

Abstract
Reference memory characterizes the long-term storage of information acquired through numerous trials. In contrast, working memory represents the short-term acquisition of trial-unique information. A number of studies in the rodent hippocampus have focused on the contribution of long-term synaptic potentiation (LTP) to long-term reference memory. In contrast, little is known about the synaptic plasticity correlates of hippocampal-based components of working memory. Here, we described a mouse with selective expression of a dominant-negative mutant of the regulatory subunit of protein kinase A (PKA) only in two regions of the hippocampus, the dentate gyrus and area CA1. This mouse showed a deficit in several forms of LTP in both hippocampal subregions and a lowered threshold for the consolidation of long-term synaptic depression (LTD). When trained with one trial per day in a water maze task, mutant mice displayed a deficit in consolidation of long-term memory. In contrast, these mice proved to be more flexible after a transfer test and also showed a delay-dependent increased performance in working memory, when repetitive information (proactive interference) was presented. We suggest that through its bidirectional control over synaptic plasticity PKA can regulate opposing forms of memory. The defect in L-LTP disrupts long-term memory consolidation. The persistence of LTD may allow acquisition of new information by restricting the body of previously stored information and suppressing interference.
Introduction
Reference memory corresponds to the long-term storage of information acquired through numerous trials. In contrast, working memory represents the short-term acquisition of trial-unique information (Baddeley, 1981; Cowan et al., 2008). It has been proposed that working memory is thought to be a short-term form of memory that, once used, is forgotten or ignored (Dudchenko, 2004; Delaney and Sahakyan, 2007). A consequence of this view is that forgetting would be deleterious to RM, but is required for working memory. However, unlike memory, forgetting processes and their biological bases are poorly understood.
Hippocampal-based memory formation is thought to require changes in synaptic efficacy (Bliss and Lomo, 1973; Bliss and Collingridge, 1993; Kandel, 2001). The two major kinds of hippocampal-based synaptic plasticity are long-term synaptic potentiation (LTP) and long-term synaptic depression (LTD) (Bear and Malenka, 1994; Bear and Abraham, 1996). Whereas studies of hippocampus-dependent memory suggest that the long-term storage of information requires LTP-like mechanisms (Kandel, 2001; Malleret et al., 2001; Martin and Morris, 2002), the role of LTD is less clear (Braunewell and Manahan-Vaughan, 2001; Malenka and Bear, 2004; Etkin et al., 2006). Novel environment exploration facilitates stimulus-induced LTD and reverses previously elicited potentiation (Manahan-Vaughan and Braunewell, 1999; Kemp and Manahan-Vaughan, 2004). In contrast, blockade of NMDA receptor-dependent forms of plasticity (which include LTD) preserves preinduced in vivo LTP and increases retention of spatial memory (Villarreal et al., 2002). It therefore has been proposed that LTD could increase the signal-to-noise ratio of a memory trace. In agreement with this notion, we recently found, using transgenic mice in which we inhibited a specific form of LTD, that LTD could function to weaken previous memory traces, thereby preventing those traces from interfering with newly encoded information when the demands of a task change as it is the case in a working memory experiment (Nicholls et al., 2008).
Here, we describe a transgenic mouse with the inverse phenotype. This mouse expresses a dominant-negative form of the PKA regulatory subunit, R(AB), known to decrease both the late phase of hippocampal LTP (L-LTP) and hippocampus-dependent long-term memory (Abel et al., 1997). We were able to limit R(AB) expression to only two areas of the brain, the hippocampus and the second and third layer of the cortex. In the hippocampus, the expression was restricted to granule cells of the dentate gyrus (DG) and pyramidal neurons of the CA1 area. The transgenic mice had deficits in the consolidation of long-lasting forms of LTP in the CA1 and dentate gyrus areas of the hippocampus, in which the transgene was expressed, but not in the CA3 area, in which the transgene was not expressed. In contrast, we found that LTD expression was enhanced in the dentate gyrus and CA1 hippocampal areas of the transgenic mice. This may be caused by attenuation of a negative constraint imposed by PKA activity over threshold mechanisms for LTD maintenance. Importantly, these physiological changes were associated with a deficit in long-term reference memory, but improved working memory abilities in the transgenic mice. However, the enhancement in working memory was observed only when repetitive information was presented across trials. This work thus provides genetic evidence that antagonistic forms of memory depend on the bidirectional regulation of synaptic plasticity, and suggests that the maintenance of the proper balance between LTP and LTD is necessary for normal cognitive processes.
Materials and Methods
Expression vector
MT-REVAB plasmid, encoding R(AB) cDNA, an inhibitory mutant of PKA RIα regulatory subunit, was generously provided by Dr. Stanley McKnight, University of Washington (Clegg et al., 1987). R(AB) cDNA was cloned into the EcoRV site of pNN265 vector containing 5′ and 3′ exon/intron splicing boundaries and a polyadenylation signal (Abel et al., 1997). The resulting NotI fragment with R(AB) and simian virus 40 (SV40) sequences was cut out from the pNN265 and ligated into the SrfI site of pGP103 vector containing 12 kb of the grp gene promoter and three copies of the neuron-restrictive silencer element (NRSE) inserted into the BstXI site (Fig. 1A).
Figure 1.

R(AB) transgene expression in the mouse brain. A, Top, Schematic representation of the 5′ end grp gene promoter sequences in front of the translation initiation site ATG, which were used to generate grp-R(AB) transgene shown in the bottom. The grp-R(AB) transgene includes 12 kb of the grp promoter, three copies of the neuron-restrictive silencer element (NRSE), an SV40-derived hybrid exon/intron splicing sequences (closed boxes) and a 5′ untranslated leader, R(AB) cDNA, and an SV40-derived 3′ exon/intron splicing sequences and a polyadenylation signal. B, RNA in situ hybridization shows expression of the R(AB) transgene in the brain. Coronal brain sections from grp-R(AB) transgenic mice were hybridized with a digoxigenin-labeled RNA probe specific to the 3′ end SV40 sequences present in the transgene. Transgene expression is limited to CA1, dentate gyrus (DG) and cortex on the brain sections corresponding to the area ranging from the dorsal to ventral hippocampus. PFC, Medial prefrontal cortex.
Transgenic mice
The 13.5 kb NotI fragment from pGP103R(AB) plasmid was isolated using Elutrap (Schleicher and Shuell) and injected into the pronuclear of BL6CBAF2/J zygotes. For genotyping, tail DNA was analyzed by Southern blotting using transgene-specific probes. Founders were backcrossed to C57BL/6J strain to more than N5 generations for electrophysiological and behavioral experiments. Mice were maintained under standard conditions, consistent with the NIH guidelines and approved by the IACUC. All animals used in our experiments were between 3 and 4 months of age.
In situ hybridization
For RNA in situ hybridization, cryostat-cut mouse coronal brain sections (20 μm thickness) were hybridized with digoxigenin-labeled RNA probes as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993).
P-CREB detection
Surgery.
Mice were implanted under general anesthesia (avertin, 300 mg/kg i.p.) with two guide cannulae (0.4 mm diameter, 8 mm long) aimed vertically toward the dorsal hippocampus. Stereotaxic coordinates were 2.0 mm posterior to the bregma, 1.3 mm each side of the sagittal suture, 0.9 mm ventral from the skull surface (Fig. 2A). Following surgery, the mice recovered for 10 d.
Figure 2.

Forskolin-induced phosphorylation of CREB is reduced in the CA1/DG-R(AB) transgenic mice in the CA1 area of the hippocampus. A, Scheme of injection sites of forskolin (FSK) in the CA1 area. B, Representative sections illustrating P-CREB immunoreactivity in the dorsal hippocampus of naive mice (B1 and B2) and mice injected with FSK (B3 and B4) for control mice (B1 and B2) and mutant mice (B3 and B4). C, Bar graphs represent means (±SEM) of the number of pCREB-positive neurons/mm2 in the CA1 for naive mice (n = 5 for both genotypes) and 1 h after bilateral intrahippocampal injections of FSK (0.3 μg in 0.2 μl) (mutant mice, n = 8; control mice, n = 7). *Significantly different (p < 0.05).
