

Abstract
Calmodulin regulates multifarious cellular processes via a panoply of target interactions. However, the central role, multiple isoforms, and complex target interactions of calmodulin make it difficult to examine its precise functions. Here, we analyzed calmodulin function in neurons using lentivirally delivered short-hairpin RNAs that suppressed expression of all calmodulin isoforms by ∼70%. Calmodulin knockdown did not significantly alter neuronal survival or synapse formation but depressed spontaneous neuronal network activity. Strikingly, calmodulin knockdown decreased the presynaptic release probability almost twofold, without altering the presynaptic readily-releasable vesicle pool or postsynaptic neurotransmitter reception. In calmodulin knockdown neurons, presynaptic release was restored to wild-type levels by expression of constitutively active calmodulin-dependent kinase-IIα (CaMKIIα); in contrast, in control neurons, expression of constitutively active CaMKIIα had no effect on presynaptic release. Viewed together, these data suggest that calmodulin performs a major function in boosting synaptic strength via direct activation of presynaptic calmodulin-dependent kinase II.
Introduction
Ca2+ ions are central signaling molecules in neurons, regulating diverse processes such as neurotransmitter release, gene expression, and long-term plasticity (Katz and Miledi, 1965; Berridge et al., 2000; Greer and Greenberg, 2008). Many cytoplasmic Ca2+-binding proteins mediate the intracellular actions of Ca2+ (Südhof and Rizo, 1996; Haeseleer et al., 2002; Burgoyne et al., 2004; Mikhaylova et al., 2006). Among these Ca2+-binding proteins, calmodulin (CaM) is likely the most important but also the most enigmatic (Chin and Means, 2000).
CaM is an abundant EF-hand Ca2+-binding protein composed of two lobes, each of which binds two Ca2+ ions with high affinity (0.5–5 μm, depending on the assay and conditions) (Gifford et al., 2007). CaM is highly conserved in all eukaryotes (Kawasaki et al., 1998), is essential for survival in all eukaryotes including yeast (Geiser et al., 1991), and acts by Ca2+-dependent and Ca2+-independent binding to innumerable target proteins (Chin and Means, 2000). Several presynaptic and postsynaptic functions of CaM were identified. Presynaptically, CaM may act as a Ca2+ sensor for release (DeLorenzo, 1981) or modulate release (Chamberlain et al., 1995; Chen et al., 1999; Junge et al., 2004). The majority of neurotransmitter release is triggered by Ca2+ binding to synaptotagmin (Fernández-Chacón et al., 2001; Maximov and Südhof, 2005), but CaM may function as an ancillary Ca2+ sensor for the asynchronous component of release, which is not mediated by synaptotagmin (Sun et al., 2007). Moreover, CaM may regulate the refilling of the readily-releasable pool (RRP) of vesicles (Sakaba and Neher, 2001) and/or modulate presynaptic release by activating calmodulin-dependent kinase-IIα (CaMKIIα) and CaMKIIβ (Llinás et al., 1985; Thiagarajan et al., 2002; Hinds et al., 2003; Dick et al., 2008). Finally, CaM controls presynaptic and postsynaptic Ca2+-channel function (Lee et al., 2000; Dick et al., 2008), mediates postsynaptic long-term plasticity by activating CaMKIIα and calcineurin (Lisman et al., 2002; Xia and Storm, 2005), and alters neuronal gene expression (Greer and Greenberg, 2008).
However, studies of the physiological function of CaM are difficult, and little is known about the physiological importance of many of its actions. CaM is abundant in brain, rendering overexpression studies uninformative. The very essentiality of CaM means that a conditional approach is needed to test its function, but no specific and effective pharmacological agents are available that act on CaM. Vertebrates contain three dispersed CaM genes that encode 100% identical proteins, in addition to more distantly related genes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), hindering genetic approaches. As a result, although innumerable brain functions have been ascribed to CaM, few direct tests of these functions were performed.
Here, we designed an RNA interference (RNAi) approach to examine neuronal CaM function. We show that knockdown (KD) of CaM does not significantly impair neuronal viability or synapse formation but reduces Ca2+-triggered release in excitatory and inhibitory synapses. The reduction in release by CaM KD can be selectively rescued by expression of constitutively active CaMKIIα, uncovering a function for CaM in maintaining presynaptic strength via activation of CaMKII.
Materials and Methods
Neuronal culture.
Primary cortical neurons were isolated from newborn pups of wild-type (WT) mice as described previously (Maximov et al., 2007a; Xu et al., 2007, 2009), with modifications. Briefly, cortices were dissociated by papain digestion (10 U/ml, with 1 μm Ca2+ and 0.5 μm EDTA) and plated on Matrigel-coated circular glass coverslips (11 mm diameter). The neurons were cultured for 14–16 d in MEM (Invitrogen) supplemented with B27 (Invitrogen), glucose, transferrin, fetal bovine serum, and AraC (Sigma).
Vector constructions.
Lentiviral vector (L309) that includes two RNA-polymerase III promoters (human H1 and human U6) was constructed based on the standard lentiviral backbone vector described by Maximov et al. (2009). The H1 and U6 promoters were cloned in tandem into the PacI site upstream of the ubiquitin C promoter (Fig. 1) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and endowed with multiple cloning sites downstream (H1 promoter: XhoI–XbaI–HpaI; U6 promoter: AscI–ClaI– RsrII–PacI). An internal ribosomal entry site (IRES)–enhanced green fluorescent protein (EGFP) or IRES–mCherry sequence was inserted downstream of the ubiquitin C promoter, leaving BamHI–EcoRI sites for inserting of rescue cDNAs upstream of the IRES sequence. The first short-hairpin sequence targeting a common sequence found in the CaM1 and CaM2 mRNAs [CTGACTGAAGAGCAGATTGC; full short-hairpin RNA (shRNA) sequence, TCGACCCCTGACTGAAGAGCAGATTGCTTCAAGAGAGCAATCTGCTCTTCAGTCAGTTTTTGGAAAT] was inserted into the XhoI–XbaI sites downstream of the H1 promoter. A second short-hairpin sequence targeting the CaM3 mRNA (sequence, CGCGCCCACGGAGCTGCAGGACATGATTATTCAAGAGATAATCATGTCCTGCAGCTCCGTTTTTTGGAAA) was inserted into the AscI–RsrII sites downstream of the U6 promoter. The targeted sequences in the rescue CaM cDNA were mutated to TTAACGGAAGAACAAATCGC and CAGAACTTCAAGATATGATCA to create a maximum difference between the shRNAs and rescue cDNA without changing the CaM protein sequence. For the transfection experiments in Figure 8, plasmids encoding the shRNAs or shRNA-resistant WT CaM were constructed as described above.
Figure 1.

