

Abstract
Protein phosphatase 2A (PP2A) is indispensable in development, and deficits of PP2A and deterioration of neuronal axons have been observed in several neurodegenerative disorders, but the direct link between PP2A and the neuronal axon development is still missing. Here, we show that PP2A is essential for axon development in transfected rat brain and the dissociated hippocampal neurons. Upregulation of PP2A catalytic subunit (PP2Ac) not only promotes formation and elongation of the functional axons but also rescues axon retardation induced by PP2A inhibition. PP2A can dephosphorylate collapsin response mediator protein-2 (CRMP2) that implements the axon polarization, whereas constitutive expression of phosphomimic-CRMP2 abrogates the effect of PP2A upregulation. We also demonstrate that PP2Ac is enriched in the distal axon of the hippocampal neurons. Our results reveal a mechanistic link between PP2A and axonogenesis/axonopathy, suggesting that upregulation of PP2A may be a promising therapeutic for some neurodegenerative disorders.
Introduction
The axonogenesis defines the generation and outgrowth of axons during neuron development (Futerman et al., 1998) and regeneration (Liedtke et al., 2007), which is critical for maintaining the polarized structure of the neurons (Arimura and Kaibuchi, 2005). The normal functions of the axon are to transport a variety of functional molecules to and from the neuronal cell body and to transmit information between the neural cells by forming synapses (Shen, 2004). Although the modes for axonal transport and the physiological functions of the synapse have been studied in detail, the mechanisms underlying axonogenesis are poorly understood and there has been no effective tool to preserve the axons.
A progressive axon dystrophy, which precedes neuron death, is a major pathological event in neurodegenerative diseases (DeWitt and Silver, 1996), neural injury (Luo and O'Leary, 2005), and motor neuron dysfunctions (Zeller and Granato, 1999). Therefore, finding the molecules responsible for axonogenesis is meaningful not only in understanding the physiological mechanisms underlying the axonogenesis but also in preserving the axons. While several protein kinases, such as glycogen synthase kinase-3 (GSK-3) (Jiang et al., 2005; Yoshimura et al., 2005), PI3 kinase (Shi et al., 2003), microtubule affinity-regulating kinase 2 (MARK2) (Chen et al., 2006), and SAD kinase (Kishi et al., 2005) have been implicated in axonogenesis, little is known about the involvement of phosphatases.
Protein phosphatase 2A (PP2A) is a principal Ser/Thr phosphatase that regulates cell cycle and differentiation (Shenolikar, 1994). The predominant form of PP2A is a heterotrimer consisting of a catalytic (PP2Ac), a scaffolding (A), and a variable regulatory subunit of B, B′, B″, or B‴ families (McCright et al., 1996). PP2Ac is indispensable for development because mice with PP2Ac knockout die at embryonic stage (Götz et al., 1998). In Alzheimer disease (AD), both the activity and mRNA of PP2A are decreased in the brain (Gong et al., 1993; Vogelsberg-Ragaglia et al., 2001), in which axonopathy is prominent. Inhibition of PP2A induces tau hyperphosphorylation and axon damages in rat brain, which is similar to that observed in AD (Sun et al., 2003; Yang et al., 2007). These data suggest an intrinsic link of PP2A and the axon. Most recently, it was demonstrated in SK-N-SH cell that PP2A could prevent okadaic acid-induced phosphorylation of collapsin response mediator protein-2 (CRMP2) (Ni et al., 2008). As dephosphorylation of CRMP2 plays a crucial role in axon development (Yoshimura et al., 2005), these data imply that PP2A may be involved in axonogenesis through dephosphorylating CRMP2. Until now, the direct evidence regarding PP2A in axonogenesis was missing.
In the present study, we demonstrate that PP2A is essential for axon development both in vivo and in vitro, and upregulation of PP2A not only promotes axonogenesis but also rescues the okadaic acid-restrained axon formation and elongation with the mechanisms involving dephosphorylation of CRMP2.
Materials and Methods
Reagents and constructs.
Neurobasal and B27 were from Invitrogen. Okadaic acid (OA) and d-erythro-S were from Calbiochem. Mouse monoclonal antibody (mAb) against Tau-1 (1:200 for immunofluorescence), mAb against MAP2 (1:200 for immunofluorescence), rabbit polyclonal antibody (pAb) against MAP2 (1:1000 for immunofluorescence), and pAb against synapsin I (SynI; 1:1000 for immunofluorescence) were from Millipore; mAb against SMI312 (1:1000 for immunofluorescence) was from Covance, pAb against PP2A C subunit (1:1000 for Western blot, 1:100 for immunoprecipitation) was from Cell Signaling Technology, mAb against methyl-PP2A C subunit (1:500 for Western blot) was from Millipore, mAb against α-Tubulin (DM1A; 1:1000 for Western blot) was from Sigma, mAb against total CRMP2 (1:1000 for Western blot) from Cell Signaling Technology, pAb against phosphorylated-CRMP2 (Thr514; 1:8000 for immunofluorescence, 1:100 for immunoprecipitation) was a generous gift from Dr. K. Kaibuchi (Nagoya University, Nagoya, Japan). Amaxa Rat Neuron Nucleofector Kit was from Lonza.
The wild-type PP2Ac (wtPP2Ac) and dominant-negative PP2Ac (dnPP2Ac; L199P mutant) were gifts of Dr. Haendeler (University of Frankfurt, Frankfurt, Germany). PP2Ac cDNA was subcloned to DsRed-N1 in EcoRI and BamHI sites. The wild-type CRMP2 (wtCRMP2) and T514D and T514A CRMP2 mutants were gifts of Dr. K. Kaibuchi. For short hairpin RNA-encoding plasmids, four sequences targeting rat PP2Ac (NM_017039) were designed. The effective sequence (CTGAGAAAGTCAGGTTTGA) and the scrambled control were then cloned into pGCSIL-EGFP or pGCsi/DsRed under an U6 promoter, followed by CMV-promoted EGFP or DsRed. By activity assay, we found that the expression of dominant-negative DsRed-PP2Ac inhibited the PP2A activity to ∼60% and the expression of wild-type DsRed-PP2Ac increased the PP2A activity to ∼150% of the control levels.
Cell culture and immunofluorescent/biochemical analyses.
The hippocampal neurons isolated from embryonic 18-d-old (E18) rats were cultured according to the established procedure (Zhu et al., 2007). The cell transfections were performed according to the manufacturer's instruction (Invitrogen), and the ratio of the plasmids to Lipofectamin 2000 was 1:2. Amaxa nucleofector kit was used for nucleotransfection of the rat neurons according to the manufacturer's procedure (Jiang et al., 2005). For immunofluorescence staining, the cells were cultured on coverslips for 3, 4, 5, or 7 d in vitro (div) for different studies, as indicated in Figures 2–7 and supplemental Figures 2–4 (available at www.jneurosci.org as supplemental material), and then fixed with 4% paraformaldehyde. Rhodamine Red-X-, Oregon Green 488- (Invitrogen), or Cy5-conjugated secondary antibodies (Millipore) were used for immunofluorescence and Hoechst 33258 (1 μg/ml) (Sigma) was used for the nuclei. The images were visualized with a laser confocal microscope (LSM510 Carl Zeiss). Western blotting was performed according to the established method (Zhu et al., 2007). PP2A activity in the cell extracts was measured using the phosphatase kit V2460 according to the manufacturer's procedure (Promega).