Drug injections.
Bilateral intrahippocampal injections of 7b-diacetyl-7b-(g-N-methylpiperazino)-butyril, dihydrochloride (forskolin, Calbiochem) were performed at the dose of 0.3 μg in 0.2 μl (2.4 mm) based on previous studies (Wong et al., 1999) in freely moving mice.
One hour after injection, an animal was deeply anesthetized with 300 mg/kg avertin and immediately perfused transcardially with ice-cold solutions of 4% paraformaldehyde in phosphate buffer (0.1 m, pH 7.4). The delay of 1 h after the forskolin injection was chosen based on our previous study that showed that it produces strong induction (Martel et al., 2006). After postfixation overnight in the same fixative at 4°C, coronal sections (40 μm) were cut on a vibratome and collected in Tris buffer (0.1 m, pH 7.4). After elimination of endogenous peroxidase activity and a blocking step, sections were incubated for 48 h with rabbit anti-phospho-CREB (Ser133) antibody (1:3000; Cat # 06-519; Millipore). Subsequently, sections were incubated with biotinylated goat anti-rabbit antibody (1:600; ABC kit; Vector) and with the ABC complex and antibody binding was visualized with diaminobenzidine (DAB, Sigma). Sections were mounted on gelatin-coated slides, air-dried, dehydrated, covered with a coverslip with mounting media, and examined using light microscopy.
The number of P-CREB-positive cells was counted in the hippocampus (CA1) using ImageJ software. Counting was performed at 63× magnification. The anteroposterior (AP) coordinates relative to bregma of the areas included for detailed analyses were AP −1.7 to −2.5 mm (Paxinos and Franklin, 2001). Nuclei were counted and expressed as the number of P-CREB-positive nuclei per square mm. Statistical analysis of the immunohistochemical data was performed using ANOVA.
Electrophysiology
Preparation and stimulation protocols.
Transverse hippocampal slices (400 mm) from control and mutant mice were incubated in an interface chamber at 28 ± 1°C, subfused with oxygenated artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 4.0 KCl, 1.5 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose), and allowed to equilibrate for at least 90 min. Field EPSPs (fEPSPs) were recorded at either CA3/Schaffer collateral-CA1 synapses, CA3/commissural-CA3 synapses or perforant pathway-dentate gyrus synapses. The stimulation intensity (square pulse, 50 ms duration) was adjusted to give fEPSP slopes of ∼40% of maximum. Baseline and after stimuli responses were sampled once per minute at this intensity.
Statistical analysis.
Data analysis consisted in Student's t test for two independent populations between series of control and transgenic traces at specific data periods (i.e., 60–90 min) using the Microcal Origin statistical tool (Microcal Software). Data in the figures represent mean ± SE and in the text mean ± SD.
Drugs.
KT5720 (Calbiochem) was used to block PKA activity.
Behavioral experiments
The water maze was a white circular swimming pool (120 cm in diameter) and its condition of use has been described previously (Malleret et al., 1999, 2001). The radial maze apparatus was an automated, elevated, eight-arm radial maze (Marighetto et al., 1993; Saxe et al., 2007). The movement of each door, located at the entrance to each arm, was controlled by the experimenter from an adjacent room who also tracked continuously the position of the subject within the maze. This enabled a real-time control of the accessibility to the maze arm(s) according to a predetermined test schedule. Food-deprived male mice (85% ad libitum weight) were habituated to retrieve 20 mg food pellets (Bioserv) from three specific baited arms of eight. On day 1, only the three baited arms were opened and food was retrieved. From day 2 to day 5, all eight arms were opened and number of errors (visit to unbaited arm) was recorded (one trial per day). On day 6, working memory tasks begun (see Fig. 8 legend and Results). Statistical analysis: The effect of genotype (control vs mutant) on path length (water maze), number of errors and score (radial maze) was evaluated by using two-way ANOVAs (Statview) when analyses across daily (or block) sessions (within-subject factors) were required (repeated measures). The effect of genotype on time spent in quadrant, number of platform crossings (water maze), score (radial maze) was evaluated by using one-way ANOVAs whenever more complex analyses were not necessary (nonrepeated measures such as quadrant type, platform type in the water maze, or delays in the radial maze).
Figure 8.

Mutant mice showed enhanced delay-dependent working memory when manipulating a limited number of items presented repetitively. A, Number of errors in a LI/HML working memory task (limited interference/high memory load). Inset shows an example for three trials (1 per day) composed of a sample phase (4 arms, randomly chosen every day, are opened; dark blue), followed by a delay phase (mouse restrained on the central platform for 5 s), followed by the test phase [all 8 arms are opened; number of errors (runs in an already visited/nonbaited arm) is scored]. No significant effect of genotype was observed. B, Score (same group of mice; n = 12/12) in a LML working memory task (LML low memory load; 1 arm to remember among 2). Inset shows an example for three trials (4 per day) composed of a sample phase (one arm and then another opposite arm, randomly chosen, are opened), followed by a delay phase (mouse restrained on the platform for 5 s (training), 1 or 2 min), followed by the test phase (two pairs of arms are opened; the same two pairs for every trials; the pairs of arms is composed by an arm already visited during the sample phase and an adjacent baited arm) in which the number of correct choices is scored (runs in an arm not visited during the sample phase). The bar graph represents the average score for 1 week of experiment at each delay. No difference was found between groups during training (5 s delay), but mutant mice showed enhanced performance when the delay increased to 1 and 2 min (p = 0.008 and 0.028 respectively) compared with control subjects. C, Score (new group: control n = 14; mutant n = 12) in a LI/LML working memory task. Only one pair (pseudorandomly determined; different pair for every trial among 8 possible pair arrangements) is now presented by trial; six trials/d; score plotted against blocks (one block = two daily sessions of 6 trials each = 12 trials). During both training (block B1 to B5; delay phase = 15 s) and testing (increased delay phase; 20–75 s), no difference was observed between groups. D, Same task as in C. However, the same pair of arms is now used for every trial (increased proactive interference; HI/LML protocol). Using this procedure, mutant mice showed enhanced performance for a delay of 55 s compared with controls (p = 0.027).
Results
The R(AB) transgene expression is limited to the CA1 and dentate gyrus areas in the hippocampus
Earlier work has provided initial pharmacological and genetic evidence for the PKA role in long-term forms of synaptic plasticity and memory in Aplysia and Drosophila as well as in the late-phase of hippocampal LTP and long-term spatial memory in the mammalian brain (Frey et al., 1993; Abel et al., 1997; Bourtchouladze et al., 1998; Abel and Nguyen, 2008). In the mouse transgenic approach used in some of these earlier studies, the R(AB) inhibitory mutant, shown to inhibit PKA function in a dominant-negative manner both in cell culture and transgenic mice (Brandon et al., 1995; Abel et al., 1997), was driven by the CaMKIIα promoter, with transgene expression distributed throughout the forebrain. In the present study, we attempted to limit regional expression of the PKA inhibitor. To this end, we have generated a transgenic mouse line that expresses the PKA mutant (R(AB)) in two specific areas of the hippocampus, the dentate gyrus and the CA1 region. In the DG/CA1-R(AB) transgenic mice, the R(AB) transgene is driven by the 12 kb-long of the upstream sequences of the gastrin-releasing-peptide (grp) gene promoter (Fig. 1A). The 3′ end of the grp promoter includes parts of the first exon and terminates immediately before the ATG translation initiation site corresponding to the GRP protein. Three copies of the neuron-restrictive silencer element (NRSE) sequences were inserted upstream of the R(AB) cDNA to increase the strength and neuron specificity of the grp promoter (Bessis et al., 1997). RNA in situ hybridization on coronal slices of the transgenic mice showed that the R(AB) transgene is limited to the dentate gyrus and the CA1 region of the hippocampus and no expression in the CA3 area (Fig. 1B). Except for the superficial layers II and III of the cerebral cortex (Fig. 1B), no other brain area expresses the transgene.