shRNA-dependent knockdown of CaM. A, Diagram of lentiviral vectors harboring the human H1 and U6 RNA-polymerase III promoters, which drive expression of shRNAs for KD of CaM1 and CaM2 (H1) and CaM3 (U6). The vector also contains a RNA-polymerase II ubiquitin promoter (Ub) that drives expression of EGFP (to visualize infected neurons, via an IRES sequence) without or with rescue cDNAs (e.g., CaM1 without or with Ca2+-binding mutations, in an shRNA-resistant form). For single polymerase III promoter vectors, see supplemental Figure 2 (available at www.jneurosci.org as supplemental material). B, Analysis of CaM KD efficiency using qRT-PCR of mRNAs encoding CaM1, CaM2, and CaM3. Cultured cortical neurons were infected with control or CaM KD vectors on DIV5 and analyzed on DIV14 (means ± SEMs; numbers in bars indicate number of experiments; statistical significance was calculated by Student's t test, ***p < 0.001). C, Analysis of CaM KD efficiency using immunoblotting. Synt1, Syntaxin-1; Syt1, synaptotagmin-1; VCP, vasolin-containing protein.
Figure 8.
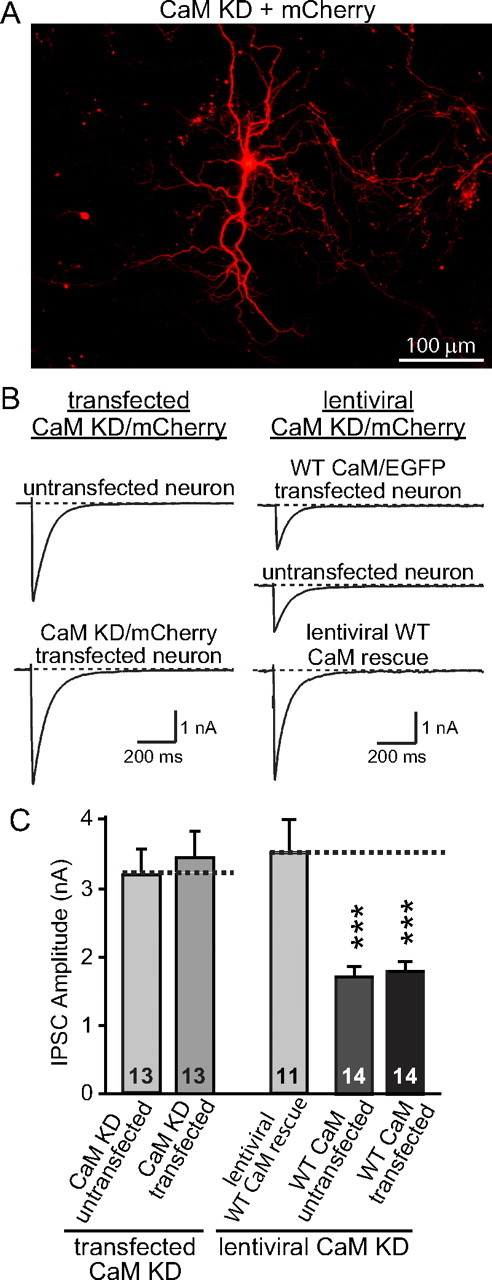
No postsynaptic contribution observed after CaM KD in reduction of synaptic strength. A, Image of sparsely transfected cultured cortical neurons expressing CaM shRNAs and mCherry. B, Representative traces of evoked IPSCs from cultured cortical neurons testing the effects of a postsynaptic CaM KD. Left traces depict experiments in which neurons were transfected with CaM shRNAs to produce a selective postsynaptic CaM KD in a few neurons in a culture dish, using mCherry as a marker for the transfected neurons. Right traces depict experiments in which all neurons were subjected to lentiviral CaM KD without or with lentivirally mediated WT CaM rescue, using mCherry as a marker for the infected neurons. Subsequently, the lentivirally mediated CaM KD neurons were transfected with shRNA-resistant WT CaM, such that only a few neurons were transfected that were identified by coexpressed EGFP. For all experiments, lentiviral infections were performed at DIV5, transfections at DIV9, and electrophysiological analyses at DIV14. Note that the two experimental designs are complementary, because the design depicted in the left traces measures the effect of a selective postsynaptic CaM KD, whereas the design depicted in the right traces measures the effect of a postsynaptic CaM rescue in CaM KD neurons. C, Summary graphs of evoked IPSC amplitudes in neurons that were manipulated as described in B. Data show means ± SEMs from three independent experiments; numbers in bars indicate number of cells analyzed. Statistical significance was calculated by Student's t test, ***p < 0.001.
Lentivirus production and infection of neuronal culture.
Lentiviruses were produced essentially as described previously (Dull et al., 1998; Logan et al., 2002; Maximov et al., 2009). The lentiviral expression vector (control vector L309 or the shRNA-expressing vectors) and three helper plasmids (pRSV–REV, pMDLg/pRRE, and vesicular stomatitis virus G protein-expressing plasmid) were cotransfected with the lentiviral vectors into human embryonic kidney 293T (HEK293T) cells (American Type Culture Collection) at 4, 2, 2, and 2 μg of DNA per 25 cm2 culture area, respectively, using FUGENE 6 transfection reagent (Roche) following the instructions of the manufacturer. At 48 h after transfection, the HEK293 cell culture medium was collected and clarified by centrifugation (500 × g for 5 min), and the supernatant containing lentiviral particles was added directly to the medium of cultured cortical neurons maintained in 24-well plates (300 μl of supernatant per well). Cortical neuronal culture was infected on 5 d in vitro (DIV5) and used for biochemical or physiological analysis on DIV14–DIV16. In all experiments, the infection rate of neurons was monitored (determined by the GFP signal driven by the ubiquitin promoter in the lentiviral vectors), and only cultures in which all neurons seemed to be infected were used for analysis. All steps were performed under level II biosafety conditions.
Quantification of mRNA levels by quantitative reverse transcription-PCR.
Cultured cortical neurons infected with lentiviruses as described above were lysed, and total RNA was extracted and purified with the RNAqueous-Micro kit (Ambion) (Cheng et al., 2007) following the instructions of the manufacturer. mRNA levels of individual genes were then analyzed by one-step quantitative reverse transcription (qRT)-PCR using TaqMan gene expression assays (Applied Biosystems). Briefly, 30 ng of RNA sample in 1 μl volume was mixed with 10 μl of TaqMan fast universal PCR master mix (2×), 0.1 μl of reverse transcriptase (50 U/μl), 0.4 μl of RNase inhibitor (20 U/μl), 7.5 μl of H2O, and 7 μl of TaqMan gene expression assay mix (including the forward and reverse primers and the TaqMan FAM–MGB probe). The reaction mixture was loaded onto an ABI7900 fast RT-PCR machine for 30 min of reverse transcription at 48°C, followed by 40 PCR amplification cycles consisting of denaturation at 95°C for 1 s, annealing and extension at 60°C for 20 s. The amplification curve was collected and analyzed with ΔΔCt methods for relative quantification of mRNAs. The mRNA levels of target genes, normalized to those of an endogenous control and relative to a calibrator sample, is calculated by 2−ΔΔCt, where ΔΔCt = ΔCtsample − ΔCtreference. The Ct value is the cycle number required for reaching the signal threshold on the quantitative RT-PCR machine, the ΔCtsample is the Ct value for any sample normalized to the endogenous housekeeping gene, and ΔCtreference is the Ct value for the calibrator also normalized to the endogenous housekeeping gene. In the current study, glyceraldehyde 3-phosphate dehydrogenase was used as the endogenous control, and RNA samples derived from neurons infected with control vector (L309) were used as the calibrator. The Taqman assay kits (Applied Biosystems) used in current study include the following: Mm00486655_m1 (for CaM1), Mm00849529_g1 (for CaM2), and Mm00482929_m1 (for CaM3).
