps focusing binding events on cardiac loci. By contrast, Ascl1’s binding is not altered by the addition of other neural reprogramming factors such as Brn2 and Mytl1, suggesting its binding is unrestrained and may occur at regulatory elements employed in the development of multiple cell types (Wapinski et al., 2013).
While reports of direct reprogramming were first documented many years ago, the tools available to dissect the precise molecular mechanism of reprogramming were limited. Advances in single cell RNA-sequencing have created avenues to identify the path a single cell can take to its endpoint, and identify the molecular determinants of these trajectories (Cacchiarelli et al., 2018; Schiebinger et al., 2019). Simultaneous advances in machine learning have improved the information gleaned from single cell RNA-sequencing data, as well as the ability to correlate changes between gene expression and chromatin remodeling (Cao et al., 2018; Deng et al., 2019; Eraslan et al., 2019; Lopez et al., 2018; Way and Greene, 2018; Welch et al., 2017). The observation that the vast majority of fibroblasts that expressed GMT proceeded into the induced cardiomyocyte trajectory suggests a higher efficiency among GMT-expressing cells than previously recognized and suggests that reprogramming efficiency might be improved by increasing the proportion of fibroblasts in which all three factors are ectopically expressed. Furthermore, our analysis suggests that other cell types, such as epicardial cells, have the potential to be partially reprogrammed, although they remain dissimilar to fibroblast-derived induced cardiomyocytes.
In conclusion, we have developed a comprehensive genomic assessment of transcription factor binding, chromatin state, and transcriptional changes, that reveals the molecular complexity involved in direct cardiac reprogramming. Mechanistic insights provided by integration of multiple datasets have started to reveal how lineage-enriched transcription factors can induce cell fate transitions in a combinatorial fashion, thereby achieving specificity of gene regulation.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deepak Srivastava (dsrivastava@gladstone.ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal work in this study was done in accordance with local institutional policies. Breeding age αMHC-GFP CD8 transgenic mice were used to generate all cells used in this study (Subramaniam et al., 1991). No influence on sex was observed.
Cell Lines and Culture
Direct cardiac reprogramming was performed on primary neonatal mouse cardiac fibroblasts as previously described (Ieda et al., 2010; Qian et al., 2013). Briefly, tissue explants from αMHC-GFP+ neonatal mouse hearts (p0-p3) were minced and cultured on gelatin-coated plates in fibroblast explant media at 37°C for ~10 days, allo wing expansion of fibroblast population prior to selection of GFP−/Thy1+ cells by FACS. After sorting, primary fibroblasts were plated at a density of 500k-700k per 10cm dish with gelatin coating.
Experiments presented in Figures 4D-E, 5, 6, and 7B-D were performed using an immortalized neonatal cardiac fibroblast cell line expressing cre-excisable large T antigen, made from αMHC-GFP+ mice. Fibroblasts were passaged the day prior to infection, and plated at a density of 500k per 10cm dish with gelatin coating.
Method Details
Medias
Fibroblast explant media: 20% fetal bovine serum, 1x penicillin-streptomycin, in IMDM Cardiomyocyte media: 10% M199, 10% FBS, 1% NEAA, 1% sodium pyruvate, 1x penicillin-streptomycin, in DMEM with Glutamax and sodium pyruvate
PlatE/293T media: 1% NEAA, 10% FBS, in DMEM with Glutamax
FACS buffer: 1% FBS, 0.5mM EDTA, 10mM HEPES, in Ca/Mg2+ free PBS
Single cell RNA-sequencing resuspension buffer: 1% BSA, in Ca/Mg2+ free PBS
Reprogramming Timeline
Day −12 (+/− 3 days): Neonatal cardiac tissue harvested and maintained in fibroblast explant media.
Day −2: Thy1+/αMHC-GFP− cells were isolated by FACS or immortalized fibroblasts were passaged.
Day −1: Fibroblasts were infected with freshly prepared pMXs-Gata4, pMXs-Mef2c, and/or pMXs-Tbx5 (GMT) retroviruses or pMXs-dsRed retrovirus.
Day 0: Fibroblast explant media was replaced with cardiomyocyte media containing sb431542 (2.6μM).
Day +1: Xav939 (5μM) was added to cells without changing media.
Indicated time points: FACS-sorted or unsorted cells were collected at time points following reprogramming initiation. We completed at least three biological replicates per condition. [See methods paper for detailed protocol (Qian et al., 2013).]
Viral Production
All retroviruses were freshly prepared. Prior to transfection with pMXs viral plasmids, platE cells were maintained under selection in 10cm dishes containing 10mL of platE/293T media with 2μl of 10 mg/mL puromycin and 10μl of 50 mg/mL blasticidin. The day before transfection, platE ~4.5 million platE were plated per 10cm dish without gelatin coating in platE/293T media without antibiotics.