Phosphorylation of CREB is reduced in the CA1 area of the transgenic mice
To examine whether PKA activity is decreased in the mutant mice we analyzed the level of phosphorylation of one of the PKA substrates, cAMP-responsive element binding protein (CREB). Free-moving mutant mice and control littermates were injected into the CA1 area of the hippocampus with the adenylate cyclase activator forskolin (Fig. 2A), which normally leads to CREB phosphorylation (Huang et al., 2000). Using an antibody that recognizes the phosphorylated form of CREB (P-CREB), we found a significant increase of P-CREB staining in the CA1 area of control mice following the forskolin injection (Fig. 2B,C). In forskolin-injected control mice, the amount of the P-CREB-positive cells was fivefold higher than that in naive control mice (F(1,10) = 18.91, p = 0.014). In contrast, mutant mice showed a significant deficit in forskolin-induced phosphorylation of CREB and no difference was detected between naive and forskolin-injected mutant animals (p > 0.1). Moreover, ANOVAs conducted on these measures indicated a significant group effect (F(1,21) = 6.01, p = 0.023). Post hoc analysis (Scheffe's F test) revealed lower P-CREB activation in mutant mice (p < 0.011). This result was consistent with previous work from our laboratory as well as others that showed that the R(AB) mutant decreased PKA activity both in cell culture and live animals (Clegg et al., 1987; Abel et al., 1997; Isiegas et al., 2006). This result suggested that PKA- and CREB-dependent transcription may be downregulated in mutant mice which in turn may lead to changes in synaptic plasticity in the CA1 and dentate gyrus areas as well as in hippocampus-dependent long-term memory.
Deficits in L-LTP correlate with R(AB) transgene expression in the CA1 and dentate gyrus areas
To determine the electrophysiological consequences of R(AB) transgene expression, we studied LTP in the Schaffer collateral to CA1 pyramidal neuron (SC-CA1) pathway. We induced protein synthesis-dependent L-LTP in mutant mice and control littermates by four trains of HFS (four trains of 1 s train at 100 Hz, 5 min intertrain interval). As described previously for mice that express R(AB) under the control of the CaMKIIα promoter (Abel et al., 1997), this stimulation protocol reliably induced L-LTP in control mice but failed to do so in mutant mice (Fig. 3A) (60–90 min: Ctr, 174 ± 10%; Mut, 141 ± 26%, p = 0.02). Furthermore, since transgene expression in these animals was restricted to the postsynaptic side at this synapse, our finding supports the existence of a postsynaptic role for PKA in L-LTP (Duffy and Nguyen, 2003). However, this impairment is not as strong as in the study by Abel et al. Since the transgene is also expressed in the dentate gyrus (DG), we examined L-LTP at perforant pathway to granule cell synapses (PP-DG). Similar to our findings in the CA1 area, L-LTP in the mutant mice was significantly reduced (Fig. 3B) (60–90 min: Ctr, 171 ± 24%; Mut, 135 ± 12%, p = 0.01). Finally, we also examined L-LTP in the CA3 to CA3 commissural pathway (CP-CA3), in which R(AB) was not expressed, and found that L-LTP at this synapse was spared in the mutant mice (Fig. 3C) (60–90 min: Ctr, 192 ± 32%; Mut, 174 ± 14%, p = 0.4). The differences we observed in L-LTP at Schaffer collateral and perforant pathway synapses were not due to changes in basal synaptic transmission as shown by similar input-output relationships of fEPSP slope at these synapses for mutant and control mice (p = 0.4, data not shown).
Figure 3.

Deficit in the late phase of LTP correlates with R(AB) expression. A–C, Four trains of HFS (1 s train at 100 Hz, 5 min intertrain interval) produced the long-lasting L-LTP in control mice that was abolished only in SC-CA1 (A) and PP-DG (B), but not CP-CA3 (C), pathways in mutant mice (n = 5/5 for each experiment). D–F, Similarly, four trains of theta bursts stimulation (TBS, 5 min intertrain interval) produced L-LTP in control mice that was also abolished in SC-CA1 (D) and PP-DG (E), but not CP-CA3 (F), pathways in mutant mice (n = 5/5 for each experiment). The early phase of LTP is unaffected in mutant mice G, H, The transient early phase of LTP (E-LTP) induced by one train of HFS (1 s train at 100 Hz) was unaffected in the SC-CA1 (G) or the PP-DG (H) pathways in mutant mice (n = 5/5 for each experiment). I, J, Whereas one train of TBS produced normal LTP in the SC-CA1 pathway (I), a deficient LTP was observed in the PP-DG pathway (J) (n = 5/5 for each experiment).
Since high-frequency stimulation (HFS) and theta burst stimulation (TBS) have been shown to evoke L-LTP by activating distinct molecular pathways (Arai et al., 1994; Huang and Kandel, 1994; Nguyen et al., 1994; Staubli and Otaky, 1994; Castro-Alamancos et al., 1995; Moody et al., 1998; Patterson et al., 2001), we next examined L-LTP induced by four trains of TBS (nine bursts of four pulses at 100 Hz, 200 ms interburst interval, 5 min intertrain interval). As with the HFS protocol, we found that repeated trains of TBS elicited impaired L-LTP in the mutant mice in both the SC-CA1 and the PP-DG pathways (Fig. 3D) (60–90 min: Ctr, 182 ± 10%; Mut, 148 ± 17%, p = 0.01) (Fig. 3E) (60–90 min: Ctr, 185 ± 10%; Mut, 144 ± 13%, p = 0.006), but not in the CP-CA3 pathway (Fig. 3F) (60–90 min: Ctr, 161 ± 26%; Mut, 171 ± 21%, p = 0.56). These findings suggest that postsynaptic PKA activity may be generally required for the expression of the late phase of LTP independent of the induction mechanism.
We next asked whether R(AB) expression might also affect the early, protein synthesis independent, phase of LTP (E-LTP). Consistent with the early findings of Abel et al. (1997), we found that one train of HFS induced similar E-LTP in both control and mutant mice in SC-CA1 synapses (Fig. 3G) (30–60 min: Ctr, 114 ± 8%; Mut, 109 ± 10%, p = 0.46) and PP-DG synapses (Fig. 3H) (30–60 min: Ctr, 128 ± 21%; Mut, 121 ± 16%, p = 0.63). Consistent with findings by Woo et al. (2000), this was also the case for LTP induced by a single train of TBS in the SC-CA1 pathway (SC-CA1 pathway) (Fig. 3I) (60–90 min: Ctr, 162 ± 15%; Mut, 152 ± 22%, p = 0.56). However, we found that a single TBS led to deficient LTP in PP-DG synapses (Fig. 3J) (60–90 min: Ctr, 153 ± 12%; Mut, 123 ± 7%, p = 0.01), and the early onset of this deficit (∼10 min) suggests a deficit in LTP induction rather than expression.
In summary, we found that postsynaptic PKA activity is crucial for the consolidation of L-LTP induced by repeated and spaced trains of HFS or TBS (e.g., 4×HFS or 4×TBS), with no noticeable contribution to E-LTP induced by a single train of HFS. LTP induced by single train of TBS stimulation was normal in the CA1 area, but impaired in the dentate gyrus area. Thus, one can speculate that the specific sensitivity to repeated and spaced patterns of stimulation, places postsynaptic PKA activity (at least in the CA1 area) as part of a molecular threshold that preferentially associates this kind of input streams of activity in a neuron.