Immunofluorescence and synapses quantitation.
Cultured cortical neurons infected with control or CaM knockdown lentivirus were fixed in cold 100% methanol, permeabilized in 0.1% saponin/PBS, and sequentially incubated with primary and secondary antibodies in PBS containing 1% goat serum and 2% bovine serum albumin. The following antibodies were used for immunocytochemistry: polyclonal rabbit anti-synapsin (E028); monoclonal mouse anti-microtubule-associated protein 2 (MAP2) (Sigma); guinea pig anti-vesicular glutamate transporter 1 (VGLUT1) (Millipore Corporation); and polyclonal rabbit anti-vesicular GABA transporter [the vesicular inhibitory amino acid transporter VIAAT and the vesicular GABA transporter VGAT (Synaptic Systems)]. Secondary antibodies (Alexa 546- or 633-labeled highly cross-adsorbed goat anti-mouse, goat anti-rabbit, or goat anti-guinea pig antisera) were from Invitrogen. Images were recorded with a Carl Zeiss LSM510 or Leica TCS2 confocal microscope. The same confocal acquisition settings were used for all samples of an experiment. NIH ImageJ 1.41 was used for the quantitations of synapse densities.
Electrophysiology.
Electrophysiology measurements were performed at DIV14–DIV16 in cultured cortical neurons as described previously (Maximov et al., 2007a; Xu et al., 2007, 2009). Evoked synaptic responses were triggered by 90 μA/1 ms current injections via a bipolar electrode (CBAEC75 Concentric Bipolar Electrode; Outer Pole, 125 μm stainless steel; Inner Pole, 25 μm platinum/iridium; FHC) placed at a distance of 100–150 μm from the soma of recorded neurons. Patch pipettes were pulled from borosilicate glass capillary tubes (catalog #64-0793; Warner Instruments) using a PC-10 pipette puller (Narishige). The resistance of pipettes filled with intracellular solution varied between 4 and 5 MΩ. After establishment of the whole-cell configuration and equilibration of the intracellular pipette solution with the cytoplasm, the series resistance was adjusted to 8–10 MΩ. Synaptic currents were monitored with a Multiclamp 700B amplifier (Molecular Devices). The frequency, duration, and magnitude of the extracellular stimulus were controlled with a model 2100 Isolated Pulse Stimulator (A-M Systems) synchronized with Clampex 9 data acquisition software (Molecular Devices). The whole-cell pipette solution contained the following (in mm): 135 CsCl, 10 HEPES, 1 EGTA, 1 Na-GTP, 4 Mg-ATP, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], pH 7.4, adjusted with CsOH. The bath solution contained the following (in mm): 140 NaCl, 5 KCl, 0.8 MgCl2, 10 HEPES-NaOH, pH 7.4, and 10 glucose. IPSCs and EPSCs were pharmacologically isolated by adding the AMPA and NMDA receptor blockers CNQX (20 μm) and d-AP-5 (50 μm), respectively, or the GABAA receptor blocker picrotoxin (50 μm) to the extracellular bath solution. MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] experiments were performed with 15 μm. Spontaneous miniature IPSCs (mIPSCs and mEPSCs) were monitored in the presence of tetrodotoxin (1 μm) to block the action potentials.
Data analysis.
Synaptic currents were sampled at 10 kHz and analyzed offline using Clampfit 9 (Molecular Devices) software. For graphic representation, the stimulus artifacts of the current traces were removed. For measurements of the frequency of spontaneous release and the amplitudes of synchronous IPSCs during stimulus trains, individual mIPSCs or IPSCs were collected using the pClamp template search function. Excitatory network activities were analyzed using threshold event search functions with a minimal duration of 200 ms.
Miscellaneous procedures.
For immunoblotting analyses from neuronal cultures, cultures were washed twice with PBS at DIV14–DIV15. Cell lysates were directly collected in SDS-gel sample buffers (50 μl of sample buffer per well for 24-well plates). Except when otherwise stated, equivalent protein amounts (25 μl) were analyzed by SDS-PAGE and immunoblotting using antibodies as follows: CaM monoclonal antibody (catalog #05-173; Millipore Corporation); synaptotagmin-1 (Cl41.1); syntaxin-1 (U6251); Rab3A (42.2); GDP-dissociation inhibitor (81.2); CaMKIIα (6G9; Sigma); and vasolin-containing protein (K330; used as an internal control).
Statistical analysis.
Data are presented as means ± SEMs. All statistical comparisons except otherwise stated were made using Student's t test as described in supplemental Table S2 (available at www.jneurosci.org as supplemental material). Kolmogorov–Smirnov test were used to compare the cumulative distributions of mIPSC and mEPSC amplitudes.
Results
An RNAi approach to CaM function
Vertebrates contain three dispersed CaM genes that encode the CaM1, CaM2, and CaM3 proteins with 100% identical protein sequences (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These are the only such CaM genes in the genome, although additional genes are present that encode more distantly CaM-related protein. The genetic complexity of CaM hinders genetic dissection of CaM function, prompting us to develop an RNAi-based method to suppress CaM expression using lentivirally synthesized shRNAs. We constructed a lentiviral RNAi vector (L309) in which two polymerase III promoters (the human H1 and U6 promoters) mediate expression of shRNAs (Fig. 1A). In the same vector, we included a ubiquitin promoter to express potential rescue cDNAs (to control for the specificity of shRNA-dependent KDs), followed by an IRES–EGFP sequence (to visualize viral infections) (Fig. 1A).
We designed multiple shRNAs to CaM and tested them with this lentiviral vector in cultured neurons, using mRNA and protein measurements of endogenous CaM as a readout for the effectiveness of the shRNAs (Fig. 1B,C). We identified one shRNA that targets a sequence common to the CaM1 and CaM2 mRNAs and that reliably suppressed expression of both CaM1 and CaM2 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We also identified a second shRNA that, when expressed in the same vector as the CaM1/CaM2 shRNA, knocked down expression of CaM3. Thus, using this construct, we can suppress expression of all endogenous CaM in neurons by ∼70%, as measured by qRT-PCR using Taqman assays for CaM mRNAs and by immunoblotting for CaM (Fig. 1B,C).
Normal spontaneous synaptic activity but impaired spontaneous network activity in CaM KD neurons
Given the many known activities of CaM, we asked whether the CaM KD alters neuronal differentiation and/or synaptogenesis. However, immunocytochemistry with antibodies to a dendritic marker MAP2 and synaptic marker synapsin uncovered no major changes in neuronal morphology and synapse density, suggesting that the CaM KD did not impair neuronal differentiation or synapse formation (Fig. 2A). We then measured the density of excitatory and inhibitory synapses to test whether the CaM KD suppressed one type of synapse but enhanced the other. Excitatory synapse density was monitored by immunohistochemistry with antibodies to VGLUT1, the vesicular glutamate transporter, and inhibitory synapse density with antibodies to VGAT, the vesicular GABA transporter. However, we observed no change in the density of excitatory or inhibitory synapses (Fig. 2B).