Figure 2.

Downregulation of PP2A retards axon development in cultured hippocampal neurons. A, B, The hippocampal neurons were treated with DMSO or 10 nm OA at 12 hiv and fixed at 72 hiv for staining of Tau-1 or anti-SynΙ (A, B, red) or anti-MAP2 (A, B, green). Scale bar, 20 μm. E, F, The neurons were transfected with DsRed or DsRed-dnPP2A (dominant-negative) at 24 hiv and fixed at 72 hiv for costaining of DsRed and Tau-1 (E, green) or anti-SynI (F, green). Scale bar, 50 μm. I, PC12 cells were transfected with EGFP-siPP2A or EGFP-ssiPP2A (scrambled control) for 48 h, and the expression of PP2Ac was confirmed by Western blotting. J, The neurons were transfected with EGFP-ssiPP2Ac or EGFP-siPP2Ac at 24 hiv and fixed at 72 hiv for costaining of EGFP and Tau-1 (red). C, D, G, H, K, Quantification of the relative neurite numbers (C, G, K; n = ∼50–60) and the proportion of the neurons with no axon (NA), single axon (SA), and multiple axons (MA) (D, H; n = ∼50–60). Mean ± SEM; *p < 0.05, **p < 0.01, versus DMSO, DsRed, or ssiRNA; t test. Scale bar, 20 μm. MW, Molecular weight.
Figure 3.

Upregulation of PP2A promotes axonogenesis before formation of the axon(s). A, B, The hippocampal neurons (12 hiv) were treated with 10 nm DES and fixed at 72 hiv for staining of Tau-1 (A, red), anti-SynΙ (B, red), or anti-MAP2 (A, B, green). E–H, The hippocampal neurons were transfected at 24 hiv with DsRed-wtPP2A and the live images were captured at 72 hiv (E), then the neurons were fixed for costaining of DsRed (red) and Tau-1 (F, single axon; G, multiple axons; green) or anti-SynI (H, green). C, D and I, J, Quantification of the relative neurite numbers (C, I; n = ∼45–55) and the proportion of the neurons with no axon (NA), single axon (SA), and multiple axons (MA) (D, J; n = ∼45–55). Mean ± SEM; **p < 0.01, versus DMSO or DsRed; t test. Scale bars: A, B, 20 μm; E–H, 50 μm.
Figure 4.
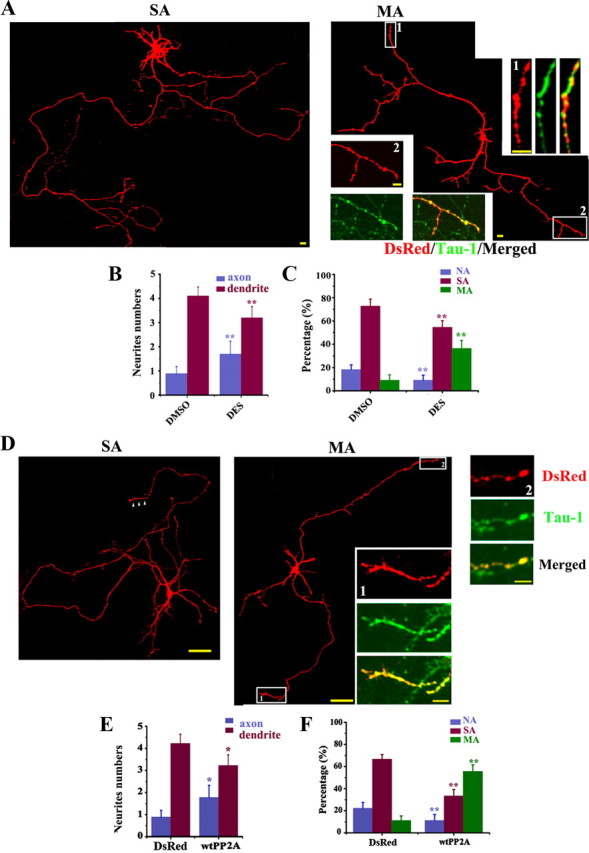
Upregulation of PP2A promotes axonogenesis after formation of the axon(s). The hippocampal neurons were transfected with DsRed at 48 hiv and cultured to 72 h to allow axon formation, then the neurons were treated with DES or the vehicle (DMSO) for 48 h and fixed at 120 hiv for costaining with Tau-1. A, The representative neurons with single axon (SA) and two axons (MA) captured at 120 hiv after DES treatment. D, The representative neurons with single axon (SA) and two axons (MA) captured at 120 hiv after transfection of DsRed-wtPP2A. B, C, E, F, Quantification of the relative neurite numbers (B, C; n = ∼35–41) and the proportion of the neurons with no axon (NA), single axon (SA), and multiple axons (MA) (E, F; n = ∼35–41). Mean ± SEM; *p < 0.05, **p < 0.01, versus DMSO or DsRed; t test. Scale bars: A, 20 μm (10 μm for insets); D, 50 μm (10 μm for insets).
Figure 5.
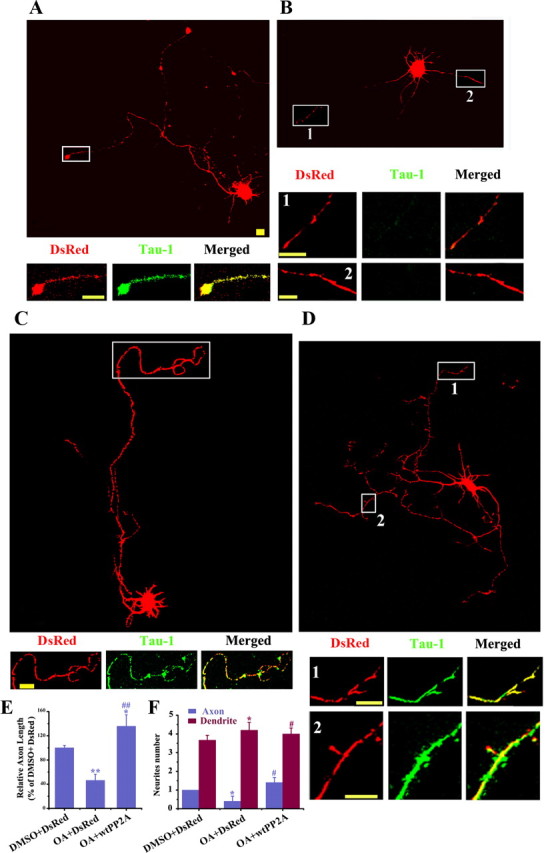
Upregulation of PP2A rescues the OA-induced inhibition of axonogenesis. The hippocampal neurons (at 12 hiv) were pretreated with 10 nm OA or DMSO for 24 h and then transfected with DsRed-wtPP2A or the vector. The neurons were fixed at 48 h after transfection and colabeled with DsRed (red) and Tau-1 (green). A, B, The representative neuron and the terminal (inset) were pretreated with DMSO (A) or OA (B) and then transfected with DsRed. C, D, The representative neuron with single axon (C) or two axons (D) was pretreated with OA and then transfected with wtPP2A. E, F, Quantification of the relative axon length (E; n = ∼25–30) and neurite numbers (F; n = ∼30–40). Mean ± SEM; *p < 0.05, **p < 0.01, versus DMSO/DsRed; #p < 0.05, ##p < 0.01, versus OA/DsRed. Scale bars: A–D and insets, 10 μm.