Mutant mice show enduring LTD
Compared with an extensive literature on the role of PKA in LTP (for review, see Nguyen and Woo, 2003), relatively fewer studies have examined its function in LTD (Brandon et al., 1995; Kameyama et al., 1998; Lee et al., 1998, 2000; Santschi et al., 2006; Smith et al., 2006). We thus studied the expression of LTD in transgenic mice in the SC-CA1 and PP-DG pathways of the hippocampus. We tested LTD induced by a long train of low-frequency stimulation (15 min at 1 Hz, 900 pulses) (Dudek and Bear, 1992; Malenka and Bear, 2004). This stimulation results in a robust early phase of synaptic depression in the CA1 area of adult mice that decays slowly back to baseline in ∼90 min (Alarcón et al., 2004; Etkin et al., 2006). We found that this 1 Hz stimulation protocol evoked transient LTD in the SC-CA1 (Fig. 4A) (90–120 min: 95 ± 8%, vs 100% p = 0.17) and the PP-DG pathways (Fig. 4B) (90–120 min: 90 ± 8%, vs 100% p = 0.45) in control mice. However, in R(AB)-expressing animals, this same protocol produced an enduring or prolonged LTD in both the SC-CA1 (Fig. 4A) (90–120 min: Mut, 81 ± 9%, p = 0.007) and the PP-DG pathways (Fig. 4B) (90–120 min: Mut, 73 ± 11%, p = 0.006). In the SC-CA1 pathway this effect was not due to enhanced LTD amplitude as the early expression of LTD was similar in control and mutant mice (Fig. 4A) (SC-CA1 10–15 min: Ctr, 63 ± 15%, Mut, 67 ± 18%, p = 0.7; SC-CA1 pathway 60–90 min: Ctr, 92 ± 10%; Mut, 83 ± 7%, p = 0.07). However, this was not the case for the PP-DG pathway, in which we did observe an enhancement in the magnitude of LTD during induction (Fig. 4B) (PP-DG 10–15 min: Ctr, 59 ± 9%; Mut, 41 ± 17%, p = 0.04) (Fig. 4B) (60–90 min: Ctr, 89 ± 8%; Mut, 77 ± 11%, p = 0.06). Interestingly, bath application of the PKA inhibitor KT5720 transitory enhanced LTD expression in the SC-CA1 pathway (Fig. 4A, inset) (10–15 min: Ctr 72 ± 9%, KT 81 ± 4%, p = 0.06; 60–90 min: Ctr 95 ± 4%, KT 78 ± 6%, p = 0.0004; 120–150 min: Ctr 96 ± 3%, KT 88 ± 16%, p = 0.33). However, that was not the case for the PP-DG pathway (Fig. 4B, inset) (10–15 min: Ctr 59 ± 4%, KT 81 ± 9%, p = 0.01; 60–90 min: Ctr 87 ± 9%, KT 85 ± 17%, p = 0.79; 120–150 min: Ctr 91 ± 6%, KT 79 ± 18%, p = 0.22). It is important to exert caution at the interpretation of this pharmacological results as this approach is different that a restricted expression of R(AB) transgene only at the postsynaptic cells.
Figure 4.
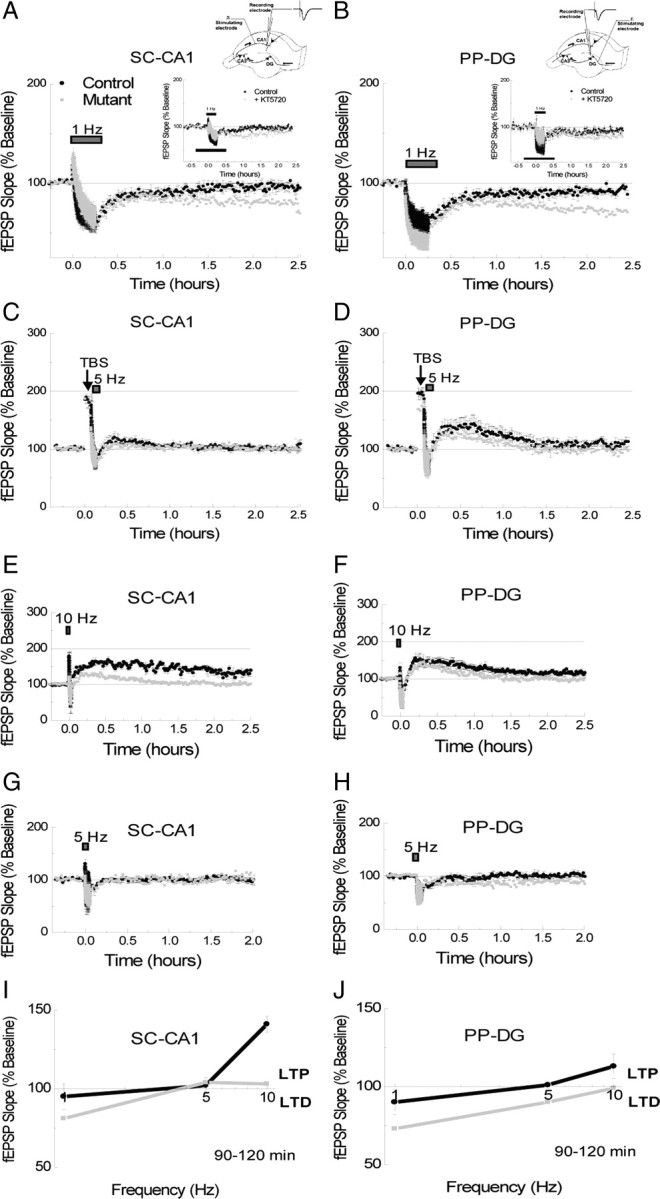
Enhanced LTD in DG/CA1-R(AB) transgenic mice. A, B, Transient LTD induced by 15 min of 1 Hz stimulation was transformed into a long-lasting LTD in mutant mice in the SC-CA1 (A) and in the PP-DG (B) pathways (n = 7/7 for each experiment). Insets, Effect of the PKA inhibitor KT5720 (1microM) on the expression of LTD in the SC-CA1 (A) and the PP-DG (B) pathways. C, D, Synaptic potentiation induced in response to one train of TBS was successfully depotentiated by 5 Hz stimulation for 3 min in both the SC-CA1 (C) and in the PP-DG (D) pathways in each group of mice (n = 5/5 for each experiment). Metaplastic shift in the PP-DG pathway. E, F, LTP induced by 90 s of 10 Hz stimulation (900 pulses) produces a long-lasting and a transient form of LTP in the SC-CA1 (E) and in the PP-DG (F) pathways respectively in control mice. Transgene expression differentially affected each pathway in mutant mice (n = 5/5 for each experiment). G, H, Three minutes of 5 Hz stimulation (900 pulses) produced no long-term plasticity change in control mice in either the SC-CA1 (G) or the PP-DG (H) pathways. However, a modest LTD was observed in the PP-DG pathway, but not in the SC-CA1 pathway, of mutant mice in response to this stimulation (n = 5/5 for each experiment). Frequency–response curves (1, 5, and 10 Hz) from SC-CA1 (I) and PP-DG (J) pathways. Mutant mice show a mild but significant metaplastic shift toward the LTD portion of the curve at the PP-DG but not the SC-CA1 pathway.
We next investigated depotentiation, a form of synaptic plasticity that produces a reduction in synaptic efficacy. However unlike LTD, depotentiation only reverses the earlier effects of potentiation, and does so through molecular mechanisms that are distinct from LTD (Barrionuevo et al., 1980; Wagner and Alger, 1996; Lee et al., 2000). Consistent with earlier studies of the role of PKA on depotentiation (Otmakhova and Lisman, 1998), we found that depotentiation of LTP by a 3 min train of 5 Hz stimulation was unaffected in mutant mice (Fig. 4C) (SC-CA1 pathway 60–90 min: Ctr, 104 ± 6%; Mut, 103 ± 11%, p = 0.83) (Fig. 4D) (PP-DG pathway 60–90 min: Ctr, 113 ± 18%; Mut, 103 ± 10%, p = 0.32).
A shift in the frequency–response curve is observed in the PP-DG pathway in mutant mice
The prolonged LTD expression observed in mutant mice could be explained by a metaplastic shift toward LTD, that is a shift in the LTP/LTD induction threshold favoring LTD expression. To examine this possibility we generated frequency–response curves (Bienenstock et al., 1982) from the synaptic responses elicited by 900 pulses at 1, 5, and 10 Hz in both the SC-CA1 and the PP-DG pathways. The effect of 10 Hz is shown in Figure 4, E and F. This stimulation induced a prolonged form of LTP in the SC-CA1 pathway (Fig. 4E), but only a transient form of LTP in the PP-DG pathway (Fig. 4F) in control mice. In mutant mice, LTP expression induced by 10 Hz stimulation was dramatically impaired in the SC-CA1 pathway (Fig. 4E) (90–120 min: Ctr, 141 ± 5%; Mut, 103 ± 8%, p = 0.00003), but only modestly affected in the PP-DG pathway (Fig. 4F) (90–120 min: Ctr, 113 ± 8%; Mut, 99 ± 9%, p = 0.04). Next, we tested the response of these pathways to 5 Hz stimulation (Fig. 4G,H). Whereas 5 Hz stimulation produced no long-term (>60 min) changes in synaptic efficacy in either synaptic pathway in control mice or in the SC-CA1 pathway in mutant mice (Fig. 4G) (90–120 min: Ctr, 102 ± 5%; Mut, 104 ± 4%, p = 0.55), it did produce a modest but significant LTD in the PP-DG pathway in mutant mice (Fig. 4H) (90–120 min: Ctr, 101 ± 2%; Mut, 90 ± 6%, p = 0.036).
