

Abstract
Endoplasmic reticulum-associated degradation (ERAD) is a system by which proteins accumulated in the endoplasmic reticulum (ER) are retrotranslocated to the cytosol and degraded by the ubiquitin–proteasome pathway. HRD1 is expressed in brain neurons and acts as an ERAD ubiquitin ligase. Amyloid precursor protein (APP) is processed into amyloid-β peptides (Aβs) that form plaque deposits in the brains of Alzheimer's disease (AD) patients. We found significantly decreased HRD1 protein levels in the cerebral cortex of AD patients. HRD1 colocalized with APP in brain neurons and interacted with APP through the proline-rich region of HRD1. HRD1 promoted APP ubiquitination and degradation, resulting in decreased generation of Aβ. Furthermore, suppression of HRD1 expression induced APP accumulation that led to increased production of Aβ associated with ER stress. Immunohistochemical analysis revealed that suppression of HRD1 expression inhibited APP aggresome formation, resulting in apoptosis. In addition, we found that the ATF6- and XBP1-induced upregulation of ERAD led to APP degradation and reduced Aβ production. These results suggest that the breakdown of HRD1-mediated ERAD causes Aβ generation and ER stress, possibly linked to AD.
Introduction
Alzheimer's disease (AD) is one of the most common neurodegenerative diseases and the major cause of dementia in the elderly. The extracellular plaques (senile plaques), consisting of amyloid-β (Aβ) deposits, are pathologically observed in the brain of AD patients (Selkoe, 2001). The Aβ peptides, including Aβ40 and the more fibrillogenic Aβ42, are produced from amyloid precursor protein (APP) by the proteolytic activity of β- and γ-secretases (Gandy, 2005). Although APP is a type-I transmembrane glycoprotein whose original functions remain controversial, it has been considered as a regulator of synapse formation and neural plasticity. APP is formed by folding and glycosylation in the endoplasmic reticulum (ER). It is transported to the Golgi apparatus for further processing and is subsequently transported to the plasma membrane. APP is processed into Aβs by β- and γ-secretases in the trans-Golgi network (TGN) and endosomes (Thinakaran and Koo, 2008).
The ER plays functional roles in the processing, glycosylation, and disulfide-bond formation of newly synthesized membrane and secretory proteins. A variety of stressors affects ER function and leads to an accumulation of unfolded proteins in the ER lumen. Under such conditions termed ER stress, the unfolded protein response (UPR), which consists of translational arrest, ER-chaperone induction, and ER-associated degradation (ERAD) are activated to unload the accumulated nonfunctional proteins (Schröder and Kaufman, 2005; Ron and Walter, 2007).
A series of complex ERAD reactions serve to remove unfolded proteins by retrograde transport from the ER to the cytosol followed by degradation through the ubiquitin-proteasome system (Bonifacino and Weissman, 1998; Tsai et al., 2002; Kaneko and Nomura, 2003). We have identified and characterized the human homolog of yeast Hrd1p/Der3p (HRD1). It is a ubiquitin-ligase E3, localized in the ER, expressed by ER stress, and it protects against ER stress-induced apoptosis (Kaneko et al., 2002; Carvalho et al., 2006; Denic et al., 2006; Ismail and Ng, 2006). Furthermore, we demonstrated that HRD1 promotes ubiquitination and degradation of Parkin-associated endothelin receptor-like receptor (Pael-R), a substrate for Parkin, and suppresses Pael-R-induced ER stress and apoptosis (Imai et al., 2001; Omura et al., 2006). We also reported that HRD1 is widely expressed in brain neurons, but not glia (Omura et al., 2008b). It has been reported that HRD1 is involved in the degradation of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, CD3-δ, and TCR-α (Kikkert et al., 2004). It is noteworthy that polyglutamine-expanded huntingtin (Htt) (Yang et al., 2007), prion protein (PrP) (Apodaca et al., 2006), and Pael-R, which are associated with neurodegenerative disease, were recently reported as HRD1 substrates. Furthermore, the tumor suppressor gene, p53, was reported to be another substrate for HRD1 (Yamasaki et al., 2007); and HRD1 has been proposed to be involved in rheumatoid arthritis (Amano et al., 2003).