Figure 6.
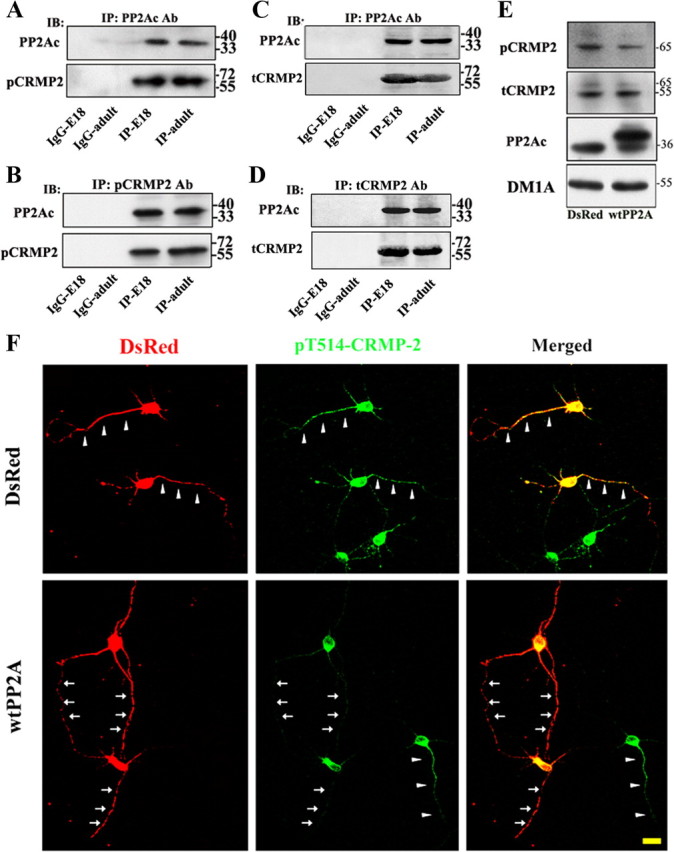
PP2A associates with and dephosphorylates CRMP2 at Thr514. A–D, PP2A associates with CRMP2. Brain extracts prepared from E18 or 3-month-old (adult) rats were immunoprecipitated (IP) with antibodies against PP2Ac (A, C) or pCRMP2 (phosphorylated, B) or tCRMP2 (total, D) and then analyzed by immunoblotting (IB). Normal IgG was used as control IPs. E, PP2A dephosphorylates CRMP2 at Thr514 measured in N2a cells. Cells were transfected with DsRed-wtPP2A or DsRed-pcDNA for 48 h and then the levels of pCRMP2, tCRMP2, and PP2Ac were measured by Western blotting. DM1A against α-tubulin was used as a loading control. F, PP2A dephosphorylates CRMP2 at Thr514 measured in hippocampal neurons. Hippocampal neurons were transfected with DsRed-pcDNA or DsRed-wtPP2A at 24 hiv and then fixed at 72 hiv for anti-pT514-CRMP2 staining. Note that the neurons expressing PP2A show weaker staining of anti-pT514-CRMP2 (arrows) than those without expressing PP2A or expressing DsRed (arrowheads). Scale bar, 20 μm.
Figure 7.
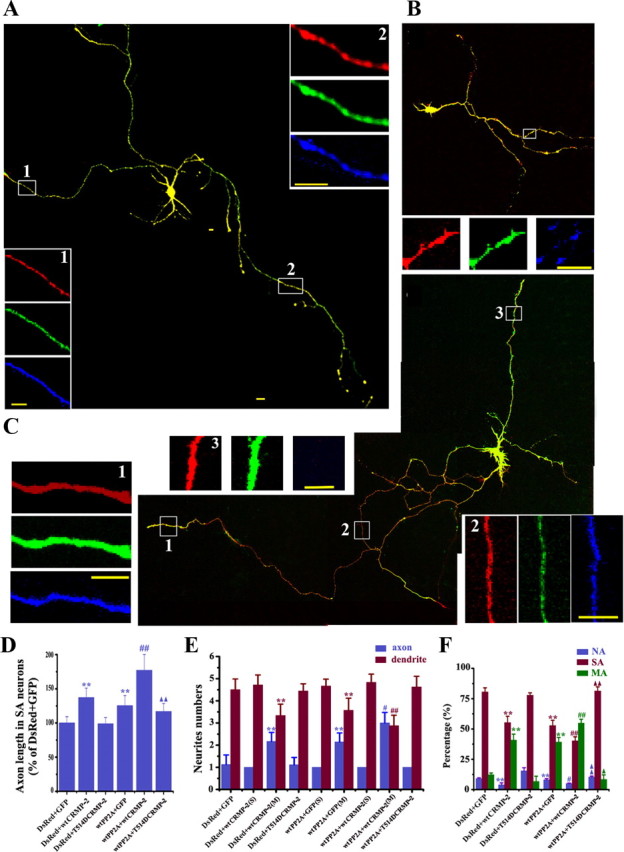
Coexpression of T514D-CRMP2 abolishes the axon-promoting effects of PP2Ac. A–C, The representative neurons were cotransfected with DsRed-wtPP2A and EGFP-vector (A), or DsRed-wtPP2A and EGFP-T514D-CRMP2 (B), or DsRed-wtPP2A and EGFP-wtCRMP2 (C) (1:1) at 24 hiv and then colabeled with Tau-1 (blue) at 3 div. D–F, Quantification of the relative axon length (D; n = ∼20–30), neurite numbers (S, neurons with single axons; M, neurons with multiple axons) (E; n = ∼26–36), and the proportion of the neurons with no axon (NA), single axon (SA), and multiple axons (MA) (F, n = ∼26–36). Mean ± SEM; **p < 0.01, versus DsRed+EGFP; ##p < 0.01, wtPP2A/wtCRMP2 versus wtPP2A/EGFP; ▲▲p < 0.01, wtPP2A/wtCRMP2 versus wtPP2A/T514D-CRMP2. Scale bars, 20 μm (10 μm for insets).