By plotting the responses we observed in the PP-DG pathway to 1, 5, and 10 Hz stimulation, we find that R(AB) expression in this pathway causes a mild shift in favor of LTD suggesting the enhanced LTD and deficient LTP we observe may result from this shift (Fig. 4J). In contrast, a similar analysis of the SC-CA1 pathway responses to 1, 5, and 10 Hz suggest that the enhanced LTD and impaired LTP we observe in this pathway in the R(AB) mice are not the result of a simple metaplastic shift (Fig. 4I; see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Because of PKA function has been shown to regulate LTP and LTD induction (Kameyama et al., 1998; Lee et al., 2000; Skeberdis et al., 2006), we investigated whether the expression of R(AB) transgene could have an impact on the extend of the depolarization envelope during the 1 s 100 Hz tetanic stimulation. Our data show that transgene expression increased the overall depolarization envelope (Fig. 5A, top traces, ACSF), with significant contribution of NMDA receptor-dependent activation (Fig. 5A, bottom traces, ACSF-APV) (cumulative area in mV/msec: Ctr: −784,088 v/s Mut: −1,030,668). The NMDA receptor-dependent increase in transgenic mice is mainly detectable during the first 300 ms and an analysis of the first 50 ms is shown in Figure 5B. This increase was not due to changes in fast inhibition as the same results were observed in the presence of the GABAA receptor blocker picrotoxin (data not shown).
Figure 5.

NMDA receptor function in mutant mice. A, NMDA receptor component is increased in mutant mice. Top traces, Depolarization envelope during 1 s at 100 Hz 1 in normal ACSF solution in slices from control (black) and mutant (gray) mice. Middle traces, Same condition but in slices treated with the NMDA receptor blocker APV (100 μm). Bottom traces, Arithmetic subtraction between the ACSF (top) and APV (middle) traces reveals the NMDA receptor activation-dependent component of the depolarization envelope. Each trace corresponds to the mean of six independent observations. B, Analysis of the first 50 ms of 1 s tetanic stimulation (bottom). The amplitude of synaptic decrease during tetanus is similar in control (black) and mutant (gray) mice (top left); however, mutant mice show larger depolarization (cumulative area under each trace) compared with control mice (top right). Each trace corresponds to the mean of eight independent observations. C, NMDA-LTD is enhanced in mutant mice. Addition of NMDA (30 μm, 10 min) induces long-lasting synaptic depression with larger amplitude in mutant mice.
Mutant yielded LTP deficit and LTD enhancement, two phenotypes that could go along with diminished NMDA receptor activity. Hence, we would have expected mutants with a smaller depolarization envelope. Our analysis demonstrates that the effect of the transgene largely affects the late, but not the early, phases of LTP and LTD. These data do not rule out a potential effect of the transgene on induction mechanisms that could contribute to the late changes in LTP or LTD maintenance or the LTP/LTD shift observed here. It is possible that the increased NMDA receptor component selectively activates or facilitates mechanisms for LTD expression but no for LTP expression (Xu et al., 2009). To further explore the role of NMDA receptor activation on the plastic properties of mutant mice, we elicited chemical-LTD by addition of NMDA. Mutant mice showed an enduring enhancement in LTD amplitude compared with control mice (Fig. 5C) (60–90 min, Ctr: 47 ± 2.16; Mut: 30 ± 3%, p < 0.05). These data suggest a connection between NMDA receptor activation and enhanced late-phase LTD in mutant mice.
In summary, we found that R(AB)-expressing animals show altered LTD. Although postsynaptic PKA could be part of the mechanism of LTD expression (Huang et al., 1999a,b), the impingement of PKA activity over LTD is thought to be indirect (Kameyama et al., 1998; Lee et al., 2000). That is, the PKA influence over LTD results from its opposing effect on phosphatase activity that leads to the induction and consolidation of LTD. Diminishing activity of this negative constraint, would therefore lower the threshold for LTD. Our data show that although the threshold for LTD induction is changed in the PP-DG pathway of mutant mice, the effect of R(AB) is general to the regulation of the threshold for LTD consolidation (i.e., prolonged LTD).
Mutant mice show normal acquisition and memory when trained with four trials per day in the classical Morris water maze task
To assess possible behavioral phenotypes that can be ascribed to the long-term synaptic changes observed in these mice, we studied a variety of memory processes using different behavioral tasks. We tested the mice in a water maze task (Morris et al., 1982) with four trials per day as previously described (Nolan et al., 2004). During the cued version of the experiment (visible platform; 2 d; hippocampal independent), or the spatial training (hidden platform; acquisition; hippocampal dependent), no significant difference in path length, latency, swimming speed, time spent floating, and thigmotaxis (time spent swimming at the periphery—like floating, thigmotaxis is an index of anxiety) was observed between mutant and controls mice indicating that the expression of the transgene did not lead to any profound behavioral deficit (Fig. 6). In fact, mutant and control mice acquired the task rather rapidly. At day 8, the average path length to reach the platform was minimal for all mice (∼200 cm; or latency ∼10 s). In addition, when tested during probe trials (performed on days 7, 12, and 19, 1 week after training) (Fig. 6C), both groups of mice spend approximately the same amount of time in the quadrant that harbored the platform, and crossed the same amount of time in this virtual location (no significant difference).
Figure 6.

Mutant mice showed normal acquisition and memory when trained with four trials per day in the water maze. Performance (path length: each point represents the average of 4 trials/d) of mutant and control (n = 15/15) mice in the water maze task. A, B, Visible platform nonspatial/hippocampal (A) and hidden platform spatial/hippocampal (B) versions of the task (no significant effect of genotype in both cases). C, Memory of the platform location was tested with a probe trial realized 1 week after training, on day 19. No significant difference in the amount spent in the target quadrant (TQ: where the platform was located during training) was found between groups (AQ: average of the two adjacent quadrants; OQ: opposite quadrant). Inset shows the amount of crossings in each virtual platform location in the corresponding quadrants. D–F, No difference was found in swim speed (D), percentage of time spent floating (E), or thigmotaxis (percentage of time spent in periphery; F), indicating that transgene expression did not lead to major behavioral deficit.
Mutant mice display increased forgetting
To further investigate long-term memory abilities in these mice, we trained a second group of mice in a new water maze task using a weak training of one trial per day (Fig. 7A). At the end of training, we observed that, even though the time spent searching in the target quadrant was equivalent in both groups of subjects (Fig. 7B), mutant mice were less accurate than their controls in locating the exact platform position, as indicated by a significantly decreased number of crossings in the target platform area (Fig. 7B, inset). More importantly, a week after training, mutant mice were not able to locate the target quadrant/platform area, suggesting they had forgotten its location (Fig. 7C). This may indicate weakened consolidation of spatial information due to hippocampal PKA inhibition that is reminiscent of the lack of consolidation of L-LTP.
Figure 7.