Because HRD1 can promote the degradation of numerous unfolded proteins induced by ER stress (Kaneko et al., 2002), it is likely that additional substrates for HRD1 exist. Accumulating evidence suggests that HRD1 plays an important role in the quality control of the ER in brain neurons. Therefore, it may be important to identify additional HRD1 substrates associated with neurodegeneration to further understand the pathogenesis of neurodegenerative disease. The metabolism of APP to Aβs and other forms has been well investigated. However, APP-holoprotein metabolism has not been elucidated. In the present study, we found that protein levels of HRD1 were significantly reduced in the cerebral cortex of examined AD patients. Furthermore, we found that HRD1 promoted the ubiquitination and degradation of APP resulting in decreased Aβ production, whereas loss of HRD1 led to an accumulation of APP and increased levels of Aβ associated with ER stress and apoptosis.
Materials and Methods
Antibodies and chemicals.
Antibodies were purchased as follows: APP (C-terminal and 6E10; Sigma-Aldrich), cleaved caspase-3 (Asp175; Cell Signaling Technology Inc.), FLAG (M2; Sigma-Aldrich), γ-tubulin (GTU-88; Sigma-Aldrich), GST (Z-5; Santa Cruz Biotechnology), HRD1 (C-terminal; Abgent; Sigma), KDEL (10C3; Stressgen Biotechnologies Corporation), c-myc (9E10; Merck), NeuN (A60; Millipore Bioscience Research Reagents), normal mouse and rabbit IgG (Millipore), PDI (RL90; Affinity BioReagents), ubiquitin (FK-2; Nippon Biotest Laboratories). Alexa Fluor antibodies were purchased from Invitrogen. Anti-HRD1 (594–606) was a gift from Otsuka GEN Research Institute, Tokushima, Japan. The reversible proteasome inhibitor MG132 was obtained from the Peptide Institute. Thapsigargin and tunicamycin were purchased from Wako Pure Chemical Industries.
Animals.
Eight-week-old male ddY mice were obtained from Japan SLC Inc. The mice were maintained in a room at 22–24°C under a constant day–night rhythm and given food and water ad libitum. All animal experiments were performed in accordance with NIH Guidelines for Care and Use of Laboratory Animals and approved by the Animal Care and Use committee at the Chiba Institute of Science.
Cell culture.
Human neuroblastoma SH-SY5Y and HEK 293 cells were maintained in DMEM (Sigma-Aldrich) with 10% (v/v) heat-inactivated fetal calf serum (Invitrogen Corp.) at 37°C in 5% CO2, 95% humidified air atmosphere.
Plasmids.
The expression vectors for wild-type (wt) human HRD1 and a series of its truncated mutants tagged with myc and polyhistidine (6× His) epitopes at the C terminus of the inserted sequence (pcDNA6; Invitrogen), have been described previously (Omura et al., 2006). The expression vector for wt human APP695, tagged with FLAG and 6× His epitopes at the C terminus (pcDNA3), was gifted by Toshiharu Suzuki (Hokkaido University, Japan). The expression vectors for the spliced form of ATF6 (amino acid region 1–373 of ATF6α) and XBP1 (spliced form), tagged with the HA epitope at the N terminus (pCR3.1, Invitrogen), have been described previously (Kaneko et al., 2007). The expression vector for RP-HRD1, fused to GST at its N terminus, was cloned into the expression vector pGEX6p-1 (GE Healthcare Bio-sciences) as described previously (Omura et al., 2006).
Analysis of protein and mRNA levels in human brains.
Human brain samples were purchased from Analytical Biological Services via KAC. Gray matter was dissected from the cerebral cortex of deceased AD patients (n = 6) and control cases (n = 8). The tissues were extracted by sonication in a buffer consisting of 10 mm Tris-HCl, pH 7.6, 420 mm NaCl, 1 mm EDTA, and 1% NP-40, with complete protease inhibitors (Roche Diagnostics). The lysates were centrifuged at 30,000 × g for 30 min. Protein equivalent samples were subjected to Western blotting using a LAS-3000 luminescent image analyzer (Fujifilm). Quantitative analysis was performed using Multi Gauge (Fujifilm).
The expression of mRNA was measured in duplicate with the TaqMan-based real-time PCR assay using a 7500 Real-Time PCR System (Applied Biosystems) as described previously (Kaneko et al., 2007).