Lentiviruses were freshly prepared and packaged with psPAX2 and pMD2.G (gifts from Didier Trono; Addgene #12260 and #12259.) Lentiviral shRNA constructs targeting candidate TFs were obtained from Sigma (Table S5). Scrambled shRNA control lentiviral construct was a gift from David Sabatini (Addgene #1864 (Sarbassov, 2005); Table S5). The day before transfection, ~600k 293T were plated per 3.5cm well (1 well of a 6-well plate) without gelatin coating in platE/293T media without antibiotics. Lentiviral constructs were transfected along with packaging vectors psPAX2 and pMD2.G at a ratio of 3:3:1 (2.5ug lentiviral plasmid + 2.5ug psPAX2 + 0.83ug pMD2.G).
Fugene HD was used for all transfections, at a ratio of 3.5:1 for Fugene:total plasmid DNA. Viral supernatant for retroviruses and lentiviruses was collected 48 hours post-transfection and used to infect cardiac fibroblasts with the addition of 0.6μg/mL polybrene.
Fluorescence Activated Cell Sorting (FACS)
Adherent cells were washed with PBS, digested in 1x TryplE for 20 minutes, then quenched with fibroblast explant media. Cells in suspension were filtered through a 70μM filter and pelleted. Pelleted cells were stained for 1 hour with Thy1-APC antibody, washed once with PBS, and resuspended in FACS buffer. APC+/GFP− cells were isolated by FACS and plated onto gelatin-coated plates (day −2) prior to infection the following day (day −1), and cell harvest at time points indicated. For sorted samples used in bulk assays, and all single cell RNA-equencing samples regardless of fluorescent selection, adherent cells were washed with PBS, digested in 1x TryplE for 20 minutes, then quenched with cardiomyocyte media. Cells in suspension were filtered through a 70μM filter, pelleted, and resuspended for FACS.
Cloning
Putative regulatory sequences for reporters used in Figure 7 were inserted into pGK:HygroR-CMV:mKate2-B_UTR (a gift from Tyler Jacks; Addgene #68480), in place of CMV after excision with AfeI and AvrII (NEB). Plasmids generated in this study have been deposited to Addgene (pGK-Ldb3-mKate2, #128766; pGK-Ptrf-mKate2, #128765).
Single Cell RNA-sequencing Library Generation
All cells undergoing single cell RNA-sequencing went through FACS, however “unsorted” samples were gated for single cells only while “sorted” samples were gated based on both singularity and fluorescence. Cells were filtered using a 70μM filter, resuspended in 1% BSA in PBS, and counted immediately prior to library preparation. Single-cell RNA-seq libraries were prepared using the Chromium Single Cell 3′ (v2) and 5’ (v1) Reagent Kits (PN-120236, PN-120237, PN-120262, PN-1000006). Libraries were constructed using 10X Genomics guidelines. All libraries were pooled and sequenced using the HiSeq 4000 to a read depth of at least 30,000 reads per cell.
Bulk RNA-Sequencing
All cells undergoing bulk RNA-sequencing went through FACS to select either αMHC-GFP+ iCMs or dsRed+ fibroblast controls. Following sorting, cells were immediately pelleted, resuspended in 200uL Qiazol, and placed at −20 degrees Celsius for at least 24 hours prior to isolation of total RNA using the miRNeasy Micro Kit (Qiagen). Bulk RNA-sequencing libraries were prepared with the Ovation RNA-seq System v2 kit (NuGEN). RNA-seq libraries were assessed by Bioanalyzer and quantified by qPCR (KAPA). Samples were sequenced at 100PE on the Illumina HiSeq 2500 at the Harvard FAS core.
ATAC-sequencing Library Preparation
We prepared iCM, single-factor, and fibroblast samples for ATAC-seq as previously described (Buenrostro et al., 2013). Aliquots of 10,000–50,000 cells were spun down (310 RCF for 3 minutes) and washed with 200 μL of chilled PBS. Samples were lysed with 200 μL of chilled lysis buffer (20 mM Tris-HCl (pH 8.0), 85 mM KCl, 0.5% NP-40) and spun down at 500 RCF for 5 minutes. Nuclear pellets were transposed with 25 μL of Tagment DNA Buffer, 2.5 μL of Tagment DNA Enzyme (Nextera Sample Prep Kit from Illumina, cat #FC-121-1030), and 22.5 μL of nuclease-free H2O. The samples were incubated at 37°C for 30 minutes and stored at −20°C. Transposed samples were purified using the QIAGEN MinElute Reaction Cleanup Kit (cat #28204). Samples were amplified using 25 μL of NEBNext High Fidelity 2x PCR Master Mix, 1.25 μM Nextera custom primer, 1.25 μM Nextera custom primers with unique barcodes, and nuclease-free H2O. We amplified samples using the following PCR conditions: 72°C for 5 minutes; 98°C for 30 seconds; and cycled at 98°C for 10 seconds, 63°C for 30 seconds and 72°C for 1 minute. Half of each sample was amplified for 12 cycles, MinElute purified and assessed by bioanalyzer for library quality. Samples concentration was quantified by Qbit before pooling. Samples shown in Figure 3 were sequenced at 100PE on the Illumina HiSeq 2500 at either the Harvard FAS core or the UCSF CAT core. ATAC-seq samples shown in Figure 6 were sequenced at 100PE on the Illumina HiSeq 4000 at the UCSF CAT core.