For FM4-64 experiments, PP2A was cotransfected with EGFP (3:1). The uptake and release function of the neurons was measured on an inverted microscope (LSM510 Carl Zeiss Axiovert 200 m). Neurons at 7 div were loaded with 10 μm FM4-64 in solution containing 45 mm K+ for 1 min, and then washed with solution containing 3 mm K+ for 15 min. After collecting FM fluorescent images, the same live neurons were subjected to 90 mm K+ solution for 5 min to evaluate the release function of the neurons.
In utero electroporation.
Plasmids were introduced into the lateral ventricle at E15.5–16.5 and specifically targeted into forebrain pyramidal neurons by using in utero electroporation as described previously (Saito, 2006). Briefly, timed-pregnant rats were anesthetized intraperitoneally with 100 μl of 3% pentobarbital sodium per 100 g of body weight. The abdominal cavity was opened and the uterine horns were exposed. DNA (3 μg in 2 μl) was injected through a glass micropipette into the lateral ventricle of the embryos. Electroporation (50 ms square pulses of 60 V at 100 ms intervals; 5 times) was then performed using an Electro Square Porator (CUY21Edit; Nepagene). The pups were perfused with 4% paraformaldehyde at postnatal day 3 (P3), P5, or P7, and then the brains were harvested and fixed in 4% paraformaldehyde for 2 h at 4°C, and cryoprotected in 30% sucrose overnight. Thereafter, the cerebrum was embedded in OCT compound (Sakura Finetek) and frozen in dry ice. Coronal cryosections (35 μm) were made and examined by confocal microscopy. Neurolucida software (MicroBrightfield) was used for the two-dimensional projection of axon tracing in the electroporated slices.
Measurement of axon length.
The measurement of neurites and axon length were performed using NIH ImageJ software as described previously (Ertürk et al., 2007). The mean length was calculated by measuring the total number of pixels in the images from which the pixels of the cell bodies had been subtracted and dividing by the average pixel number per micrometer of the axons and the total number of cell bodies in the image [total length per neuron = total number of pixels (only axons) × (pixel number per micrometer)−1 × (number of neurons)−1]. The fluorescence intensity of immunostaining was calculated by ImageJ software.
Statistic analysis.
Data were analyzed using SPSS 11.0 statistical software. All data were expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA procedure followed by Student-Newman-Keuls post hoc test with 95% confidence and Student's two-tailed t test.
Results
PP2A is required for axonogenesis both in vivo and in vitro
To explore the in vivo role of PP2A in axon development, we injected the EGFP-labeled siPP2Ac (a small interfering RNA construct designed specifically for PP2Ac) or ssiPP2Ac (a scrambled control) into the lateral ventricle of the embryonic rat brains and transfected the plasmid into neuronal progenitor cells by in utero electroporation (Saito, 2006). After allowing in vivo development, the pups' brain slices were prepared at P0, P3, P5, and P7 and the development of cortical axons was studied. The in vivo knockdown efficiency of PP2Ac by siRNA was confirmed by coimmunofluorescence staining of EGFP and PP2Ac (Fig. 1A). At P0, we could see a decreased number of migrating cells between subventricular zone and marginal zone (Fig. 1B). The growing neurofibers were seen in ssiPP2Ac-injected rats from P3 to P7, whereas largely migrating cells with shorter processes were observed in rats with PP2Ac knockdown by siPP2Ac (Fig. 1C–E). At P7, the long axons derived from migrated cortical neurons crossed the midline (ML) of the brain in the controls, whereas much shorter processes were observed in the neurons expressing siPP2Ac (Fig. 1F,G). To confirm that the neurofibers were axons, we labeled the brain slices at P3 with SMI312, an axon marker. We observed that the neurofibers in ssiPP2Ac-expressing neurons were nicely costained with SMI312 (Fig. 1H, boxes 1 and 2 and the enlarged). When siPP2Ac was expressed, shorter neurofibers in the proximal end of the neurons were not colabeled with SMI312 (Fig. 1H, box 3 and the enlarged), and rarely neurofibers but largely cell bodies were detected in distal end of the neurons (subplate region) (Fig. 1H, box 4 and the enlarged). These data suggest that PP2A may be a prerequisite factor in axon development.
Figure 1.

Downregulation of PP2A retards axonogenesis in embryonic rat brain. The EGFP-siPP2Ac or EGFP-ssiPP2Ac was injected into the embryonic rat brain by in utero electroporation. A, The in vivo expression of siPP2Ac diminished the immunoreactivity of PP2Ac measured by costaining with EGFP at P3. B–E, The pup's brain slices were prepared at various postnatal stages. At P0, decreased numbers of migrating cells between the subventricular zone (SVZ) and marginal zone (MZ) were shown by siPP2Ac (B). The growing neurofibers (arrowheads) were seen in ssiPP2Ac-injected rats from P3 to P7, whereas largely migrating cells with shorter processes were observed in rats with PP2A knockdown (C–E). F, The fluorescence images show retarded axon outgrowth evaluated by whether the fluorescence-labeled neurofibers could cross the ML of the brain at P7. G, The enlarged two-dimensional images from B show the morphology and the length of individual axons. H, Costaining of EGFP (green) and SMI312 (red), showing that the control neurons grow long axons which are costained with SMI312 (boxes 1 and 2), whereas the neurons expressing siPP2Ac only grow shorter processes that were not costained with SMI312 (box 3), or, in the region of subplate (SP), almost no neural processes but only cell bodies were detected (box 4). CP, Cortical plate; IZ, intermediate zone. Scale bars: A, 10 μm; B–F, 200 μm; H, 100 μm (10 μm for boxes 1–4).