Immunohistochemistry and immunocytochemistry.
Coronal sections of the mouse brains embedded in paraffin were subjected to immunohistochemistry, as described previously (Omura et al., 2008b). Immunofluorescence staining was performed with anti-HRD1 (C-terminal, Abgent) and either anti-APP (6E10) or anti-NeuN antibody, and then with anti-mouse conjugated with Alexa Fluor 546 and anti-rabbit with Alexa Fluor 488. Fluorescence images were acquired using an LSM 510 META confocal microscope (Carl Zeiss AG).
For subcellular localization of endogenous HRD1, APP, and γ-tubulin, SH-SY5Y cells were subjected to immunocytochemistry as described previously (Omura et al., 2006). The cells were treated overnight with anti-HRD1 (594–606, Otsuka), anti-APP (6E10), and anti-γ-tubulin antibodies at 4°C. The cells were incubated with anti-rabbit Alexa Fluor 546 and anti-mouse Fluor 488 antibodies.
Immunoprecipitation.
For overexpressed APP-FLAG immunoprecipitation, transfected HEK293 cells were incubated in a lysis buffer (20 mm HEPES, pH 7.4, 120 mm NaCl, 5 mm EDTA, 10% glycerol, and 1% Triton X-100 with complete protease inhibitors). Five hundred μg of total protein of the supernatant was incubated overnight with anti-FLAG M2 affinity gel (Sigma-Aldrich). For endogenous APP immunoprecipitation, SH-SY5Y cells were treated with 5 μg/ml tunicamycin (Tm) for 12 h. The cells were incubated with 2 mm dithiobis(succinimidylpropionate) (Sigma-Aldrich) in PBS for 2 h, followed by 20 mm Tris-HCl, pH 7.6, for 15 min. Cell lysates were incubated overnight with anti-APP antibody (C-terminal). The immune complexes were incubated with Protein G Magnetic Beads (Millipore) for 1 h, and rinsed three times with wash buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10% glycerol, and 0.2% Triton X-100) and boiled with Laemmli SDS-PAGE sample buffer for 5 min. Total cell extracts and immunoprecipitates were subjected to Western blotting using Clean-Blot IP Detection Reagent (HRP; Thermo Fisher Scientific) and ECL Plus Western Blotting Detection Reagents (GE Healthcare).
In vitro ubiquitination assay.
GST-RP-HRD1, lacking the transmembrane domains, was expressed in cultured Escherichia coli and purified, as described previously (Omura et al., 2006). APP-FLAG were produced by TNT-quick coupled transcription/translation systems (Promega Corporation). The reaction products (16 μl) containing APP-FLAG were mixed with RP-HRD1 (E3; 3 ng), rabbit ubiquitin activating enzyme (E1; 25 ng), GST-UbcH5c (E2; 400 ng), and GST-ubiquitin (7 ng) purchased from Boston Biochem, in 100 μl of reaction buffer (containing, in mm: 40 Tris-HCl, pH 7.6, 5 MgCl2, 2 ATP, and 2 DTT). The reaction solution was incubated at 30°C for 90 min, immunoprecipitated with anti-FLAG M2 affinity gel, and subjected to Western blotting using anti-GST antibody.
Aβ ELISA.
Aβs secreted in the culture medium were measured by a standard sandwich ELISA using a human amyloid β (1–40) and (1–42) assay kit from IBL. For overexpression of HRD1 and APP, HEK293 cells and those expressing wt and M-HRD1 were transfected with APP-FLAG using LipofectAMINE 2000 reagent (Invitrogen). For the knockdown experiments of HRD1, SH-SY5Y cells stably expressing APP were transfected with HRD1-siRNA, siGENOME-Individual Duplex targeted against human HRD1 (M-007090-01; Dharmacon Research), using LipofectAMINE RNAiMAX reagent (Invitrogen), and incubated for 72 h.
Apoptosis assay.
SH-SY5Y cells stably expressing APP-FLAG were transiently transfected with non-target control (NC) or HRD1 siRNA. At 72 h after transfection, the cells were fixed and subjected to immunofluorescence staining with anti-FLAG and anti-cleaved caspase-3 (recognizing its active form) antibodies. The cells were incubated with anti-mouse Alexa Fluor 488 and anti-rabbit Fluor 546 antibodies. The cells were counted in three different areas in three independent experiments.