Figure 2.

CaM KD does not alter synapse density in cultured neurons. A, Representative images of neurons infected with control or CaM KD lentiviruses and immunolabeled with antibodies to MAP2 and synapsin (Syn). Synapse density was quantified by counting the number of synapsin-stained puncta per dendrite area identified by MAP2 staining using NIH ImageJ. In control condition, the average density of synapses is 5.73 ± 0.27/μm2 dendritic area; in the CaM KD condition, the average synaptic density is 5.73 ± 0.22/μm2 (n = 51 and 52 in control and CaM KD, respectively). Three batches of cultures were analyzed. B, Representative images of excitatory and inhibitory synapses in cultured neurons infected with control lentivirus or the CaM KD lentivirus. Synapses were immunolabeled with antibodies to the vesicular glutamate transporter (specific for excitatory synapses; VGLUT1) and to the vesicular GABA transporter (specific for inhibitory synapses; VGAT). Insets show magnified images from the areas indicated by dashed squares. There is no difference in the number of either VGAT or VGLUT1 puncta between control and CaM KD (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The ratios of VGLUT1/VGAT puncta are 1.01 ± 0.06 and 0.99 ± 0.06 in control (n = 41) and CaM KD (n = 43). Four different batches of cultured neurons were analyzed.
In the next set of experiments, we tested whether the CaM KD altered the spontaneous synaptic activity of neurons in which action potentials were suppressed with tetrodotoxin. We detected no obvious changes in the frequency or amplitude of mEPSCs and mIPSCs, respectively (Fig. 3) The cumulative distribution of mEPSC and mIPSC amplitudes also uncovered no significant effect of the CaM KD, suggesting that neurotransmitter loading of synaptic vesicles and postsynaptic receptor function are not altered by the CaM KD. Moreover, there is no significant change in the kinetics of both mEPSC and mIPSCs (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Figure 3.

CaM KD does not alter spontaneous synaptic “mini” release in cultured neurons. A, Representative traces of mEPSCs in control and CaM KD synapses. B, Cumulative probability of the distribution of the mEPSC frequency. The average frequency of mEPSCs in control is 1.34 ± 0.20 Hz (n = 22) and in CaM KD is 1.26 ± 0.21 Hz (n = 28). C, Cumulative probability of the distribution of the mEPSC amplitudes. The average amplitudes of mEPSCs are 10.46 ± 0.49 (n = 22) and 11.22 ± 0.72 (n = 28) in control and CaM KD, respectively. D, Representative traces of mIPSCs in control and CaM KD synapses. E, Cumulative probability of the distribution of the mIPSC frequency. The average frequencies of mEPSCs in control and CaM KD are 2.58 ± 0.32 Hz (n = 24) and 2.26 ± 0.27 Hz (n = 34), respectively. F, Cumulative probability of the distribution of the mIPSC amplitudes. The average amplitudes of mIPSCs are 48.52 ± 2.82 (n = 24) and 44.13 ± 2.63 (n = 34) in control and CaM KD, respectively. Data presented are means ± SEMs; statistical significance was calculated by Student's t test and Kolmogorov–Smirnov test.
We next examined whether the CaM KD alters spontaneous network activity, a sensitive measure of synaptic function and neuronal excitability that is produced in cultured neurons when inhibitory synaptic transmission is attenuated by addition of the GABAA receptor blocker picrotoxin (Fig. 4A). In control cultures, we observed bursts of action potentials, reflecting synaptic events lasting ∼1 s, with two to three bursts per minute (Fig. 4B). Knockdown of CaM did not change the frequency of synaptic bursts but slightly increased the duration. CaM KD decreased the strength of synaptic events more than twofold, as evidenced by a decrease of the transfer rate of the synaptic charge during the bursts (Fig. 4B). Thus, the CaM KD did not significantly impair synapse formation or spontaneous synaptic activity and did not inactivate signaling or block synaptic transmission in neurons but did alter their synaptic network properties. Moreover, there are no significant changes in membrane input resistance and membrane capacitance after CaM KD (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Figure 4.

CaM KD suppresses network activity in cultured neurons. A, Representative electrophysiological traces of excitatory network activity in high-density cultures of cortical neurons, infected with control lentivirus or lentivirus expressing the CaM shRNAs without (CaM KD) or with the wild-type CaM proteins (CaM KD + WT CaM). B, Quantitation of four parameters of the excitatory network activity as a function of the CaM KD: burst frequency (far left), burst duration (middle left), synaptic charge transfer during bursts (middle right), and charge transfer rate (far right). Data are means ± SEMs; numbers in bars indicate number of cells analyzed in at least three independent experiments. Statistical significance was calculated by Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001.
CaM KD reduces evoked excitatory synaptic strength
The reduction in the excitatory network activity in CaM KD neurons suggests that synaptic transmission is altered (Fig. 4). To probe whether evoked synaptic transmission was impaired by the CaM KD, we measured the amplitude of EPSCs evoked by isolated action potentials. We first monitored evoked AMPA-receptor-mediated EPSCs in the presence of picrotoxin and d-APV, using a concentric bipolar electrode for stimulation (Maximov et al., 2007a). We observed a large reduction in the size of the synaptic responses (∼45%) (Fig. 5A). This reduction of evoked EPSCs was rescued by coexpression of wild-type CaM, suggesting that it is specific (Fig. 5A).
Figure 5.

CaM KD decreases the strength and release probability of excitatory synapses. A, Representative traces (left) and summary graphs of the amplitudes (right) of evoked AMPA-receptor-mediated EPSCs in neurons infected with lentiviruses that express only EGFP (Control), coexpress EGFP with the CaM KD shRNAs (CaM KD), or with CaM shRNAs and wild-type CaM (CaM KD + WT CaM). B, Same as A but for NMDA-receptor-mediated evoked responses. C, Summary graph of relative NMDA-receptor-mediated EPSC amplitudes as a function of stimulus number in the presence of MK-801; representative traces are shown in the inset. Measurements of the presynaptic release probability using progressive blockade of NMDA-receptor-mediated EPSCs with the irreversible open-channel inhibitor MK-801. The decay constant, express as the Tau value of the decay in stimulus numbers, is inversely proportional to release probability (Hessler et al., 1993; Rosenmund et al., 1993). Data are means ± SEMs; numbers in bars indicate number of cells analyzed in at least three independent experiments. Statistical significance was calculated by Student's t test, ***p < 0.001.
In a second set of experiments, we monitored evoked NMDA-receptor-mediated EPSCs, recorded in the presence of picrotoxin and CNQX at a holding potential of +40 mV (Fig. 5B). Again, we observed a ∼45% reduction. For both AMPA- and NMDA-receptor-mediated EPSCs, we detected no apparent change in kinetics.