The primary hippocampal neuron is a well defined model for studying the axon development (Barnes et al., 2007). To further verify the role of PP2A in axonogenesis, we treated hippocampal neurons [(12 h in vitro (hiv)] with OA, an inhibitor of PP2A, for 60 h and measured the alterations of axons and dendrites by using axon markers Tau-1 and anti-SynI, or dendrite marker anti-MAP2. We observed that most neurons treated with vehicle (DMSO) grew a single axon with average length of ∼183.7 ± 10.8 μm, in which the distal was positively labeled by Tau-1 (Fig. 2A, red) and anti-SynI (Fig. 2B, red), but the proximal segments of the axons and dendrites were mainly labeled by anti-MAP2 (Fig. 2A,B, green). The neurites became blunted with uniformed staining of Tau-1 and anti-SynI after OA treatment (Fig. 2A,B). Quantitative data showed that the average axon length was ∼87.5 ± 2.7 μm and the axon number was ∼0.6/neuron (Fig. 2C) and almost no multiple-axon (Fig. 2D) was observed after OA treatment. The alteration of PP2A activity by OA was confirmed by phosphatase activity assay (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and the activity-dependent methylation of PP2A (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
To confirm the role of PP2A, we expressed dominant-negative PP2Ac (DsRed-dnPP2Ac) into the hippocampal neurons at 24 hiv to downregulate PP2A. L199P substitution inhibits PP2Ac activity by disrupting binding and/or the substrate specificity of the holoenzyme (Evans et al., 1999). We then measured the alterations of neuronal processes at 72 hiv. Most neurons transfected with vector grew single long axon (220.4 ± 2.8 μm) that was colabeled with Tau-1 (Fig. 2E, green) and anti-SynI (Fig. 2F, green), especially in the distal segment (Fig. 2E,F, arrows). The shorter neurites were no longer colabeled with Tau-1 (Fig. 2E) or anti-SynI (Fig. 2F) and significantly decreased length (90.9 ± 5.0 μm), numbers (Fig. 2G), and the percentage of multiple axons (Fig. 2H) were detected when dnPP2A was expressed.
We also constructed EGFP-labeled siRNA of PP2A, which could respectively suppress the expression (Fig. 2I) and the activity of PP2A to ∼45% of the control levels in PC12 cells. Then, we expressed siPP2Ac or ssiPP2Ac in hippocampal neurons at 24 hiv and measured the axon length and numbers at 72 hiv by colabeling with Tau-1 (Fig. 2J, red). A remarkable suppression of axon development, demonstrated by the decreased axon length (from ∼178.3 μm to 110.2 μm) and the axon numbers (from ∼1.1 to ∼0.6/neuron) were also observed by siPP2Ac (Fig. 2K). These in vitro data together further support the critical role of PP2A in axon development.
Upregulation of PP2A facilitates functional axonogenesis before formation of the axon(s)
The activity of PP2A is decreased in the brains with AD and other neurodegenerative disorders, in which progressive axon deterioration has been observed. To explore whether upregulation of PP2A could be beneficial for axonogenesis, we first treated the hippocampal neurons with d-erythro-S (DES) to activate PP2A (Dobrowsky et al., 1993) at 12 hiv and measured the axon at 72 hiv (Fig. 3A,B). We found that the axon length and the axon number/neuron (Fig. 3C) increased respectively to ∼1.8- and ∼1.6-fold of the control levels, and ∼37% of the neurons grew multiple axons (Fig. 3D) after DES treatment, suggesting that upregulation of PP2A can promote axonogenesis. We also noticed that the numbers of dendrites decreased with increase of axons in DES group (Fig. 3C,D), suggesting that the formation of multiple axons must have been paid by expending dendrites as reported (Jiang et al., 2005). The alteration of PP2A activity by DES were confirmed by phosphatase activity assay (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and the activity-dependent methylation of PP2A (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material).
To confirm the stimulating role of PP2A, we generated and expressed the DsRed-wtPP2Ac in hippocampal neurons at 24 hiv and measured the axon alterations at 72 hiv by DsRed (Fig. 3E), or colabeling with Tau-1 (Fig. 3F, single axon; G, multiple axons) or anti-SynI (Fig. 3H). Compared with the vector transfected neurons, the axon length (300.4 ± 7.0 μm), the numbers (1.7 ± 0.7) (Fig. 3I), and the percentage of multiple axons (Fig. 3J) were all increased in the neurons expressing wtPP2A.
These data together demonstrate that upregulation of PP2A pharmacologically or genetically can stimulate functional axonogenesis before the establishment of neuronal polarity.
Upregulation of PP2A facilitates axonogenesis after formation of the axon(s)
To explore whether upregulating PP2A could promote axonogenesis in hippocampal neurons with an already formed axon, such as seen on the adult neurons, we labeled the neurons with DsRed at 48 hiv and cultured to 72 hiv to allow axon formation. At this time point, most neurons grew a single axon. Then, we treated the neurons with DES or DMSO for 48 h (to 120 hiv) before the measurement of axon length and numbers. Compared with DMSO-treated controls (data not shown), most neurons cultured with DES grew a longer single axon (Fig. 4A, left) or more multiple axons (Fig. 4A, right) that colabeled with Tau-1 (amplified inserts). By quantitative analysis, the prominently increased axon length (563.95 ± 10.9 μm), axon numbers (Fig. 4B), and the percentage of multiple axons (Fig. 4C) after DES treatment were shown.
We also expressed DsRed-wtPP2A (Fig. 4D) or its vector as a control (data not shown) in hippocampal neurons at 72 hiv and measured the axon length and the numbers at 120 hiv. Compared with the vector transfected neurons, the axon length was increased to ∼600.7 ± 14.2 μm. Similar results about axon numbers and the percentage of multiple axons were observed by quantitative analysis (Fig. 4E, F), except that the ratio of the multiple axon neurons was even higher than that induced by pharmacological activation of PP2A shown in Figure 4C.
To evaluate whether the axons induced by PP2A possess synaptic function, we transfected into the primary hippocampal neurons at 72 hiv with EGFP/wtPP2A (1:3) and cultured the neurons for another 6 div, then measured the uptake and release functions of the axons by time-lapse recording. The neurons expressing wtPP2A were visualized by EGFP (supplemental Fig. 2, green, available at www.jneurosci.org as supplemental material). Upon an initial stimulation with 45 mm K+ for 1 min, the single axon and the branches were positively loaded with FM4-64 (supplemental Fig. 2A, insets, red, pre, available at www.jneurosci.org as supplemental material), suggesting a normal uptake function of the axons. The FM4-64 was almost completely depleted when the same neuron was further treated with 90 mm K+ (supplemental Fig. 2A, insets, red, post, available at www.jneurosci.org as supplemental material), suggesting the release function of the axons. The similar uptake and release functions were also detected in neurons with multiple axons (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
These data reveal that upregulation of PP2A can facilitate axonogenesis even after the formation of neuronal polarity, which suggests a therapeutic potential of PP2A upregulation in preserving the axons.
Upregulation of PP2A rescues the OA-induced inhibition of axonogenesis
To explore whether upregulation of PP2A could rescue the AD-like axon deficits, we treated the primary hippocampal neurons (12 hiv) with OA for 24 h, and then transfected into the OA-treated neurons with DsRed-wtPP2A or the vector. At 48 h after the transfection, the axons were visualized by costaining of Tau-1 (green) and DsRed (red). Compared with the control neurons (301.4 ± 9.0 μm) (Fig. 5A), the neuronal processes became much shorter (139.3 ± 22.0 μm) with negative Tau-1 staining after OA treatment (Fig. 5B). However, long single axon (408.8 ± 42.3 μm) (Fig. 5C) and multiple axons (Fig. 5D) with costaining of Tau-1 were detected when wtPP2A was expressed in OA-pretreated neurons. Quantitative data showed that compared with OA plus vector group, both axon length and the number were significantly increased in OA plus wtPP2A group (Fig. 5E,F). These results demonstrate that upregulation of PP2A can rescue the OA-induced inhibition of axonogenesis of the neurons.