ChIP-Seq Protocol and Library Generation
Cells (107 per ChIP) were crosslinked in 1% formaldehyde in suspension at room temperature for 10 minutes with gentle rotation. Crosslinking was quenched by addition of glycine (final 125 mM), followed by incubation at room temperature for 5 minutes with gentle rotation. Cell pellets were incubated in cell lysis buffer (20 mM Tris-HCl, pH 8, 85 mM KCl, 0.5% NP-40, protease inhibitors) for 10 minutes on a rotator at 4°C. Nuc lei were isolated by centrifugation (2,500 x g, 5 minutes, 4°C), resuspended in nuclear lysis buffer (50 mM Tris-HCl, pH 8, 10 mM EDTA, pH 8, 1% SDS, protease inhibitors) and incubated on a rotator for 30 minutes at 4°C. Chromatin was sheared using a Covaris S2 sonicator for 15 minutes (60-second cycles, 5% duty cycle, 200 cycles/burst, intensity = 5) until DNA was in the 200–700 base-pair range. Chromatin was diluted five-fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8, 167 mM NaCl, protease inhibitors) and incubated with antibody (2 mg/million cells) at 4°C overnight under rotation. Antibodies used are Santa Cruz, Gata4, sc-1237x; Cell Signal Tech, Mef2c 5030; Santa Cruz, Tbx5 (C-20) sc-17866x. Antibody-protein complexes were immunoprecipitated using Pierce Protein A/G magnetic beads at 4°C for 2 hours under rotation. Beads were washed five times (2-minute washes under rotation) with cold RIPA buffer (50 mM HEPES- KOH, pH 7.5, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na-deoxycholate), followed by one wash in cold final wash buffer (1xTE, 50 mM NaCl). Immunoprecipitated chromatin was eluted at 65°C with agitation for 30 minutes in elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). High-salt buffer (250 mM Tris-HCl, pH 7.5, 32.5 mM EDTA, pH 8, 1.25M NaCl) and Proteinase K were added and crosslinks were reversed overnight at 65°C. Samples were treated with RNase A, and DNA was purified with Agencourt AMPure XP beads (Beckman Coulter cat #A63881). Fragmented ChIP and input DNA was end-repaired, 5′ phosphorylated and dA-tailed with NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB E7645). Samples were ligated to adaptor oligos for multiplex sequencing (NEB E7335), PCR amplified, and sequenced on an Illumina NextSeq 500 at the Gladstone Institutes.
Quantification and Statistical Analysis
Single Cell RNA-Sequencing Analysis
The 10X Genomics Cell Ranger pipeline was used to demultiplex raw data, align reads, count transcripts and aggregate multiple samples and timepoints. The R packages Seurat v2.3 and Monocle v3 were used for all downstream analyses (Butler et al., 2018; Cao et al., 2019). Cells that met unique molecular index (UMI) and gene thresholds were included in subsequent analyses. Clustering was performed using the top principal components and visualized using UMAP (McInnes et al., 2018). Differential expression between the clusters was calculated using the negative binomial test implemented in Seurat (Figures 1 and 2) and Moran’s I test implemented in Monocle (Figure 2) (Butler et al., 2018; Cao et al., 2019).
Bulk RNA-Sequencing Analysis
Trimming of reads in raw fastq files for known adapters and low-quality regions of reads was performed using Fastq-mcf (https://github.com/ExpressionAnalysis/ea-utils). Sequence quality control was assessed using the program FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and RSeQC (Wang et al., 2012). Alignment of the reads to the reference mm9 genome was performed using STAR 2.5.2a. Reads were assigned to genes and summarized as gene-level counts using "featureCounts" (Liao et al., 2014), part of the Subread suite (http://subread.sourceforge.net/), with Ensembl gene annotations, in GTF format. The association of the expression of genes with the day of reprogramming was estimated using a linear model in edgeR with statistical significance determined using a likelihood ratio test. The expression of 8,327 genes were significantly associated with the day of reprogramming using an FDR < 0.05 threshold.
ATAC-Sequencing Analysis
Alignment to the mm9 reference genome was performed using Bowtie 2.2.4 (Langmead and Salzberg, 2012) with options: -X 600 --no-mixed --no-discordant. Duplicate reads were removed using Picard MarkDuplicates (http://broadinstitute.github.io/picard). Peaks were called using macs2 callpeak with options: -p 0.1 --nomodel --shift 100 --extsize 200 -B --SPMR --call-summits. Peaks concordant between at least two of three replicates were considered for further analysis. Clustering was performed using the bioconductor package HOPACH and visualized using pheatmap in R (Laan et al., 2003). Regions of open chromatin are determined by first estimating counts in each of the replicates for each of the time-points across a merged set of 307,204 peaks called by MACS2 (Zhang et al., 2008), then normalizing the counts for differences in sequencing depths and estimating the association with time using the likelihood ratio tests based on negative binomial generalized log-linear model in bioconductor package edgeR (Robinson and Oshlack, 2010; Robinson et al., 2010). At an FDR of 5%, 100,691 regions were significantly associated with time. Of these, we selected the 10,000 most significantly dynamic regions for downstream analysis. We associated biological processes to these regions using GREAT (McLean et al., 2010). Motif enrichment analysis was performed using HOMER (Heinz et al., 2010).