The similar reduction in AMPA- and NMDA-receptor-mediated EPSCs without a change in mEPSC amplitude or frequency and without a change in synapse density suggest that the CaM KD decreases synaptic strength by depressing neurotransmitter release. To confirm this hypothesis, we monitored the effect of the CaM KD on presynaptic release probability, using as an assay of the rate of blockade of NMDA receptor-mediated responses in the presence of the open-channel NMDA-receptor inhibitor MK-801 (Hessler et al., 1993; Rosenmund et al., 1993) (Fig. 5C). Because MK-801 is an irreversible open-channel blocker of NMDA receptors, all NMDA-receptor-dependent responses in an excitatory synapse become inactivated by MK-801 whenever a synapse releases glutamate. As a result, the rate of blockade of synaptic NMDA-receptor-mediated responses in the presence of MK-801 during low-frequency stimulation is directly proportional to the distribution of release probabilities of the synapses on the neuron that is being analyzed. We found that the CaM KD dramatically decreased the rate at which NMDA receptors were blocked by MK-801, as evidenced by the 60% increase in the time constant of decay (Fig. 5C). This result, in conjunction with the lack of change in kinetics of NMDA-receptor-mediated responses, demonstrates that the CaM KD decreases the presynaptic release probability.
CaM KD reduces evoked release from inhibitory synapses
To explore whether inhibitory synapses are also affected by KD of CaM, we monitored evoked IPSCs. Similar to the changes in EPSCs, the KD of CaM caused a large decrease in IPSC amplitudes (∼50%;) (Fig. 6A). Again, the decrease in inhibitory synaptic strength was rescued by wild-type CaM. Moreover, we compared the kinetics of evoked IPSCs between CaM KD and control neurons and found no major change (Fig. 6B). Thus, KD of CaM causes a uniform decrease in synaptic strength in excitatory and inhibitory synapses, presumably, based on the MK-801 measurements, because of a decrease in release probability.
Figure 6.

Inhibitory synaptic strength is reduced in CaM KD synapses. A, Representative traces (left), and summary graphs of the amplitudes (middle), and charge transfer (right) of evoked IPSCs in neurons infected with lentiviruses that express only EGFP (Control), coexpress EGFP with the CaM KD shRNAs (CaM KD), or with CaM shRNAs and wild-type CaM (CaM KD + WT CaM). B, Kinetics of the IPSCs as a function of CaM KD, calculated as the cumulative normalized charge transfer (Maximov and Südhof, 2005). C, Representative traces (left) and summary graphs of the charge transfer (right) of IPSCs induced by hypertonic sucrose (0.5 m for 30 s) in neurons infected with lentiviruses that express only EGFP (Control), coexpress EGFP with the CaM KD shRNAs (CaM KD), or with CaM shRNAs and wild-type CaM (CaM KD + WT rescue). Data are means ± SEMs; numbers in bars indicate number of cells analyzed in at least three independent experiments. Statistical significance was calculated by Student's t test, ***p < 0.001.
One cause of a decrease in release probability could be a decrease in the size of the RRP of synaptic vesicles, suggested by previous studies implicating CaM in the replenishment of the RRP (Sakaba and Neher, 2001). To test this hypothesis, we applied hypertonic sucrose [which elicits exocytosis to the entire RRP in a synapse (Rosenmund and Stevens, 1996)] to cultured neurons and measured synaptic responses but detected no obvious change in RRP size as a function of the CaM KD (Fig. 6C).
Short-term synaptic plasticity
In excitatory synapses of CaM KD neurons, the decrease in EPSC amplitudes is caused by a decrease in release probability. To test whether the same applies to inhibitory synapses and whether this decrease is uniformly present independent of the number of action potentials applied, we explored the effect of the CaM KD on synaptic transmission induced by action potential trains at high frequency (10 Hz). This experiment allowed us to confirm that the release probability is decreased (which would lead to decreased depression/increased facilitation of synaptic responses during high-frequency stimulus trains), as indicated by the MK-801 experiments, and whether the dynamics of release change during the train as a function of the CaM KD.
Consistent with a reduction in release probability, we observed a large decrease in the initial IPSC amplitudes during the stimulus train in CaM KD neurons and an equally large alleviation of synaptic depression (Fig. 7A). The decrease in the IPSCs translated into a decrease of synaptic strength throughout the stimulus train; even delayed release, a measure of asynchronous neurotransmitter release (Maximov and Südhof, 2005), was decreased correspondingly. However, we uncovered no significant difference between the various phases of release during the stimulus train after the CaM KD. This result, consistent with the lack of a change in IPSC kinetics (Fig. 6), indicates that the CaM KD equally decreases synchronous and asynchronous release. Quantitation of the relative synaptic responses during prolonged stimulus trains uncovered a continuing effect of the CaM KD on use-dependent depression throughout the stimulus train (Fig. 7C).
Figure 7.

Effect of CaM KD on synaptic transmission during 10 Hz stimulus trains. A, Representative traces of synaptic responses induced by a 10 Hz, 1 s stimulus train in neurons infected with lentiviruses that express only EGFP (Control), coexpress EGFP with the CaM KD shRNAs (CaM KD), or with CaM shRNAs and wild-type CaM (CaM KD + WT CaM). B, Quantitation of the effect of CaM KDs on the synaptic charge transfer during the initial response of the 10 Hz stimulus train (left), total train (middle), and delayed release (right). C, Representative traces of IPSCs during 10 s, 10 Hz stimulus trains (left) and normalized responses to quantify the degree of depression (right). Note that only the synchronous component of the responses is included in the normalization, which increases the apparent degree of depression. Data are means ± SEMs; numbers in bars in B indicate number of cells analyzed in at least three independent experiments. Statistical significance was calculated by Student's t test, ***p < 0.001. Numbers of recordings presented in C are 27 and 30 for control and CaM KD, respectively. Curves in C are significantly different (p < 0.0001) using two-way ANOVA.
Postsynaptic CaM KD has no effect on the presynaptic inputs onto a given neuron
At least three mechanisms appear possible to account for the general effect of the CaM KD on synaptic transmission. (1) CaM may be required for the overall growth of neurons; smaller neurons would have fewer synapses even if the synapse density per dendrite length is unaltered. (2) CaM may be directly required in the presynaptic neuron to maintain the normal efficacy of neurotransmitter release. (3) CaM may be required for the postsynaptic synthesis of a retrogradely synthesized signal that then maintains a normal level of presynaptic function.
The first mechanisms is effectively ruled out by the observation that the capacitance and input resistance remain unchanged after CaM KD, suggesting that the overall size and properties of the neurons do not change (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). The second mechanism appears most likely given previous studies (Llinás et al., 1985; Sakaba and Neher, 2001; Thiagarajan et al., 2002; Hinds et al., 2003; Dick et al., 2008), but the third mechanism cannot be excluded with the data up to now. Thus, to test the third mechanism, we performed two complementary sets of experiments (Fig. 8).
First, we transfected isolated neurons in a dense culture of cortical neurons with a plasmid expressing the CaM KD shRNAs and then measured the size of evoked IPSCs in transfected and nontransfected neurons in the culture (Fig. 8A,B). We observed no decrease of the IPSC size as a function of the postsynaptic CaM KD (Fig. 8C).