PP2A facilitates axonogenesis by dephosphorylating CRMP2
Dephosphorylation of CRMP2 at Thr514 plays a crucial stimulating role in the formation of neuronal axons (Arimura et al., 2004; Yoshimura et al., 2005). To explore whether PP2A could dephosphorylate CRMP2, we first examined the in vivo association of PP2A and pCRMP2 or total CRMP2 (tCRMP2) by coimmunoprecipitation. We found that both pCRMP2 (Fig. 6A) and tCRMP2 (Fig. 6C) were coprecipitated by anti-PP2Ac, and conversely, PP2Ac was coprecipitated by anti-pCRMP2 (Fig. 6B) and anti-tCRMP2 (Fig. 6D) in the brain extracts of E18 and 3-month-old rats, suggesting a direct interaction of PP2Ac with pCRMP2. In N2a cells, expression of wtPP2A resulted in ∼38% decrement of CRMP2 phosphorylation at Thr514 (pT514-CRMP2) (Fig. 6E). In hippocampal neurons, the fluorescent signal of pT514-CRMP2 in the shaft of the neurites became much weaker in neurons overexpressing wtPP2A (Fig. 6F, arrows) than those failing to express wtPP2A or those expressing the control vector (Fig. 6F, arrowheads). Quantitative data showed that overexpression of PP2Ac reduced the phosphorylation of CRMP2 to ∼32% of the control level. In contrast, knockdown of PP2Ac increased the phosphorylation of CRMP2 at Thr514 to ∼140% of the control level (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These data suggest that PP2Ac can interact with and dephosphorylate CRMP2.
To further verify the role of CRMP2 dephosphorylation in PP2A-induced axonogenesis, we coexpressed in the hippocampal neurons with DsRed-wtPP2A (red) and EGFP (green) (Fig. 7A), or Thr514-phosphomimic CRMP2 (EGFP-T514D-CRMP2) (Yoshimura et al., 2005) (Fig. 7B), or EGFP-wtCRMP2 (Fig. 7C) (1:1) at 24 hiv and measured the axon alterations at 72 hiv. We found that formation of multiple axons labeled by Tau-1 (blue) was commonly seen in neurons overexpressing wtPP2A (Fig. 7A) and in the neurons with coexpression of wtPP2A and wtCRMP2 (Fig. 7C), whereas less axon (blue) was detected when T514D-CRMP2 was coexpressed with wtPP2A (Fig. 7B). Quantitative data showed that the axon length increased ∼1.4-fold by coexpression of wtCRMP2 and wtPP2A compared with wtPP2A alone, but the increase was no longer detected when T514D-CRMP2 was expressed (Fig. 7D). Furthermore, the axon numbers increased 2.1-, 2.2-, and 3.0-fold by wtPP2A, wtCRMP2, and wtPP2A plus wtCRMP2, respectively, but the increase was not detected when T514D-CRMP2 was coexpressed with wtPP2A (Fig. 7E). Approximately 55% of the neurons grew multiple axons and <10% of the neurons did not grow axon when wtCRMP2 and wtPP2A were coexpressed, whereas only ∼8% of the neurons grew multiple axons when T514D-CRMP2 was coexpressed (Fig. 7F). We also found that coexpression of T514A-CRMP2, the dephosphorylated mutant, could rescue the siPP2Ac-induced retardation of axonogenesis (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material), whereas expression of T514A alone could augment axon outgrowth as reported previously (Yoshimura et al., 2005). Quantitative data showed that compared with siPP2A plus vector group, both axon length and the number were significantly increased in siPP2A plus T514A-CRMP2 group (supplemental Fig. 4B,C, available at www.jneurosci.org as supplemental material). These data further support that PP2A facilitates axonogenesis by dephosphorylating CRMP2 at Thr514.
We also coexpressed in the hippocampal neurons EGFP, EGFP-wtCRMP2, or the T514D-CRMP2 mutant with DsRed at 24 hiv and measured the role of CRMP2 in axonogenesis at 3 div. We found that wtCRMP2 could enhance axon elongation, whereas expression of T514D mutant did not show any stimulating effects (data not shown), which further confirmed the role of CRMP2 dephosphorylation at Thr514 in axonogenesis in our culture system.
PP2Ac is enriched in the distal axon of the neurons
The polarized localization of specific guidance molecules plays a crucial role in axonogenesis (Arimura and Kaibuchi, 2007). Therefore, we detected the distribution of PP2Ac in the growing axons. We expressed EGFP in the dissociated hippocampal neurons before plating, and colabeled the neurons with EGFP and anti-PP2Ac at 12 hiv, 24 hiv, and 48 hiv (Fig. 8A), respectively. We found that PP2Ac was initially present in all neurites of the unpolarized neurons (Fig. 8B), but it was preferentially enriched in the distal segment of the nascent (Fig. 8C) and maturing axons, extending into the tips of axons (Fig. 8D). This polarized localization of PP2Ac in the axon was further confirmed by Tau-1 staining (Fig. 8E,F).
Figure 8.

PP2A is enriched in the distal axons of the neurons. A–F, A polarized distribution of PP2Ac in differentiated neurons. The dissociated hippocampal neurons were transfected with EGFP before plating and then fixed at 12, 24, or 48 hiv for costaining with PP2Ac (A), the relative fluorescence intensity of PP2Ac in the axon (B–D), and costaining of PP2Ac with Tau-1 (E–F). Scale bar, 20 μm.
Discussion
A general axon dystrophy has been observed in several neurodegenerative disorders, including AD (Martin-Rehrmann et al., 2005). Therefore, preservation of the axons may help to prevent and rescue neurodegeneration. Recent studies have shown that the asymmetric neurite outgrowth and axon specification involve cytoskeleton rearrangements and protein trafficking, which is regulated by diverse signaling molecules (Jiang et al., 2005; Oliva et al., 2006; Sosa et al., 2006; Yan et al., 2006). PP2A plays a crucial role in dephosphorylating microtubule-associated protein tau (Wang et al., 1995), an important cytoskeleton protein. Knockout of PP2A is lethal in the early embryonic stage (Götz et al., 1998). Moreover, the activity of PP2A is significantly decreased with a concomitant degeneration of the neuronal axons in AD brain (Gong et al., 1993; Vogelsberg-Ragaglia et al., 2001). All these suggest a critical role of PP2A in axon development and maintenance. However, the direct link of PP2A with axonogenesis has not been reported. By pharmacological inhibition, dominant-negative downregulation, and siRNA knockdown of PP2A, we demonstrate here that PP2A is essential for axon formation and outgrowth in hippocampus and cortex neurons both in vivo and in vitro. Moreover, both pharmacological activation and genetic overexpression of wtPP2A remarkably promote axonogenesis in cultured hippocampal neurons. Previous studies demonstrated that suppression of PP2A led to neurite retraction (Leuba et al., 2008) and inhibited bipolar projection in N2a cells (Tanaka et al., 1995), whereas the two neuron-enriched B′ regulatory subunits, B′β and B′δ, promote neuritogenesis and differentiation of PC12 cells (Van Kanegan and Strack, 2009). Our results here provide the first evidence showing that PP2A is essential for axon development and upregulation of PP2A facilitates axonogenesis.