Statistics.
All data are expressed as mean ± SEM Statistical evaluation was performed by ANOVA followed by two-tailed Bonferroni correction (multiple comparison) or two-tailed Student's t tests. Each experiment was performed ≥3 times.
Results
Protein levels of HRD1 are reduced in AD patients
To clarify the involvement of HRD1 in AD pathology, we initially investigated protein and mRNA HRD1 expression levels in the cerebral cortex of AD patients and compared them with non-AD controls. There were no significant differences in age, postmortem interval, or gender among subjects in this study (supplemental Table 1, available at www.jneurosci.org as supplemental material). Moreover, we biochemically confirmed that Aβ40 and Aβ42 levels were elevated in the AD brains (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Surprisingly, HRD1 protein levels in most of the AD brains were significantly lower compared with non-AD brains (Fig. 1a, first panel, b; supplemental Fig. 2a,b, available at www.jneurosci.org as supplemental material). In contrast, SEL1, which colocalizes and interacts with HRD1 to stabilize it (supplemental Fig. 3a,b, available at www.jneurosci.org as supplemental material), and ER-resident chaperone protein-disulfide isomerase (PDI) were not significantly different between AD and control brains (Fig. 1a, second and third panels, b; supplemental Fig. 2a,b, available at www.jneurosci.org as supplemental material).
Figure 1.

Protein and mRNA levels of HRD1 in the AD brain. a, Protein levels of HRD1, SEL1, PDI, and β-actin in the AD brains. The cerebral cortex of AD patients and non-AD controls were subjected to Western blotting. b, Statistical analysis of a. Data are normalized to the amount of β-actin; results are expressed as a box plot (control, n = 8; AD, n = 6). Asterisk represents a significant difference (Student's t test, *p < 0.05). c, mRNA levels of HRD1and ER stress-responsive genes in the AD brain. The mRNA expression levels in the brains were quantified by real-time PCR. Data are normalized to the amount of β-actin. Results are expressed as a box plot (Student's t test; *p < 0.05, **p < 0.01).
To determine whether the low HRD1 expression was due to a decrease in mRNA or protein, we examined mRNA expression in AD and control brains. PCR analysis demonstrated that HRD1 mRNA expression levels were not reduced in AD brains compared with controls indicating that low levels of HRD1 protein in AD do not result from a decrease in mRNA expression (Fig. 1c). Because AD pathology is reportedly associated with ER stress (Lindholm et al., 2006), we investigated mRNA expression of the ER stress-responsive genes sel1, chop, and grp78 (also known as bip) (Kaneko et al., 2007). In AD brains, chop and grp78 expression was significantly higher than in controls, whereas hrd1 and sel1 only tended to increase (Fig. 1c), suggesting that the AD brains were under ER-stress conditions. Thus, to elucidate the involvement of HRD1 in AD pathology, we investigated whether HRD1 acts as an E3 ligase for APP.
HRD1 and APP are colocalized in the brain neurons and cellular ER
We have previously reported that HRD1 is expressed in neurons, but not glia, of the mouse brain. In particular, HRDI was strongly expressed in the hippocampus (dentate gyrus and CA3-1 regions) and cerebral cortex (Omura et al., 2008b), where neuronal degeneration is observed in the AD brain. Using immunofluorescence staining, we examined whether HRD1 and APP were coexpressed in these regions. Consistent with previous observations, HRD1 (green) coexisted with mouse hippocampal neurons (NeuN; red) (Fig. 2a, top) and was coexpressed with APP in the hippocampus (Fig. 2a, middle) and cerebral cortex (Fig. 2a, bottom; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Furthermore, we immunocytochemically examined the cellular colocalization of HRD1 and APP with PDI in SH-SY5Y neuroblastoma cells. Endogenous HRD1 and APP were colocalized in the ER of SH-SY5Y cells (Fig. 2b).
Figure 2.