Second, we infected all neurons in a cortical culture with the lentiviruses mediating the CaM KD, without or with encoding the WT CaM rescue. We then transfected the CaM KD neurons without lentiviral CaM rescue with a plasmid encoding WT shRNA-resistant CaM. Finally, we measured the size of IPSCs in the lentivirally rescued CaM KD neurons (as a control) and in the CaM KD neurons that were transfected with the CaM plasmid, measuring IPSCs from both transfected and nontransfected neurons (Fig. 8B). Quantitations revealed that the IPSC size in the lentivirally rescued CaM KD neurons was not significantly different from that of controls, whereas the nonrescued CaM KD neurons exhibited a greatly suppressed IPSC size independent of whether they expressed postsynaptic CaM or not (Fig. 8C). Thus, the postsynaptic CaM does not account for the presynaptic CaM KD phenotype.
CaMKIIα overexpression rescues synaptic strength in CaM KD synapses
We next examined whether lack of the CaM-dependent activation of two presynaptic CaM-binding proteins that were implicated in regulating synaptic strength may be responsible for the synaptic phenotype in CaM KD neurons: adenylate cyclase, which plays a central role in setting presynaptic and postsynaptic strength (Xia and Storm, 2005; Pelkey et al., 2008) and may be constitutively activated by CaM in cultured neurons (Maximov et al., 2007b), and CaMKIIα, which may also be constitutively activated by CaM in presynaptic terminals and regulates synaptic strength (Llinás et al., 1985; Chen et al., 1999; Hinds et al., 2003). Direct stimulation of adenylate cyclase in CaM KD neurons with forskolin did not reverse the decrease in synaptic strength induced by the CaM KD (data not shown). Strikingly, however, overexpression of wild-type CaMKIIα in CaM KD neurons partly rescued the synaptic phenotype induced by the CaM KD (Fig. 9A,C,D). Moreover, overexpression of the constitutively active T286D mutant of CaMKIIα (CaMKIIαT286D), in which CaM is no longer required for activation (Brickey et al., 1994), completely rescued the synaptic phenotype in CaM KD neurons (Fig. 9A,C,D).
Figure 9.
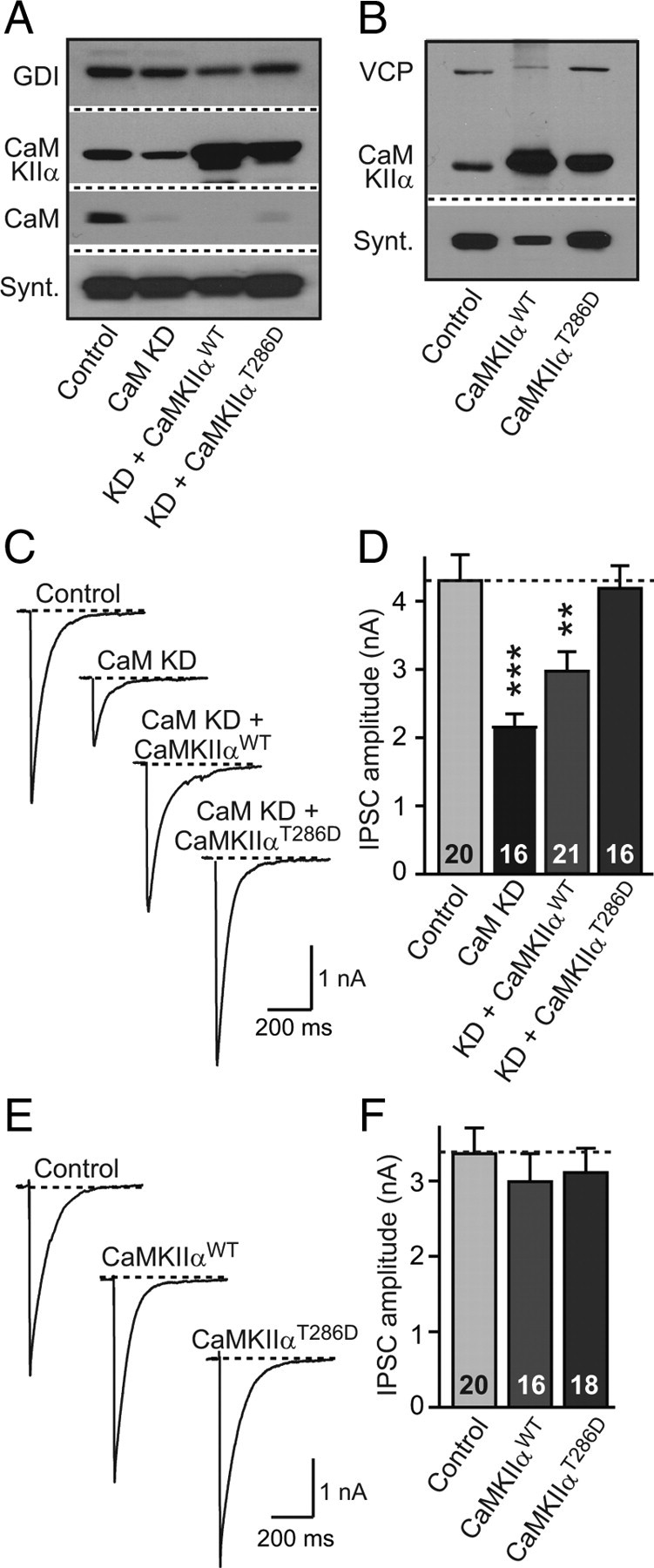
CaMKIIa overexpression rescues synaptic strength in CaM KD synapses. A, Neurons were infected with lentivirus expressing only EGFP (Control); coexpressing EGFP with CaM KD shRNAs (CaM KD); coexpressing EGFP, CaM KD shRNAs, and wild-type CaM Kinase IIα (KD + CaMKIIαWT), and coexpressing EGFP, CaM KD shRNAs, and the T286D constitutively active mutant of CaMKIIα (KD + CaMKIIαT286D). Representative immunoblots of CaMKIIα, CaM, syntaxin (Synt), and GDI (GDP-dissociation inhibitor) were used as loading control. B, Immunoblots of cultures (without CaM KD shRNAs) expressing EGFP (Control); coexpressing EGFP with CaMKIIαWT (CaMKIIαWT); and coexpressing of EGFP with CaMKIIαT286D (CaMKIIαT286D). VCP (vasolin-containing protein) was used as loading control. C, Representative traces of evoked IPSCs in CaM KD cultures with manipulations described in A. D, Summary graphs of the mean IPSC amplitudes in neurons that were manipulated as described in A. E, Representative traces of evoked IPSCs in control cultures with or without CaMKIIαWT or CaMKIIαT286D (i.e., the effect of CaMKIIα overexpression on neurons expressing normal CaM levels was investigated). F, Summary graphs of the mean IPSC amplitudes in neurons that were manipulated as described in E. Data are means ± SEM; numbers in bars indicate number of cells analyzed in at least three independent experiments; statistical significance was calculated by Student's t test. **p < 0.01, ***p < 0.001.