Mutant mice showed increased forgetting and cognitive flexibility. A–C, Performance (A) and probe (B) trials performed at the end of training (probe 1) and 1 week after (probe 2) (C) of a second group of naive mice when trained with a one-trial-per-day regimen. Even though no difference was initially seen between groups during training (A), the statistical analysis realized on probe trial (B) showed that mutant mice were not able to locate as accurately as controls the target platform location (significant genotype effect: P TQ p = 0.042). A week later (C), they also show a total loss of memory for this position as they were not even able to preferentially swim in the target quadrant as controls did (TQ p = 0.001; P TQ p = 0.015) suggesting impaired ability to consolidate information in long-term memory. We thus observed a decrease in the time spent searching the TQ (percentage of time spent in TQ during probe 2 − percentage of time spent during probe 1) in mutant, but not control mice (genotype effect; p = 0.008), indicating forgetting of the platform location. D, Path length (same group of mice as in Fig. 6) during a transfer task (platform placed in the opposite quadrant) plotted against day (one trial per day); no significant difference between groups. E, F, Statistical analysis of the probe trials realized on day 9 (E) and a week later on day 16 (F) revealed that mutant mice were able to flexibly learn the new platform position more efficiently than controls [Significant genotype effect for probe (F) p = 0.017, and probe (F) p = 0.004] as they spent more time in the new TQ (TQ: new target quadrant; OQ: old target quadrant). Mutant mice were also crossing significantly more than controls in the platform target location on day 16 (p = 0.003) but not on day 9 (insets E and F).
Improved cognitive flexibility in mutant mice
To further evaluate the behavioral phenotype of the mutant mice, we next tested cognitive flexibility, an adaptive form of behavior that requires that an animal does not perseverate in choosing a wrong solution (i.e., the one that was previously correct but is no longer appropriate). To do so, we relocated the platform to a new quadrant (in the same experimental context) after the initial training phase (seen previously in Fig. 6A–C). Both groups of mice learned to locate the new position of the platform as suggested by the decrease in path length observed over days of transfer training (Fig. 7D). However, when tested during probe trials, mutant subjects spent more time searching for this platform in the new quadrant/platform area (Fig. 7E,F), suggesting that they were more flexible and adapted their search pattern to the new platform position more accurately than controls.
Intact working memory in the radial maze when submitted to one daily trial
To investigate the consequence of increased cognitive flexibility, we next tested working memory in a radial maze apparatus. This form of memory heavily relies on the short-term storage of information and the forgetting of previously learned information (Dudchenko, 2004) that is no longer relevant (proactive interference). In a first experiment (Floresco et al., 1997), the mice were submitted to one trial per day (limited number of interference LI). During a sample phase, they first had to explore four arms randomly chosen to obtain food rewards. After a short delay (5 s), in a subsequent choice phase, they were allowed to explore all eight arms, but only the four arms not explored previously during the sample phase were baited. Entry into an already visited arm was counted as a working memory error. The ANOVA revealed a significant session effect (F(14,308) = 2.966; p = 0.0003), indicating that all mice improved their performance over days and acquired the rule necessary to execute the task (Fig. 8A). However, no significant difference in weight, food-deprivation regimen, latency to visit the first arm, time to perform the task or number of errors were found between mutant and control mice. This suggests that the mutant and control mice were equivalent in motivation, locomotion, and even cognitive abilities to remember once-a-day multiple information. Thus, in a high memory load task (HML—four arms to be remembered among eight), control and mutant mice were indistinguishable.
Mutant mice exhibit improved performance in a working memory task when submitted to multiple trials per day and long intratrial delays
By using a different protocol (low memory load; LML; working memory task), we then tested the mutant mice in a multiple-trials/2-choices/radial-maze task. The mice were given four trials per day with an intertrial (ITI) delay of 50 min. During a sample phase (Fig. 8B, inset), they were presented one randomly chosen arm among a pair of baited arms (pair #1). After this first rewarded visit, they came back to the central platform and were allowed to visit a second arm (randomly chosen—pair of arms #2). After a variable delay (intratrial delay), the mice were then submitted to the choice phase were they were allowed to enter one arm only of pair #1 (both arms of the pair now being opened) and then of pair #2. A run into an arm not previously visited, and still baited, was scored as a correct choice. The maximum score for a daily session was therefore eight (four trials × two correct choices/trial). Both groups of mice quickly learned this new rule when the delay used between sample and choice phase was short (<5 s). However, with a delay equal or >1 min, controls showed a dramatic decrease in performance compared with mutant mice which strikingly seemed not to be affected by the increasing delay (Fig. 8B) (delay 5 s: cont = 5.25 ± 0.24, mut = 5.38 ± 0.24; 1 min: cont = 4.45 ± 0.15, mut = 5.33 ± 0.27; 2 min: cont = 4.53 ± 0.15, mut = 5.28 ± 0.28; chance = 4).
The improvement in working memory is specifically observed in task requiring the manipulation of repetitive information
To elucidate the nature of such working memory enhancement in mutant mice, we decided to perform an additional LML working memory experiment. We used a modified version of the protocol described above with the only difference being that only one pair of arms is visited during each trial (Lee and Kesner, 2003). To avoid postural mediation strategy (Dudchenko, 2004), the mice were removed from the maze during the delay period, and were submitted to 6-trials per day (ITI = 5 min). During stage 1 (Fig. 8C), each trial (sample phase followed by choice phase) consisted of the visit into a different pair of adjacent arms (limited number of proactive interference LI). During stage 2 (Fig. 8D), the same pair of arms was used every day, for every trial. This procedure allowed us to assess the influence of an increasing number of interference on the performance of the animals, with stage 2 (repetitive presentation of the same pair of arms) representing the higher level (HI) of conflicting information. Two weeks of training were required for all mice to reach an acceptable level of performance when confronted with the LI/LML task (stage 1) with a 15 s delay. No difference between groups was observed. When confronted with increasing delay, both groups exhibited decreased performance in a similar way. Two weeks were also necessary to acquire the stage 2 HI/LML task. However, when confronted with a 55″ delay, once again, mutant mice showed better performance than their controls. This significant effect was not due to any experimental artifact as we replicated this result over two additional blocks of trials (4 d—significant effect of genotype; p = 0.016). In addition, there were no contributions from any aging or building effect during the experimental procedure because mutant animals returned to controls level when they were submitted for a second time to the LI/LML task (data not shown). Transgenic mice are therefore more apt at manipulating repetitive information over an intermediate delay, suggesting they are less sensitive to proactive interference than their control littermates.
Discussion
We find that the DG/CA1-R(AB) mutant mice have opposite changes in long-term synaptic plasticity. Whereas consolidation of LTP is impaired, consolidation of LTD is facilitated. Behaviorally, these mice are more flexible in memory tasks, which accounts for a delay-dependent improvement of working memory when repetitive information is presented. This improvement may be due to increased forgetting, since the mutant mice also exhibit a deficit in consolidation of long-term memory.
There is now clear evidence that PKA signaling has a key role in a variety of implicit and explicit memory processes in both invertebrate and in mammals (Ghirardi et al., 1992; Kandel and Squire, 2000; Schafe and LeDoux, 2000; Kaplan and Abel, 2003). For example, PKA activation is crucial for the consolidation of long-term memories in both the hippocampus and amygdala. While PKA activation enhances LTP in the hippocampus and long-term memory consolidation, its inhibition disrupts LTP and memory consolidation (Frey et al., 1993; Huang and Kandel, 1994; Abel et al., 1997; Lee et al., 2000; Arnsten et al., 2005). Our results provide further evidence for the role of PKA in long-term memory processes. While training the mice with 4 trials/d in the water maze did not impair the performance of mutant mice, a more difficult training using only 1 trial per day caused a deficit in the long-term storage of spatial information. This indicates a role for PKA in reference memory that can be compensated with intense training.
At the biochemical level, PKA is crucial for the dynamic balance between protein kinases and phosphatases which regulates the consolidation of LTP and LTD. Previous genetic and pharmacological studies show that changes in PKA function differentially affect expression of LTP and LTD (Brandon et al., 1995; Abel et al., 1997; Hensch et al., 1998; Rao et al., 2004). Because reducing kinase activity results in decreased phosphorylation of AMPA receptors (Lee et al., 1998, 2000), thereby sustaining LTD (Kameyama et al., 1998), changes in PKA function can have an effect not only on LTP but also on LTD. Our study in mutant mice provides independent genetic support for this idea. We found that restricted expression of a dominant-negative mutant of the PKA regulatory subunit produces bidirectional changes in hippocampal synaptic plasticity, impaired LTP and facilitated LTD consolidation that affect not only reference memory but also working memory.