ChIP-Sequencing Analysis
Trimming of known adapters and low-quality regions of reads was performed using Fastq-mcf (http://code.google.com/p/ea-utils). Sequence quality control was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and RSeQC (Wang et al., 2012). Alignment to the mm9 reference genome was performed using Bowtie 2.2.4 (Langmead and Salzberg, 2012). Peaks were called using GEM (Guo et al., 2012; Langmead and Salzberg, 2012). Read counts per peak were generated with featureCounts (Liao et al., 2014) and normalized to account for differences in sequencing depth between samples using upper quartile normalization separately for the ChIP and input samples of each of the three transcription factors. Regions bound by each transcription factor in the Single Factor (SF) and/or All Factor (AF) setting were determined using empirical Bayes F-tests for a quasi-likelihood negative binomial generalized log-linear model of the count data as implemented in edgeR. Specifically, we tested for a significant (i.e. non-zero at FDR < 5%) log2 fold-increase in normalized peak signal for ChIP versus the corresponding input sample in at least one of the SF and AF setting, adjusting for sample identifier to account for the inclusion of technical replicates for some Gata4 AF ChIP samples. These log2 fold-changes were clustered using HOPACH with the following settings: clusters="best", initord="clust". Region intersections were found using BEDTools (Quinlan and Hall, 2010). Motif enrichment analysis was performed with HOMER (Heinz et al., 2010). Known motifs and sequence logos were generated from de novo motifs matched to the JASPAR CORE non-redundant vertebrate database (Heinz et al., 2010; Mathelier et al., 2015) using Tomtom from the MEME suite (Bailey et al., 2009). Significant differences in peak distances from gene groups was determined by the Wilcoxon-Mann-Whitney rank sum test.
Summary of Machine Learning Model
We implemented a biophysically inspired machine-learning model to explain how rates of gene expression change between two time points and differ across genes as a function of changes in transcription factor binding motifs in open chromatin near each gene’s transcription start site (TSS). Due to chromatin opening/closing during differentiation, a gene can have different binding motifs in nearby open chromatin between two time points. To learn how these chromatin accessibility dynamics relate to the temporal regulation of cardiomyocyte differentiation, we focused on associating binding motif content to the rate of gene expression change between each pair of time points (i.e., gene expression slope), rather than gene expression level per se.
To accomplish this modeling task, we first needed to encode how chromatin accessibility dynamics change the transcription factor motif content nearby each gene whose expression is associated with the day of reprogramming using the bulk RNA-seq data. For every time-point, we counted the number of times each transcription factor motif occurs within 2kb of each gene’s TSS and down-weighted these counts by their distances from the TSS. Then, for every pair of time points and every gene, we ranked transcription factors by how much their weighted motif occurrences changed nearby that gene, with the lowest rank encoding the greatest loss of motifs and the highest rank encoding the greatest gain of motifs. For a second version of the model, we repeated this procedure using all motifs between 2kb and 500kb from the TSS.
Next we associated gains and losses of accessible transcription factor motifs with gene expression dynamics, by employing targeted maximum likelihood estimation (tmle) methodology (Laan et al., 2006). This approach allows us to combine the effects of binding motif gain and loss into a single association, while leveraging a theoretically optimal, data-adaptive model selection procedure called the SuperLearner (Laan et al., 2006, 2007) to determine which transcription factors are important for the rate of gene expression change. Specifically, for a given pair of time points and for each transcription factor, we estimated the “gain” effect as the average gene expression slope for genes in the top 10% of positive weighted motif changes for that transcription factor minus the average slope for genes in the bottom 25% of absolute weighted motif changes. Similarly, we estimated the transcription factor’s “loss” effect as the average gene expression slope for genes in the bottom 10% of negative weighted motifs changes for that transcription factor minus the average slope for genes in the bottom 25% of absolute weighted motif changes. These two estimates account for potential confounding due to all other transcription factors (i.e., there are “marginal” associations estimated from a model including all transcription factors). Finally, we estimated a transcription factor’s net importance score (see below) for a pair of time points as the difference between its gain and loss effects.
We used random forests and generalized linear models as the underlying machine learning algorithms for SuperLearner. The fitted model was validated using a ten-fold crossvalidation framework where the estimation of the differences in the rates of changes gene expression is viewed as a prediction problem that is solved using random forests (Breiman, 2001) with the Pearson correlation between the observed and predicted differences as the performance metric. These methods are described in greater detail below.
Notation for Machine Learning Model
Let Tt denote the tth time-point, t ∈ {0,1,2,3,4,5}, where gene expression and chromatin state are assayed (T0 = 0 (Fibroblast stage before the GMT vectors are added), T1 =2 (day 2), T2 =3 (day 3), T3 =7 (week 1), T4 =14 (week 2) and T5 =21 (week 3)).