The rescue of the synaptic phenotype by CaMKIIα overexpression suggests that CaM normally boosts synaptic strength by stimulating presynaptic CaMKIIα. A possible concern with these experiments, however, is that the apparent rescue may be unrelated to the CaM KD, i.e., that the rescue simply reflects an independent, positive effect of CaMKIIα overexpression on synaptic strength. If this were the case, CaMKIIα overexpression in neurons with normal CaM levels under the same conditions used for the CaM KD experiments should result in an increase in synaptic strength. Thus, to address this concern, we measured the effect of CaMKIIα overexpression on synaptic responses in synapses containing normal CaM levels, using the identical vector used for the KD experiments (Fig. 9B,E,F). We found that CaMKIIα overexpression had no effect on synaptic responses in the presence of normal CaM concentrations, demonstrating that the rescue effects of CaMKIIα specifically operate in the absence of CaM, i.e., that this is the pathway that mediates the CaM-dependent regulation of synaptic strength.
Discussion
Using a newly developed, lentivirally mediated RNAi approach, we knocked down expression of CaM in cortical neurons and examined the effect of the CaM KD on synaptic transmission. The knockdown approach allowed us to examine the overall role of CaM in synaptic transmission, as opposed to the role of specific CaM-binding proteins independently studied in many previous papers (Llinás et al., 1985; Malinow et al., 1989; Mayford et al., 1995; Keen et al., 1999; Lee et al., 2000; DeMaria et al., 2001; Thiagarajan et al., 2002; Hinds et al., 2003; Junge et al., 2004; Dick et al., 2008). Examining the overall role of CaM in synaptic transmission would have been difficult with a pharmacological approach because no effective and specific drugs against CaM exist or with a conditional knockout approach because CaM in brain is expressed from three dispersed CaM genes that encode identical proteins and that would have to be deleted simultaneously by a conditional knockout. Future developments of drugs that allow a stronger inhibition of CaM or future generation of mutant mice that allow conditional deletions of all three CaM genes would improve the present analysis but could also engender new pitfalls attributable to the pervasive cellular functions of CaM in many different processes. Thus, short of new technical developments, we feel that the present approach is the best available to directly examine the overall functions of CaM at the synapse.
Our approach yielded two major results. First, a ∼70% loss of CaM protein (Fig. 1) causes an exclusively presynaptic impairment in basal synaptic transmission that manifests as a uniform reduction of the strength of excitatory and inhibitory synapses (Figs. 5–7). As a result, spontaneous network activity is impaired (Fig. 4), without a change in synapse density or spontaneous synaptic activity (Figs. 2, 3). The mechanism involved appears to operate at the level of Ca2+ triggering of release, as shown by the MK-801 studies and short-term synaptic plasticity experiments, which revealed a decrease in release probability (Figs. 5, 7). Moreover, the hypertonic sucrose stimulations suggested that the RRP is unchanged after CaM KD (Fig. 6). Please note that our results show that CaM does not act as an ancillary Ca2+ sensor for release because it cannot trigger release on its own in the absence of synaptotagmin-1, the Ca2+ sensor for release (Maximov and Südhof, 2005). Please also note that we ruled out a postsynaptic effect of the CaM KD (Fig. 8) or a nonspecific general effect on the neurons (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Thus, in cultured neurons, CaM constitutively boosts synaptic strength by enhancing Ca2+-triggered release mediated by synaptotagmin.
Although our observed phenotype is major, it is not as large as would have been expected based on the many known presynaptic and postsynaptic targets for CaM. This difference is most likely attributable, at least in part, to the partial nature of the KD of CaM we analyzed. Thus, our data reveal the functions of CaM that require its physiologically high concentrations but do not allow conclusions about potential essential functions of CaM that require lower CaM concentrations. The partial nature of the CaM KD using RNAi reported here actually may be somewhat advantageous because the universal cellular functions of CaM (Geiser et al., 1991; Chin and Means, 2000) suggest that a more complete KD may have led to cytotoxicity. One concern of our study may be that RNAi approaches are prone to artifacts, especially off-target effects (Alvarez et al., 2006). To control for such effects, we have confirmed the specificity of the salient effects of the CaM KD using rescue experiments. CaM is particularly enriched in brain, and our results thus suggest that one of the reasons for the very high concentrations of CaM in brain is to maintain synaptic transmission by boosting neurotransmitter release from presynaptic nerve terminals.
Our second major result is the finding that the decrease in release caused by the CaM KD is completely rescued by overexpression of mutant, CaM-independent CaMKIIαT286D (Fig. 9). This result suggests that lack of activation of a single target, CaMKII, is responsible for the decreased release induced by the CaM KD. Overexpression of wild-type CaMKIIα produced a partial rescue of the CaM KD phenotype, probably because, according to the mass action law, the residual CaM in the KD neurons is more effective in activating CaMKIIα when its concentrations are increased. We examined only CaMKIIα in the current study because it is the most abundant and best examined isoform of CaMKII (Braun and Schulman, 1995). However, our results likely apply to CaMKIIβ in addition to CaMKIIα because these two isoforms are highly homologous (∼76% sequence identity) and structurally and functionally similar (Hoelz et al., 2003) and because the CaM KD would have affected all CaMKII isoforms equally. CaMKIIα is primarily expressed in glutamatergic neurons (Liu and Jones, 1996; Sík et al., 1998; Thiagarajan et al., 2002), whereas CaMKIIβ is expressed in both excitatory and inhibitory neurons (Sík et al., 1998; Thiagarajan et al., 2002). Selective expression of CaMKIIα versus CaMKIIβ in subsets of neurons may confer subtle differences to the CaM-dependent regulation of neurotransmitter release (Thiagarajan et al., 2002).
Overexpressed CaMKIIαT286D did not boost synaptic strength by a mechanism that operates separately from, and independently of, CaM because, in control synapses with normal CaM concentrations, CaMKIIα T286D did not increase synaptic strength (Fig. 9). Our finding that CaMKII enhances the presynaptic release probability is in line with a large body of literature investigating the synaptic functions of CaMKII (Lin et al., 1990; Chapman et al., 1995; Ohyama et al., 2002; Ninan and Arancio, 2004; Jiang et al., 2008; Wang, 2008). CaMKIIα likely boosts presynaptic release probability by phosphorylating one or multiple targets, possibly synapsin-1a and rabphilin, which are CaMKII substrates that have been implicated previously in neurotransmitter release (Benfenati et al., 1992; Li et al., 1994). However, knock-outs of both of these molecules have only very small effects on release, if any (Rosahl et al., 1993; Schlüter et al., 1999), indicating that they are unlikely to be solely responsible and that other presynaptic targets for CaMKIIα may also be involved. CaMKII was also reported to bind to syntaxin-1A (Ohyama et al., 2002) or CaV2.1 channels (Jiang et al., 2008) to exert presynaptic regulation. As one of the most abundant synaptic proteins, CaMKII may also play some scaffolding roles in addition to its kinase activity. Consistent with this idea, it was proposed that CaMKII regulates presynaptic short-term plasticity in the absence of its kinase activity (Hojjati et al., 2007). Identifying the precise targets of CaMKII in regulating neurotransmitter release and their mechanism of action will provide significant insights into neurotransmitter release as a whole but will require a major research effort in the future.