Working memory is trial-unique-specific (Baddeley, 1981) and is particularly sensitive to interference (Dudchenko, 2004). If the interferences are of similar nature as the information to be remembered, or represent previously stored information that has to be discarded (proactive interference), the interference may cause a disruption during a working memory task (e.g., calculation, word processing). Until recently, the role of the hippocampus in working memory has been underestimated. While the hippocampus has been thought to have a restrictive, but nonetheless significant role in reference long-term memory (Scoville and Milner, 1957), the prefrontal cortex was thought to be involved exclusively in cognitive flexibility, action planning, and in working memory functions (Fuster, 2000). Activation of PKA in prefrontal cortex was found to have a deleterious effect on working memory as assessed in a delayed alternation task (Taylor et al., 1999). Interestingly, inhibition of PKA activity by prefrontal injection of Rp-cAMPS led to working memory impairment in a delayed spatial win-shift task in the radial maze (Aujla and Beninger, 2001). These studies are in agreement with earlier studies showing that too much or too little activation of the prefrontal dopamine D1 receptor pathway (which regulates PKA activity) can lead to prefrontal-dependent cognitive functions (Seamans and Yang, 2004). Thus, we cannot rule out the possibility that the improved performance in working memory task observed in our mutant mice can be affected by the limited cortical expression of the R(AB) transgene.
Recently however, the hippocampus has gained recognition as an anatomical site for information storage during working memory tasks. To begin with, there is now good evidence for parallel processing of information between the prefrontal cortex and the hippocampus in the storage of a multiple number of trial-unique items during a working memory task (Floresco et al., 1997). For example, Lee and Kesner have documented this point using single and double inactivation of the prefrontal cortex and the hippocampus in a delayed nonmatch to place task (Lee and Kesner, 2003). When the working memory rule is applied to a small number of trial-unique items, these authors found that double inactivation provoked a deficit in such task, but single inactivation has no impact on the performance of pretrained animals for delays up to 10 s, indicating that the inactivation of one structure can be compensated by the activity of the other. However, when the delay exceeded 10 s, single inactivation of the hippocampus, but not of the prefrontal cortex, was sufficient to impair working memory. Together, these results indicate that for short-term delays the hippocampus and the prefrontal cortex might process working memory information in parallel; but as soon as the system detects a longer delay, hippocampal memory may become essential, demonstrating more persistence than the prefrontal cortex. Our findings are consistent with this view. We find that our mutant mice display an improvement of performance specifically when the delay used was 55 s and therefore was hippocampus-dependent. Interestingly, we recently found that manipulation specifically restricted to the hippocampus (ablation of adult hippocampal neurogenesis in the dentate gyrus) yields the same delay-dependent phenotype (Saxe et al., 2007). Therefore, although we cannot rule out a prefrontal cortex influence, we favor the hippocampal interpretation for the enhanced working memory performance in our mice.
Previous genetic manipulations in animals have focused on either increased LTP and improved long-term memory (Tang et al., 1999; Malleret et al., 2001) or diminished LTD and impaired working memory (Zeng et al., 2001). Here, we have here tried to examine both LTP and LTD concomitantly. This allowed us to investigate the cooperative relationship between these processes and different forms of learning and memory. While hippocampal LTP is largely correlated with long-term reference memory, little is known about the memory correlates of hippocampal LTD (Kemp and Manahan-Vaughan, 2004; Etkin et al., 2006). Recently, Nakao and colleagues found that performance of rats in a spatial working memory task tightly correlates with the magnitude of LTD observed in the dentate gyrus (Nakao et al., 2002). Similarly, inhibition of protein phosphatase calcineurin in the forebrain causes both impairment in a spatial working memory task and a decrease in hippocampal LTD (Zeng et al., 2001). These results suggest that LTD has a positive role in working memory. Our results support this idea and provide an explanation to this role of LTD in working memory. In our working memory task, the high degree of interference from previous trials requires the subject to dampen or inhibit the memory formed during the preceding trials. Mechanisms associated to the facilitated persistence of LTD may improve the ability of the animal to forget what occurred during previous trials (proactive interference), thus enabling it to commit fewer errors. Although the interplay between LTP and LTD may well take place during the manifestation of each memory process, the association between LTD and improvement in working memory performance in these specific conditions defines one role of LTD-like mechanisms as an essential process to remove or inhibit extraneous information and thus improve the processing of relevant information. In agreement with this view, we recently showed that forebrain inhibition of the phosphatase PP2A impaired an NMDA-dependent form of LTD, reversal learning in the water maze (transfer), and working memory performance by inhibiting forgetting processes (Nicholls et al., 2008). We here show that inhibiting PKA and protein-phosphorylation has the opposite effect.
Whereas the “remembering” aspect of memory has been well studied, mechanisms involved in forgetting are far less explored (Markowitch and Scott, 1988; Wixted, 2004). Here, we report an antagonism between forms of memory that require (working memory), or are impaired by (long-term/reference memory), forgetting of previously encoded information. Interestingly, we showed that ablation of hippocampal neurogenesis yields a similar outcome-deficient long-term memory and enhanced working memory performance (Saxe et al., 2006, 2007). These results are in agreement with studies that find rich contextual encoding can be an impediment to working memory (Delaney and Sahakyan, 2007; Cowan et al., 2008). Forgetting is thought to occur as a result of low-frequency stimulation of synapses in the theta or delta range (Staubli and Lynch, 1990), which are firing frequencies that are naturally observed in sleep or restful states in neuronal networks (Buzsáki and Draguhn, 2004; Wolansky et al., 2006). It was recently suggested that the role of slow-wave sleep is to downscale synaptic strength to a baseline level that is beneficial for learning and memory, and that downscaling is likely to use many of the same molecular mechanisms involved in LTD (Tononi and Cirelli, 2006). Sleep-induced synaptic depression could thus promote some sort of reset of neuronal network necessary for working memory and high cognitive functions in general. Our data allow exploration of the causal relationship between bidirectional synaptic plasticity and two antagonistic types of memory that might differentially rely on these processes.
Footnotes
G.P.S. is supported by the Whitehall Foundation, the National Alliance for Research on Schizophrenia and Depression, the Charles and Johanna Busch Memorial Fund, the New Jersey Governor's Council on Autism, and the National Science Foundation Grant IOS-0752811. E.R.K., G.M., and J.M.A. are supported by the Howard Hughes Medical Institute and the Kavli Institute for Brain Sciences. We are thankful to Russell Nicholls, Joe Rayman, and Christoph Kellendonk for valuable comments on an early version of this manuscript and Isabel Muzzio for her help with statistical analysis.