Let Xt,i denote the log2 mean (across the 3 replicates) normalized (over all replicates over time) expression of gene i at time t. Let M denote the number of genes (this corresponds to the set of genes whose mean expression is associated with time or the day of reprogramming). Let Yt,i denote the rate of change of the logarithm of the mean of expression of gene i at time t.
| (1) |
Let ΔYt,i denote the difference between the rates of change of the log2 expression of gene i at time t and at time t-1.
| (2) |
We assume that the changes in rate of change of the expression of gene i between time t and time t-1 can be explained by some subset of N sequence motifs. Each sequence motif is associated with a transcription factor. Let denote the change in the strength of association of motif j, j ∈ {l,2,⋯,N} with gene i between time t and time t-1 resulting from the differential opening and closing of chromatin.
A common model of gene expression regulation across all genes between times t-1 and t (denoted by Ft) is assumed.
| (3) |
The functional form of Ft is non-parametrically identified using a supervised learning approach (see next sections).
Biophysical Motivation for Modeling Approach
The above gene regulation model has a biophysical motivation. The rate of change of the log of the expression of a given gene at a given time is the difference between the rate at which it is transcribed and the rate at which the corresponding mRNA decays. The rate of transcription of this gene is assumed to be a (unknown) function of the strengths of association of transcription factors/proteins with this gene at this time-point. The rate of decay of the log of the expression of the gene is a fixed constant (independent of time) if one assumes a first-order rate of decay for the corresponding mRNA. Therefore the differences in the rate of change of the log of the expression of gene at the two time points should be a function of the difference in the strengths of association of the transcription factors between these time points. From a biophysics perspective, regulation depends on strength and counts of binding motifs, transcription factor concentrations, protein interactions, epigenetics, and other parameters. Since we do not have data on each of these parameters, we focus on the number of transcription factor motifs in open chromatin nearby the regulated gene (details below). Note the analysis assumes the same model, Ft (Equation 3), for all genes. This is a simplification necessitated by a grossly smaller number of samples (order 1) versus the number of possible interacting motifs/transcription factors (order 100) and will result in the identification of modes of regulation that are apparent across a relatively large proportion of genes while potentially missing modes that operate on small subsets of genes. On the other hand this simplification has the advantage of not directly requiring the concentrations of the regulating transcription factors corresponding to over-represented sequence motifs.
Transcription Factors for Machine Learning
The list of transcription factors to use for the model between consecutive time points are identified using the open chromatin regions for each replicate ATAC-seq sample at these time points (see ATAC-sequencing Analysis section for peak calling). The findMotifsGenome.pl function (using the options –size given and hypergeometric enrichment scoring) in Homer (Heinz et al., 2010) is used to identify motifs enriched (p-value < 1e-1000) at time t, using the open chromatin regions at time t as foreground and the open chromatin regions at time t-1 as background. Similarly, this function is used to identify motifs enriched at time t-1 using the open chromatin regions at time, t as background. The list of transcription factors used in the analysis at time t is the union of the motifs from the two above enrichment analyses.
Motif Strength with Gene for Machine Learning
The location of each of the above identified motifs in the open chromatin regions at each time-point is obtained using the –find option of the findMotifsGenome.pl function. Assume that there are motif locations of motif j in the open chromatin regions at time t. Then the strength of association of motif j with gene i is given by,
| (4) |
where is the distance of the kth location of the motif from the transcription start site (TSS) of gene i. D corresponds to a distance domain. In the analysis, two distance domains are considered – (0, 2kb) and (2kb, 500kb) corresponding to promoter proximal and distal (potential enhancer) associations.
The change in motif gene association is then defined as,
| (5) |
Validation of Machine Learning Model
The gene regulation model stated in Equation (3) is fit across a set of genes using the random forests (Breiman, 2001) supervised learning approach. This is done using the rfsrc function that is part of the randomForestSRC package (Ishwaran and Lu, 2019), in R (R package version 1.6; Team RC, 2015). The set of genes whose mean expression is associated with the time or the day of reprogramming is randomly divided into ten groups. The data for the genes corresponding to nine of the ten groups are used to learn the model given in Equation (3). The correlation between the observed ΔYt,i for the set of the genes in the remaining group and the predicted ΔYt,i for these genes using the model learnt is computed.
Importance of Transcription Factors
The importance of each of the transcription factors in explaining changes in rate of change of expression across all the genes between time-point t-1 and t, is defined here. This is followed by its estimation procedure.
In words, the importance of a given transcription factor at a given time-point, t is defined as the change in the mean difference in the rates of changes of expression of genes which is associated with this transcription factor from the mean difference in the rates of changes of expression of genes which are not associated with this transcription factor after accounting for effects from all other transcription factors on this difference.
Denote the set of positive values of by,
| (6) |
the second of negative values of , by
| (7) |
and the set of absolute values of , by
| (8) |
Define as the 90th quantile of , as the 10th quantile of and as the 25th quantile of .