The neuronal axons can be evaluated by the relative length and specific molecular markers, such as SynI and Tau-1, and the function of the axons is usually examined by the vesicle recycle (Jiang et al., 2005). To explore whether the neogenetic axons did possess normal synaptic functions, we measured the uptake and release functions of the axons in cultured hippocampal neurons. We found that FM4-64 was quickly taken into the axons in normal neurons by 45 mm K+ treatment and it was depleted by a subsequent stimulation of 90 mm K+. With stimulation of PP2A, the neogenetic axons could uptake and release the dye as efficiently as the control neurons. These data reveal that the neogenetic axons formed by upregulating PP2A not only fit in with the structural characteristics of the axons but also possess the vesicle uptake and release functions, which makes PP2A a promising target for the regeneration of functional synapses. Furthermore, as upregulation of PP2A could rescue the okadaic acid-restrained axonogenesis, which mimics the pathological changes observed in AD brain, it implies that PP2A may serve as a therapeutic target in preventing AD neurodegeneration.
The axon outgrowth involves intricate processes. Therefore, the underlying mechanism for the axonogenesis induced by upregulation of PP2A is not fully understood. As a microtubule-binding protein, CRMP2 is expressed exclusively in the developing nervous system (Wang and Strittmatter, 1996), and it plays an important role in the regulation of axon formation during neuronal polarization (Arimura et al., 2004; Arimura and Kaibuchi, 2007). Recent studies show that overexpression of constitutively active GSK-3β causes impairment of neuronal polarization, whereas the unphosphorylated CRMP2 counteracts the inhibitory effects of GSK-3β, which indicates that GSK-3β regulates neuronal polarity through phosphorylation of CRMP2. Furthermore, suppressing the phosphorylation of CRMP2 by NT-3 and brain-derived neurotrophic factor (BDNF) promotes the axon outgrowth (Yoshimura et al., 2005). According to these data, we speculate that PP2A may exert the function through dephosphorylating CRMP2. Therefore, we detected the phosphorylation status of CRMP2 while manipulating the activity of PP2A. We found that the CRMP2 was dephosphorylated at Thr-514 in neurites of the neurons overexpressing wtPP2A and downregulation of PP2A increased the phosphorylation of CRMP2, which is the first direct evidence showing dephosphorylation of CRMP2 by PP2A. Furthermore, coexpression of wtCRMP2 with wtPP2A induced more robust axon outgrowth than expressing PP2A or CRMP2 alone, and expression of the dephosphomimic CRMP2 (T514A) could rescue the axon retardation induced by PP2A inhibition. CRMP2 can regulate microtubule assembly, actin filament reorganization, and protein trafficking in the axons (Arimura and Kaibuchi, 2005). The phosphorylated CRMP2 at Thr-514 loses the ability to interact with tubulin dimer and thus disrupts the formation of microtubules (Fukata et al., 2002), whereas overexpression of an unphosphorylated CRMP2 efficiently induces the formation of multiple axons (Yoshimura et al., 2005). By mass spectrometry, it has been demonstrated that inhibition of PP2A by OA can induce phosphorylation of CRMP2 (Hill et al., 2006). The phosphorylated CRMP2 was also detected in the AD brains (Gu et al., 2000), in which the activity of PP2A decreased prominently (Gong et al., 1993; Vogelsberg-Ragaglia et al., 2001). These data together strongly suggest that PP2A promotes axonogenesis by dephosphorylating CRMP2. PP2A is the most active phosphatase in dephosphorylating tau (Wang et al., 1996), however, tau dephosphorylation by PP2A should lead to inhibition of axonogenesis because phosphorylation of tau at Ser262 promotes axon formation (Kishi et al., 2005). Additionally, PP2A also dephosphorylates axonal neurofilament proteins (Veeranna et al., 1995) and overexpression of PP2ABγ promotes differentiation of PC12 cells by activating the mitogen-activated protein kinase cascade (Strack, 2002), which may impact the axon development. Whether these effects of PP2A are also on the pathway of the observed neuronal axonogenesis deserves further investigations.
As the mature neurons are not repleviable, the axon outgrowth of the neurons in the adult brain must have been continuously tuned to sustain physiological plasticity and repair of the damages. The observation that the axons from adult central nerves system can regenerate into peripheral nerve implants (Wictorin et al., 1990), together with the discovery that myelinating oligodendrocytes express neurite growth-inhibitory proteins (Schwab et al., 1993), has led to the notion that the neurons from CNS may be intrinsically able to regenerate their neurites, but fail to do so because of deficits of the promoting factors, such as PP2A in AD brain. The upstream factors that regulate PP2A activity are currently not very clear. A recent study demonstrates that the BDNF can activate PP2A (Takei et al., 2009). We propose that PP2A may be one of these intrinsic neuronal determinants for promoting axon outgrowth; therefore, the deficits of PP2A observed in the AD brains may be a critical factor for the neurodegeneration. Different from PP2A, GSK-3β and MARK2 may play a negative role in axonogenesis because inactivation of the kinases facilitates the formation of the axons (Jiang et al., 2005; Yoshimura et al., 2005; Chen et al., 2006), whereas upregulation of GSK-3β inhibits the axon formation (Jiang et al., 2005). Currently, one of the major approaches aimed to regenerate the injured neurons is to promote recovery of the connectivity. We believe that upregulation of PP2A may be at least a worthwhile try for the regeneration of the axons, especially in AD patients, in which the activity of PP2A is significantly downregulated.
Footnotes
This work was supported in part by the National Natural Science Foundation of China (Grants 30800342 and 30971478) and the National Science and Technology Committee of China (Grant 2006CB500703). We thank Dr. J. Haendeler for wtPP2A and dnPP2A plasmids, Drs. K. Kaibuchi and T. Yoshimura for pT514-CRMP-2 antibody and CRMP2 plasmids, Dr. S. M. Duan for the guidance of in utero electroporation, Dr. K. Shen for the constructive suggestions, and Dr. Z. L. Chen for the technical support.