References
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Arai A, Black J, Lynch G. Origins of the variations in long-term potentiation between synapses in the basal versus apical dendrites of hippocampal neurons. Hippocampus. 1994;4:1–9. doi: 10.1002/hipo.450040103. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Ramos BP, Birnbaum SG, Taylor JR. Protein kinase A as a therapeutic target for memory disorders: rationale and challenges. Trends Mol Med. 2005;11:121–128. doi: 10.1016/j.molmed.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Aujla H, Beninger RJ. Hippocampal-prefrontocortical circuits: PKA inhibition in the prefrontal cortex impairs delayed nonmatching in the radial maze in rats. Behav Neurosci. 2001;115:1204–1211. [PubMed] [Google Scholar]
- Baddeley A. The concept of working memory: a view of its current state and probable future development. Cognition. 1981;10:17–23. doi: 10.1016/0010-0277(81)90020-2. [DOI] [PubMed] [Google Scholar]
- Barrionuevo G, Schottler F, Lynch G. The effects of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci. 1980;27:2385–2391. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bessis A, Champtiaux N, Chatelin L, Changeux JP. The neuron-restrictive silencer element: a dual enhancer/silencer crucial for patterned expression of a nicotinic receptor gene in the brain. Proc Natl Acad Sci U S A. 1997;94:5906–5911. doi: 10.1073/pnas.94.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunewell KH, Manahan-Vaughan D. Long-term depression: a cellular basis for learning? Rev Neurosci. 2001;12:121–140. doi: 10.1515/revneuro.2001.12.2.121. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci. 1995;15:5324–5333. doi: 10.1523/JNEUROSCI.15-07-05324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg CH, Correll LA, Cadd GG, McKnight GS. Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]
- Cowan N, Morey CC, Chen Z, Gilchrist AL, Saults JS. Theory and measurement of working memory. Psychol Learn Motivation. 2008;49:49–103. [Google Scholar]
- Delaney PF, Sahakyan L. Unexpected costs of high working memory capacity following directed forgetting and contextual change manipulations. Mem Cogn. 2007;35:1074–1082. doi: 10.3758/bf03193479. [DOI] [PubMed] [Google Scholar]
- Dudchenko PA. An overview of the tasks used to test working memory in rodents. Neurosci Biobehav Rev. 2004;28:699–709. doi: 10.1016/j.neubiorev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci U S A. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy SN, Nguyen PV. Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci. 2003;23:1142–1150. doi: 10.1523/JNEUROSCI.23-04-01142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Alarcón JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, Kandel ER. A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron. 2006;50:127–143. doi: 10.1016/j.neuron.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Seamans JK, Phillips AG. Selective roles for hippocampal, prefrontal cortical, and ventral striatal circuits in radial-arm maze tasks with or without a delay. J Neurosci. 1997;17:1880–1890. doi: 10.1523/JNEUROSCI.17-05-01880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Fuster JM. Executive frontal functions. Exp Brain Res. 2000;133:66–70. doi: 10.1007/s002210000401. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N. Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron. 1992;9:479–489. doi: 10.1016/0896-6273(92)90185-g. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Gordon JA, Brandon EP, McKnight GS, Idzerda RL, Stryker MP. Comparison of plasticity in vivo and in vitro in the developing visual cortex of normal and protein kinase A RIβ-deficient mice. J Neurosci. 1998;18:2108–2117. doi: 10.1523/JNEUROSCI.18-06-02108.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999a;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- Huang LQ, Rowan MJ, Anwyl R. Role of protein kinases A and C in the induction of mGluR-dependent long-term depression in the medial perforant path of the rat dentate gyrus in vitro. Neurosci Lett. 1999b;274:71–74. doi: 10.1016/s0304-3940(99)00561-3. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci. 2000;20:6317–6325. doi: 10.1523/JNEUROSCI.20-17-06317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isiegas C, Park A, Kandel ER, Abel T, Lattal KM. Transgenic inhibition of neuronal protein kinase A activity facilitates fear extinction. J Neurosci. 2006;26:12700–12707. doi: 10.1523/JNEUROSCI.2743-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Squire LR. Neuroscience: breaking down scientific barriers to the study of brain and mind. Science. 2000;290:1113–1120. doi: 10.1126/science.290.5494.1113. [DOI] [PubMed] [Google Scholar]
- Kaplan MP, Abel T. Genetic approaches to the study of synaptic plasticity and memory storage. CNS Spectr. 2003;8:597–610. doi: 10.1017/s1092852900018873. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc Natl Acad Sci U S A. 2004;101:8192–8197. doi: 10.1073/pnas.0402650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP. Time-dependent relationship between the dorsal hippocampus and the prefrontal cortex in spatial memory. J Neurosci. 2003;23:1517–1523. doi: 10.1523/JNEUROSCI.23-04-01517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malleret G, Hen R, Guillou JL, Segu L, Buhot MC. 5-HT1B receptor knock-out mice exhibit increased exploratory activity and enhanced spatial memory performance in the Morris water maze. J Neurosci. 1999;19:6157–6168. doi: 10.1523/JNEUROSCI.19-14-06157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Braunewell KH. Novelty acquisition is associated with induction of hippocampal long-term depression. Proc Natl Acad Sci U S A. 1999;96:8739–8744. doi: 10.1073/pnas.96.15.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marighetto A, Micheau J, Jaffard R. Relationships between testing-induced alterations of hippocampal cholinergic activity and memory performance on two spatial tasks in mice. Behav Brain Res. 1993;56:133–144. doi: 10.1016/0166-4328(93)90031-k. [DOI] [PubMed] [Google Scholar]
- Markowitch S, Scott PD. The role of forgetting in learning. Proceedings of the fifth international conference on machine learning; Ann Arbor, MI: Morgan Kaufmann; 1988. [Google Scholar]
- Martel G, Millard A, Jaffard R, Guillou JL. Stimulation of hippocampal adenylyl cyclase activity dissociates memory consolidation processes for response and place learning. Learn Mem. 2006;13:342–348. doi: 10.1101/lm.149506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Morris RG. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–636. doi: 10.1002/hipo.10107. [DOI] [PubMed] [Google Scholar]
- Moody TD, Thomas MJ, Makhinson M, O'Dell TJ. 5-Hz stimulation of CA3 pyramidal cell axons induces a beta-adrenergic modulated potentiation at synapses on CA1, but not CA3, pyramidal cells. Brain Res. 1998;794:75–79. doi: 10.1016/s0006-8993(98)00217-0. [DOI] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nakao K, Ikegaya Y, Yamada MK, Nishiyama N, Matsuki N. Hippocampal long-term depression as an index of spatial working memory. Eur J Neurosci. 2002;16:970–974. doi: 10.1046/j.1460-9568.2002.02159.x. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, Kandel ER. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron. 2008;58:104–117. doi: 10.1016/j.neuron.2008.01.039. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed. 2. San Diego: Academic; 2001. [Google Scholar]
- Rao Y, Fischer QS, Yang Y, McKnight GS, LaRue A, Daw NW. Reduced ocular dominance plasticity and long-term potentiation in the developing visual cortex of protein kinase A RII alpha mutant mice. Eur J Neurosci. 2004;20:837–842. doi: 10.1111/j.1460-9568.2004.03499.x. [DOI] [PubMed] [Google Scholar]
- Santschi LA, Zhang XL, Stanton PK. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. J Neurobiol. 2006;66:205–219. doi: 10.1002/neu.20213. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Malleret G, Vronskaya S, Mendez I, Garcia AD, Sofroniew MV, Kandel ER, Hen R. Paradoxical influence of hippocampal neurogenesis on working memory. Proc Natl Acad Sci U S A. 2007;104:4642–4646. doi: 10.1073/pnas.0611718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML. cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci. 2006;26:2391–2402. doi: 10.1523/JNEUROSCI.3092-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Lynch G. Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res. 1990;513:113–118. doi: 10.1016/0006-8993(90)91096-y. [DOI] [PubMed] [Google Scholar]
- Staubli U, Otaky N. Serotonin controls the magnitude of LTP induced by theta bursts via an action on NMDA-receptor-mediated responses. Brain Res. 1994;643:10–16. doi: 10.1016/0006-8993(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Birnbaum S, Ubriani R, Arnsten AF. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci. 1999;19:RC23. doi: 10.1523/JNEUROSCI.19-18-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Villarreal DM, Do V, Haddad E, Derrick BE. NMDA receptor antagonists sustain LTP and spatial memory: active processes mediate LTP decay. Nat Neurosci. 2002;5:48–52. doi: 10.1038/nn776. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE. Homosynaptic LTD and depotentiation: do they differ in name only? Hippocampus. 1996;6:24–29. doi: 10.1002/(SICI)1098-1063(1996)6:1<24::AID-HIPO5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Wixted JT. The psychology and neuroscience of forgetting. Annu Rev Psychol. 2004;55:235–269. doi: 10.1146/annurev.psych.55.090902.141555. [DOI] [PubMed] [Google Scholar]
- Wolansky T, Clement EA, Peters SR, Palczak MA, Dickson CT. Hippocampal slow oscillation: a novel EEG state and its coordination with ongoing neocortical activity. J Neurosci. 2006;26:6213–6229. doi: 10.1523/JNEUROSCI.5594-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Woo NH, Duffy SN, Abel T, Nguyen PV. Genetic and pharmacological demonstration of differential recruitment of cAMP-dependent protein kinases by synaptic activity. J Neurophysiol. 2000;84:2739–2745. doi: 10.1152/jn.2000.84.6.2739. [DOI] [PubMed] [Google Scholar]
- Xu Z, Chen RQ, Gu QH, Yan JZ, Wang SH, Liu SY, Lu W. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J Neurosci. 2009;29:8764–8773. doi: 10.1523/JNEUROSCI.1014-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]