Denote as a binary variable that is equal 1 if motif j is associated with gene i at time t.
| (9) |
Denote as a binary variable that is equal −1 if motif j is associated with gene i at time t-1.
| (10) |
Denote as a binary variable that is equal 2 if motif j is not associated with gene i at either time-point.
| (11) |
Let denote the vector of changes in motif association with gene i across all motifs except motif j.
| (12) |
Then the marginal mean difference in rate of change of expression between time points t and t-1 across genes associated with motif j at time t is defined as,
| (13) |
The symbol E denotes expectation while its subscript denotes the values over which the expectation is taken. The marginal mean difference in rate of change of expression between time points t and t-1 across genes associated with motif j at time t-1 is defined as,
| (14) |
The marginal mean difference in rate of change of expression between time points t and t-1 across genes not associated with motif j at either time is defined as,
| (15) |
, and are estimated using the targeted Maximum Likelihood Estimation (tMLE) approach using the tmle package (Gruber and van der Laan, 2012) in R. Random forests and Generalized Linear Models (glm) are the two models specified for use by the SuperLearner (Laan et al., 2006, 2007) for estimation in the tmle function.
The importance of motif j at time point t is then defined as,
| (16) |
The importance of motif j at time point t-1 is defined as,
| (17) |
The statistical significance of and are determined from the standard errors of , and estimated using the tmle function. The significant motifs for time points t and t-1 are identified using a Bonferroni-defined threshold of 0.05/(2N), where N is the number of identified enriched motifs in the open chromatin regions at these time points.
The net importance of each motif is then defined as,
The bar plot in Figure 4B represents the net importance values of the transcription factors. The net importance of a given transcription factor motif captures the mean fold-change (day 2 versus fibroblast stage) across all genes that gain occurrences of this motif resulting from chromatin changes in a 2kb- 500kb neighborhood around their transcription start sites during this transition versus the mean fold-change across all genes that loose occurrences of this motif during the same transition, after accounting for the effects for all other transcription factor motifs. While both positive and negative scores indicate associations with dynamically expressed genes, a positive score for a transcription factor would be consistent with an activating influence on gene expression while a negative score with a repressing influence on gene expression.
Data and Code Availability
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar, 2002) and are accessible through GEO Series accession number GSE131328 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131328).
Supplementary Material
Table S1. Differential Gene Expression for Each Cluster, Related to Figure 1.
Table S2. Differential Gene Expression for Each Cluster, Related to Figure 2.
Table S3. Dynamic ATAC-seq Regions, Related to Figure 3.
Table S4. Bulk RNA-seq Gene Expression during Reprogramming, Related to Figure 3 and Figure 4.
Table S5. Predicted Co-regulatory Interactions of Transcription Factor Candidates, Related to Figure 4.
Table S6. Motif Enrichment Related to Figure 5.
Table S7. Cluster, Fold Change and Motif Information, Related to Figure 6.
Highlights:
Integrated analyses of scRNA-, ATAC-, and ChIP-seq data interrogate cardiac reprogramming
Context-specific cooperative mechanisms guide cardiac reprogramming
Mef2c and Tbx5 bind to inaccessible chromatin and promote its remodeling
Gata4, Mef2c, and Tbx5 both facilitate and limit one another’s ability to bind to DNA
Acknowledgments
We thank members of the Srivastava lab for helpful discussions; T.R. Roberts, G. Maki, and K. Claiborn for critical comments and editing on the manuscript; A.G. Williams, T.A. Friedrich, and S. Thomas in the Gladstone Bioinformatics Core; Y. Hao and J. McGuire in the Gladstone Genomics Core; M. Cavrois, M. Gesner, and N. Raman in the Gladstone Flow Cytometry Core; S. Elmes in the UCSF Flow Cytometry Core; E. Chow and D. Bogdanoff in the UCSF Center for Advanced Technology Core; and C. Benitez and I. Espineda in the Gladstone Laboratory Animal Resource Center. All protocols concerning animal use were approved by the IACUC at the University of California San Francisco and conducted in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. N.R.S was supported by a National Science Foundation Graduate Research Fellowship, R01 Research Supplement to Promote Diversity in Health-Related Research, and a Ruth L. Kirschstein NRSA Institutional Research Training Grant. C.A.G. is an HHMI fellow of the Damon Runyon Cancer Research Foundation (DRG-2206-14). K.S.P. was supported by NHLBI/NIH grants P01 HL089707 and UM1 HL098179. D.S. is supported by NHLBI/NIH grants R01 HL057181, P01 HL089707, and UM1 HL098179; the Roddenberry Foundation; the L.K. Whittier Foundation; and the Younger Family Fund. This work was also supported by NIH/NCRR grant C06 RR018928 to the Gladstone Institutes and NIH P30 AI027763, NIH S10 RR028962, and the James B. Pendleton Charitable Trust to the Gladstone FACS core.
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE131328.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
D.S. is a co-founder and member of the board of directors of Tenaya Therapeutics. D.S., K.I., K.S.P., and T.M.A.M. have equity in Tenaya Therapeutics.
D.S. holds a patent related to this work: U.S. Patent 9,517,250 entitled “Methods for Generating Cardiomyocytes” issued on October 19, 2012. Inventors: Deepak Srivastava and Masaki leda.