References
- Arimura N, Kaibuchi K. Key regulators in neuronal polarity. Neuron. 2005;48:881–884. doi: 10.1016/j.neuron.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci. 2007;8:194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- Arimura N, Menager C, Fukata Y, Kaibuchi K. Role of CRMP-2 in neuronal polarity. J Neurobiol. 2004;58:34–47. doi: 10.1002/neu.10269. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Chen YM, Wang QJ, Hu HS, Yu PC, Zhu J, Drewes G, Piwnica-Worms H, Luo ZG. Microtubule affinity-regulating kinase 2 functions downstream of the PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal polarity. Proc Natl Acad Sci U S A. 2006;103:8534–8539. doi: 10.1073/pnas.0509955103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt DA, Silver J. Regenerative failure: a potential mechanism for neuritic dystrophy in Alzheimer's disease. Exp Neurol. 1996;142:103–110. doi: 10.1006/exnr.1996.0182. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DR, Myles T, Hofsteenge J, Hemmings BA. Functional expression of human PP2Ac in yeast permits the identification of novel C-terminal and dominant-negative mutant forms. J Biol Chem. 1999;274:24038–24046. doi: 10.1074/jbc.274.34.24038. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Ménager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Futerman AH, Boldin S, Brann AB, Schwarz A, Zisling R. Regulatory roles for sphingolipids in the growth of polarized neurons. Ann N Y Acad Sci. 1998;845:176–187. doi: 10.1111/j.1749-6632.1998.tb09670.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- Götz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc Natl Acad Sci U S A. 1998;95:12370–12375. doi: 10.1073/pnas.95.21.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Hamajima N, Ihara Y. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry. 2000;39:4267–4275. doi: 10.1021/bi992323h. [DOI] [PubMed] [Google Scholar]
- Hill JJ, Callaghan DA, Ding W, Kelly JF, Chakravarthy BR. Identification of okadaic acid-induced phosphorylation events by a mass spectrometry approach. Biochem Biophys Res Commun. 2006;342:791–799. doi: 10.1016/j.bbrc.2006.02.029. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–932. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, Marin P, Bouras C, Savioz A. Postsynaptic density protein PSD-95 expression in Alzheimer's disease and okadaic acid induced neuritic retraction. Neurobiol Dis. 2008;30:408–419. doi: 10.1016/j.nbd.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Liedtke T, Schwamborn JC, Schröer U, Thanos S. Elongation of axons during regeneration involves retinal crystallin beta b2 (crybb2) Mol Cell Proteomics. 2007;6:895–907. doi: 10.1074/mcp.M600245-MCP200. [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Martin-Rehrmann MD, Hoe HS, Capuani EM, Rebeck GW. Association of apolipoprotein J-positive beta-amyloid plaques with dystrophic neurites in Alzheimer's disease brain. Neurotox Res. 2005;7:231–242. doi: 10.1007/BF03036452. [DOI] [PubMed] [Google Scholar]
- McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271:22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- Ni MH, Wu CC, Chan WH, Chien KY, Yu JS. GSK-3 mediates the okadaic acid-induced modification of collapsin response mediator protein-2 in human SK-N-SH neuroblastoma cells. J Cell Biochem. 2008;103:1833–1848. doi: 10.1002/jcb.21575. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Kapfhammer JP, Bandtlow CE. Inhibitors of neurite growth. Annu Rev Neurosci. 1993;16:565–595. doi: 10.1146/annurev.ne.16.030193.003025. [DOI] [PubMed] [Google Scholar]
- Shen K. Molecular mechanisms of target specificity during synapse formation. Curr Opin Neurobiol. 2004;14:83–88. doi: 10.1016/j.conb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Shenolikar S. Protein serine/threonine phosphatases–new avenues for cell regulation. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Sosa L, Dupraz S, Laurino L, Bollati F, Bisbal M, Cáceres A, Pfenninger KH, Quiroga S. IGF-1 receptor is essential for the establishment of hippocampal neuronal polarity. Nat Neurosci. 2006;9:993–995. doi: 10.1038/nn1742. [DOI] [PubMed] [Google Scholar]
- Strack S. Overexpression of the protein phosphatase 2A regulatory subunit Bgamma promotes neuronal differentiation by activating the MAP kinase (MAPK) cascade. J Biol Chem. 2002;277:41525–41532. doi: 10.1074/jbc.M203767200. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. 2003;118:1175–1182. doi: 10.1016/s0306-4522(02)00697-8. [DOI] [PubMed] [Google Scholar]
- Takei N, Kawamura M, Ishizuka Y, Kakiya N, Inamura N, Namba H, Nawa H. Brain-derived neurotrophic factor enhances the basal rate of protein synthesis by increasing active eukaryotic elongation factor 2 levels and promoting translation elongation in cortical neurons. J Biol Chem. 2009;284:26340–26348. doi: 10.1074/jbc.M109.023010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Katagiri M, Arima S, Matsuzaki K, Inokoshi J, Omura S. Neuronal differentiation of Neuro 2a cells by lactacystin and its partial inhibition by the protein phosphatase inhibitors calyculin A and okadaic acid. Biochem Biophys Res Commun. 1995;216:291–297. doi: 10.1006/bbrc.1995.2623. [DOI] [PubMed] [Google Scholar]
- Van Kanegan MJ, Strack S. The protein phosphatase 2A regulatory subunits B′beta and B′delta mediate sustained TrkA neurotrophin receptor autophosphorylation and neuronal differentiation. Mol Cell Biol. 2009;29:662–674. doi: 10.1128/MCB.01242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna, Shetty KT, Link WT, Jaffe H, Wang J, Pant HC. Neuronal cyclin-dependent kinase-5 phosphorylation sites in neurofilament protein (NF-H) are dephosphorylated by protein phosphatase 2A. J Neurochem. 1995;64:2681–2690. doi: 10.1046/j.1471-4159.1995.64062681.x. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol. 2001;168:402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–4860. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996;16:6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wictorin K, Brundin P, Gustavii B, Lindvall O, Björklund A. Reformation of long axon pathways in adult rat central nervous system by human forebrain neuroblasts. Nature. 1990;347:556–558. doi: 10.1038/347556a0. [DOI] [PubMed] [Google Scholar]
- Yan D, Guo L, Wang Y. Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J Cell Biol. 2006;174:415–424. doi: 10.1083/jcb.200511028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yang XF, Wang YP, Tian Q, Wang XC, Li HL, Wang Q, Wang JZ. Inhibition of protein phosphatases induces transport deficits and axonopathy. J Neurochem. 2007;102:878–886. doi: 10.1111/j.1471-4159.2007.04603.x. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Zeller J, Granato M. The zebrafish diwanka gene controls an early step of motor growth cone migration. Development. 1999;126:3461–3472. doi: 10.1242/dev.126.15.3461. [DOI] [PubMed] [Google Scholar]
- Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, Wang Q, Chen JG, Wang JZ. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci. 2007;27:12211–12220. doi: 10.1523/JNEUROSCI.3321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]