The fact that the CaM KD impairs neurotransmitter release even during single isolated action potentials in cultured neurons indicates that, in these neurons, CaM and CaMKIIα are tonically activated in what is the resting state of these neurons. It seems likely that the resting state in high-density cultures of neurons involves some degree of spontaneous activity, similar to many neurons in situ in brain, which are also subject to continuous firing. Thus, it is possible that the presynaptic potentiation of neurotransmitter release by activation of CaM and CaMKII normally operates as a positive feedforward mechanism to regulate synaptic strength, i.e., a mechanism that increases neurotransmitter release in neurons that are used frequently. Such a mechanism may increase the efficiency of heavily used synapses and counteract use-dependent depression. Moreover, in physiologically more quiet neurons, presynaptic receptor activation may raise Ca2+ levels to boost release via CaM, thereby providing a presynaptic modulatory pathway that enhances release.
Footnotes
Z.P.P is a recipient of a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression and supported by National Institutes of Health/National Institutes of Neurological Disorders and Stroke Epilepsy Training Grant 5T32NS007280. We thank Kaishan Xian and Iryna Huryeva for their excellent technical support and the Südhof laboratory for valuable discussions.
References
- Alvarez VA, Ridenour DA, Sabatini BL. Retraction of synapses and dendritic spines induced by off-target effects of RNA interference. J Neurosci. 2006;26:7820–7825. doi: 10.1523/JNEUROSCI.1957-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- Brickey DA, Bann JG, Fong YL, Perrino L, Brennan RG, Soderling TR. Mutational analysis of the autoinhibitory domain of calmodulin kinase II. J Biol Chem. 1994;269:29047–29054. [PubMed] [Google Scholar]
- Burgoyne RD, O'Callaghan DW, Hasdemir B, Haynes LP, Tepikin AV. Neuronal Ca2+-sensor proteins: multitalented regulators of neuronal function. Trends Neurosci. 2004;27:203–209. doi: 10.1016/j.tins.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Chamberlain LH, Roth D, Morgan A, Burgoyne RD. Distinct effects of alpha-SNAP, 14-3-3 proteins, and calmodulin on priming and triggering of regulated exocytosis. J Cell Biol. 1995;130:1063–1070. doi: 10.1083/jcb.130.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ. The alpha-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron. 1995;14:591–597. doi: 10.1016/0896-6273(95)90315-1. [DOI] [PubMed] [Google Scholar]
- Chen YA, Duvvuri V, Schulman H, Scheller RH. Calmodulin and protein kinase C increase Ca2+-stimulated secretion by modulating membrane-attached exocytic machinery. J Biol Chem. 1999;274:26469–26476. doi: 10.1074/jbc.274.37.26469. [DOI] [PubMed] [Google Scholar]
- Cheng J, Alkayed NJ, Hurn PD. Deleterious effects of dihydrotestosterone on cerebral ischemic injury. J Cereb Blood Flow Metab. 2007;27:1553–1562. doi: 10.1038/sj.jcbfm.9600457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ. The calmodulin hypothesis of neurotransmission. Cell Calcium. 1981;2:365–385. doi: 10.1016/0143-4160(81)90026-9. [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–49. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- Geiser JR, van Tuinen D, Brockerhoff SE, Neff MM, Davis TN. Can calmodulin function without binding calcium? Cell. 1991;65:949–959. doi: 10.1016/0092-8674(91)90547-c. [DOI] [PubMed] [Google Scholar]
- Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K. Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Hinds HL, Goussakov I, Nakazawa K, Tonegawa S, Bolshakov VY. Essential function of alpha-calcium/calmodulin-dependent protein kinase II in neurotransmitter release at a glutamatergic central synapse. Proc Natl Acad Sci U S A. 2003;100:4275–4280. doi: 10.1073/pnas.0530202100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–1251. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- Hojjati MR, van Woerden GM, Tyler WJ, Giese KP, Silva AJ, Pozzo-Miller L, Elgersma Y. Kinase activity is not required for alphaCaMKII-dependent presynaptic plasticity at CA3-CA1 synapses. Nat Neurosci. 2007;10:1125–1127. doi: 10.1038/nn1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A. 2008;105:341–346. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form Ca2+-sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The effect of calcium on acetylcholine release from motor nerve terminals. Proc R Soc Lond B Biol Sci. 1965;161:496–503. doi: 10.1098/rspb.1965.0017. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Nakayama S, Kretsinger RH. Classification and evolution of EF-hand proteins. Biometals. 1998;11:277–295. doi: 10.1023/a:1009282307967. [DOI] [PubMed] [Google Scholar]
- Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J Neurosci. 1999;19:8830–8838. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Takei K, Geppert M, Daniell L, Stenius K, Chapman ER, Jahn R, De Camilli P, Südhof TC. Synaptic targeting of rabphilin-3A, a synaptic vesicle Ca2+/phospholipid-binding protein, depends on rab3A/3C. Neuron. 1994;13:885–898. doi: 10.1016/0896-6273(94)90254-2. [DOI] [PubMed] [Google Scholar]
- Lin JW, Sugimori M, Llinás RR, McGuinness TL, Greengard P. Effects of synapsin I and calcium/calmodulin-dependent protein kinase II on spontaneous neurotransmitter release in the squid giant synapse. Proc Natl Acad Sci U S A. 1990;87:8257–8261. doi: 10.1073/pnas.87.21.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Liu XB, Jones EG. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci U S A. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan AC, Lutzko C, Kohn DB. Advances in lentiviral vector design for gene-modification of hematopoietic stem cells. Curr Opin Biotechnol. 2002;13:429–436. doi: 10.1016/s0958-1669(02)00346-4. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Maximov A, Südhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DG, Südhof TC. Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods. 2007a;161:75–87. doi: 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Südhof TC. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol. 2007b;176:113–124. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Mikhaylova M, Sharma Y, Reissner C, Nagel F, Aravind P, Rajini B, Smalla KH, Gundelfinger ED, Kreutz MR. Neuronal Ca2+ signaling via caldendrin and calneurons. Biochim Biophys Acta. 2006;1763:1229–1237. doi: 10.1016/j.bbamcr.2006.08.047. [DOI] [PubMed] [Google Scholar]
- Ninan I, Arancio O. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 2004;42:129–141. doi: 10.1016/s0896-6273(04)00143-6. [DOI] [PubMed] [Google Scholar]
- Ohyama A, Hosaka K, Komiya Y, Akagawa K, Yamauchi E, Taniguchi H, Sasagawa N, Kumakura K, Mochida S, Yamauchi T, Igarashi M. Regulation of exocytosis through Ca2+/ATP-dependent binding of autophosphorylated Ca2+/calmodulin-activated protein kinase II to syntaxin 1A. J Neurosci. 2002;22:3342–3351. doi: 10.1523/JNEUROSCI.22-09-03342.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC, McBain CJ. State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron. 2008;60:980–987. doi: 10.1016/j.neuron.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Südhof TC. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell. 1993;75:661–670. doi: 10.1016/0092-8674(93)90487-b. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Schnell E, Verhage M, Tzonopoulos T, Nicoll RA, Janz R, Malenka RC, Geppert M, Südhof TC. Rabphilin knock-out mice reveal that rabphilin is not required for rab3 function in regulating neurotransmitter release. J Neurosci. 1999;19:5834–5846. doi: 10.1523/JNEUROSCI.19-14-05834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sík A, Hájos N, Gulácsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci U S A. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca2+-sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+-sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]