REFERENCES
- Achen MG, Jeltsch M, Kukk E, Mäkinen T, Vitali A, Wilks AF, Alitalo K, and Stacker SA (1998). Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. U. S. A. 95, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, Christoforou N, Epstein JA, and Gearhart JD (2013). Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol. 60, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P, Wylie JN, Galceran J, Arkhitko O, Li C, Deng C, Grosschedl R, and Bruneau BG (2003). Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 130, 623–633. [DOI] [PubMed] [Google Scholar]
- Ahn D-G, Kourakis MJ, Rohde LA, Silver LM, and Ho RK (2002). T-box gene tbx5 is essential for formation of the pectoral limb bud. Nature 417, 754–758. [DOI] [PubMed] [Google Scholar]
- Andzelm MM, Cherry TJ, Harmin DA, Boeke AC, Lee C, Hemberg M, Pawlyk B, Malik AN, Flavell SW, Sandberg MA, et al. (2015). MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 86, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang Y-S, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TMA, Fu J-D, Spencer CI, et al. (2016). Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 167, 1734–1749.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E (2013). Comparison of Sequencing Utility Programs. The Open Bioinformatics Journal 7, 1–8. [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Evaluation of UMAP as an alternative to t-SNE for single-cell data. [Google Scholar]
- Breiman L (2001). Machine Learning. 45, 261–277. [Google Scholar]
- Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. (2001). A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106, 709–721. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Qiu X, Srivatsan S, Manfredi A, Ziller M, Overbey E, Grimaldi A, Grimsby J, Pokharel P, Livak KJ, et al. (2018). Aligning Single-Cell Developmental and Reprogramming Trajectories Identifies Molecular Determinants of Myogenic Reprogramming Outcome. Cell Syst 7, 258–268.e3. [DOI] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallero S, Shen H, Yi C, Lien C-L, Kumar SR, and Sucov HM (2015). CXCL12 Signaling Is Essential for Maturation of the Ventricular Coronary Endothelial Plexus and Establishment of Functional Coronary Circulation. Dev. Cell 33, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou N, Chellappan M, Adler AF, Kirkton RD, Wu T, Addis RC, Bursac N, and Leong KW (2013). Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PLoS One 8, e63577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, and Zaret KS (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. [DOI] [PubMed] [Google Scholar]
- Deng Y, Bao F, Dai Q, Wu LF, and Altschuler SJ (2019). Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nature Methods 16, 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghey J, Thakurela S, Charlton J, Chen JS, Smith ZD, Gu H, Pop R, Clement K, Stamenova EK, Karnik R, et al. (2018). Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat. Genet. 50, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eraslan G, Avsec Ž, Gagneur J, and Theis FJ (2019). Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. [DOI] [PubMed] [Google Scholar]
- Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, and Graf T (2008). PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc. Natl. Acad. Sci. U. S. A. 105, 6057–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J-D, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, and Srivastava D (2013). Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Reports 1, 235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S and van der Laan MJ (2009). Targeted Maximum Likelihood Estimation: A Gentle Introduction. U.C. Berkeley Division of Biostatistics Working Paper Series. WP252. https://biostats.bepress.com/ucbbiostat/paper252 [Google Scholar]
- Gruber S, van der Laan MJ (2012). tmle: An R Package for Targeted Maximum Likelihood Estimation. Journal of Statistical Software 51, 1–35.23504300 [Google Scholar]
- Guo Y, Mahony S, and Gifford DK (2012). High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput. Biol. 8, e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzinger A, and Evans T (2005). Gata4 regulates the formation of multiple organs. Development 132, 4005–4014. [DOI] [PubMed] [Google Scholar]
- leda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifkovits JL, Addis RC, Epstein JA, and Gearhart JD (2014). Inhibition of TGFβ Signaling Increases Direct Conversion of Fibroblasts to Induced Cardiomyocytes. PLoS ONE 9, e89678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishwaran H, and Lu M (2019). Standard errors and confidence intervals for variable importance in random forest regression, classification, and survival. Stat. Med. 38, 558–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson CP, Pratt RE, Rosenberg PB, Mirotsou M, and Dzau VJ (2015). MicroRNA induced cardiac reprogramming in vivo: evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 116, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan MJ, van der Laan MJ, and Pollard KS (2003). A new algorithm for hybrid hierarchical clustering with visualization and the bootstrap. Journal of Statistical Planning and Inference 117, 275–303. [Google Scholar]
- van der Laan MJ, van der Laan MJ, and Rubin D (2006). Targeted Maximum Likelihood Learning. The International Journal of Biostatistics 2. [Google Scholar]
- van der Laan MJ, van der Laan MJ, Polley EC, and Hubbard AE (2007). Super Learner. Statistical Applications in Genetics and Molecular Biology 6. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, Soussou W, Nie Z, Kang Y-J, Nakanishi N, et al. (2008). Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl. Acad. Sci. U. S. A. 105, 9397–9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin Q, Schwarz J, Bucana C, and Olson EN (1997). Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276, 1404–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J, Kist R, Scherer G, and Yutzey KE (2007). Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 305, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. (2017). Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R, Regier J, Cole MB, Jordan MI, and Yosef N (2018). Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MAH, He D, Small EM, Mileikovsky M, et al. (2016). Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 164, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra M, Schluterman MK, Nichols HA, Richardson JA, Lo CW, Srivastava D, and Garg V (2009). Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev. Biol. 326, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen C-Y, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. (2015). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Research, 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. Journal of Open Source Software 3, 861. [Google Scholar]
- McLane LM, Banerjee PP, Cosma GL, Makedonas G, Wherry EJ, Orange JS, and Betts MR (2013). Differential localization of T-bet and Eomes in CD8 T cell memory populations. J. Immunol. 190, 3207–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed TMA, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, Ang Y-S, Yu P, Wang H, Tang S, et al. (2017). Chemical Enhancement of In Vitro and In Vivo Direct Cardiac Reprogramming. Circulation 135, 978–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, and Olson EN (1997). Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 11, 1061–1072. [DOI] [PubMed] [Google Scholar]
- Nam Y-J,-J Nam Y, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, et al. (2013). Reprogramming of human fibroblasts toward a cardiac fate. Proceedings of the National Academy of Sciences 110, 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y-J, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, Bassel-Duby R, Olson EN, and Munshi NV (2014). Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development 141, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Scott Kuehn M, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. (2012). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda M, Kumaki Y, Shigeta M, Jakt LM, Matsuoka C, Yamagiwa A, Niwa H, and Okano M (2013). DNA methylation restricts lineage-specific functions of transcription factor Gata4 during embryonic stem cell differentiation. PLoS Genet. 9, e1003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protze S, Khattak S, Poulet C, Lindemann D, Tanaka EM, and Ravens U (2012). A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell. Cardiol. 53, 323–332. [DOI] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J-D, and Srivastava D (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Berry EC, Fu J-D, Ieda M, and Srivastava D (2013). Reprogramming of mouse fibroblasts into cardiomyocyte-like cells in vitro. Nat. Protoc. 8, 1204–1215. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing; https://www.R-project.org/ [Google Scholar]
- Rackham OJL, The FANTOM Consortium, Firas J, Fang H, Oates ME, Holmes ML, Knaupp AS, Suzuki H, Nefzger CM, Daub CO, et al. (2016). A predictive computational framework for direct reprogramming between human cell types. Nature Genetics 48, 331–335. [DOI] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD (2005). Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Sauls K, Greco TM, Wang L, Zou M, Villasmil M, Qian L, Cristea IM, and Conlon FL (2018). Initiating Events in Direct Cardiomyocyte Reprogramming. Cell Rep. 22, 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, Gould J, Liu S, Lin S, Berube P, et al. (2019). Optimal-Transport Analysis of Single-Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176, 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, and Bonni A (2006). A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 3l1, 1012–1017. [DOI] [PubMed] [Google Scholar]
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Maeda S, Furuhata E, Shimizu Y, Nishimura H, Kishima M, and Suzuki H (2017). A screening system to identify transcription factors that induce binding site-directed DNA demethylation. Epigenetics Chromatin 10, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, McKnight SL, and Russell DW (1997). Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11, 72–82. [DOI] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan MJ and Pollard KS (2003). Hybrid clustering of gene expression data with visualization and the bootstrap. Journal of Statistical Planning and Infererence 117, 275–303. [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Lee QY, Chen AC, Li R, Corces MR, Ang CE, Treutlein B, Xiang C, Baubet V, Suchy FP, et al. (2017). Rapid Chromatin Switch in the Direct Reprogramming of Fibroblasts to Neurons. Cell Rep. 20, 3236–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way GP, and Greene CS (2018). Bayesian deep learning for single-cell analysis. Nat. Methods 15, 1009–1010. [DOI] [PubMed] [Google Scholar]
- Welch JD, Hartemink AJ, and Prins JF (2017). MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 18, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Zhao J-P, Pruszak J, Hedlund E, Fu D, Soldner F, Broccoli V, Constantine-Paton M, Isacson O, and Jaenisch R (2008). Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 105, 5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Swaby LA, and Massagué J (1999). Multiple modes of repression by the Smad transcriptional corepressor TGIF. J. Biol. Chem. 274, 37105–37110. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, Moise AR, Wythe JD, and Martin JF (2018). Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 45, 153–169.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wei X, Wang M, Zhang R, Fu Y, Xing M, Hua Q, and Xie X (2013). Proliferation rate of somatic cells affects reprogramming efficiency. J. Biol. Chem. 288, 9767–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Morales MG, Hashimoto H, Dickson ME, Song K, Ye W, Kim MS, Niederstrasser H, Wang Z, Chen B, et al. (2017). ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev. 31, 1770–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu J-D, Wang GG, Liu J, et al. (2016). Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differential Gene Expression for Each Cluster, Related to Figure 1.
Table S2. Differential Gene Expression for Each Cluster, Related to Figure 2.
Table S3. Dynamic ATAC-seq Regions, Related to Figure 3.
Table S4. Bulk RNA-seq Gene Expression during Reprogramming, Related to Figure 3 and Figure 4.
Table S5. Predicted Co-regulatory Interactions of Transcription Factor Candidates, Related to Figure 4.
Table S6. Motif Enrichment Related to Figure 5.
Table S7. Cluster, Fold Change and Motif Information, Related to Figure 6.
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar, 2002) and are accessible through GEO Series accession number GSE131328 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131328).