Colocalization of HRD1 and APP in the brain and neuronal cells. a, Colocalization of HRD1 and APP in the murine hippocampus and cerebral cortex. The coronal sections of the murine hippocampus (top and middle) and cerebral cortex (bottom) were subjected to immunofluorescence staining with HRD1 (Abgent), APP (6E10), and NeuN antibodies. DG, dentate gyrus; Cortex, cerebral cortex. Scale bars, 200 μm. b, Colocalization of HRD1 and APP in the ER. SH-SY5Y cells were subjected to immunofluorescence staining with HRD1 (Otsuka), APP (C-terminal), and PDI antibodies. Scale bars, 20 μm.
HRD1 interacts with APP through the proline-rich region
To examine whether HRD1 interacts with APP, human myc-tagged HRD1 and FLAG-tagged APP were transiently cotransfected into HEK293 cells and immunoprecipitated with anti-FLAG antibody. Overexpressed HRD1 protein coprecipitated with overexpressed APP (Fig. 3a, lane 3), whereas HRD1 was not observed in the normal-IgG precipitate (Fig. 3a, lane 6). We further demonstrated physiological interaction between endogenous HRD1 and APP in SH-SY5Y cells. Although the interaction between endogenous HRD1 and APP was not observed under steady-state conditions (Fig. 3b, top, lane 3), endogenous HRD1 was coprecipitated with endogenous APP in cells treated with Tm, a glycosylation inhibitor (Fig. 3b, top, lane 4).
Figure 3.

Interaction of HRD1 with APP. a, Interaction of overexpressed APP and HRD1 in HEK293 cells. Coimmunoprecipitation of HRD1 and APP was performed in HEK293 cells transiently transfected with APP-FLAG and HRD1-myc. The cell lysates were immunoprecipitated with anti-FLAG antibody or normal mouse IgG. The immune complexes were analyzed by Western blotting with anti-myc or anti-FLAG antibodies. b, Interaction of endogenous HRD1 and APP in SH-SY5Y cells. SH-SY5Y cells were treated with 5 μg/ml Tm for 12 h. The cells were incubated with 2 mm dithiobis(succinimidylpropionate) for 2 h, followed by 20 mm Tris-HCl, pH 7.6, for 15 min. Cell lysates were immunoprecipitated with anti-APP (C-terminal) antibody. Immune complexes were analyzed by Western blotting using anti-HRD1 (Sigma) and APP (C-terminal) antibodies. c, Schematic diagrams of HRD1 deletion mutants. The panel graphically represents wt-HRD1 and a variety of HRD1 mutants. d, Interaction of APP with HRD1 and its mutants. Coimmunoprecipitation of APP and HRD1 mutants was performed in HEK293 cells transiently transfected with APP-FLAG and either an empty vector (mock), wt-, M-, ΔM-, or MR-HRD1-myc. The cell lysates were immunoprecipitated with anti-FLAG antibody, and then analyzed by Western blotting using anti-myc antibody.
Using Pael-R, a substrate for HRD1, we previously determined that the proline-rich region of human HRD1 is required for interaction with its substrates (Omura et al., 2006, 2008a). Similarly, we confirmed that the proline-rich region is also necessary for the interaction with APP. A variety of HRD1 mutants and wt-HRD1 tagged with myc epitope were transfected into HEK293 cells and immunoprecipitated with FLAG-tagged APP (Fig. 3c). Full-length (1-616 aa) wt-HRD1 and ΔM-HRD1 (ΔMembrane, Δ42-204 aa; lacking four of five membrane-spanning domains) coprecipitated with APP (Fig. 3d, lanes 2 and 4), whereas M-HRD1 (1-290 aa; defect in cytoplasmic domains including RING-finger and proline-rich regions) and MR-HRD1 (Membrane-RING, 1-340 aa; defect in a proline-rich region) were not contained in either precipitate (Fig. 3d, lanes 3 and 5). This result indicated that HRD1 interacts with APP through the proline-rich region.
HRD1 promotes the ubiquitination and degradation of APP resulting in decreased Aβ generation
To evaluate the ubiquitin ligase activity of HRD1 for APP in vitro, an APP-ubiquitin conjugation reaction was reconstituted with glutathione S-transferase (GST)-tagged recombinant RING-Proline (RP)-HRD1, E1, E2 (UbcH5c), and ubiquitin proteins. A previous report has demonstrated UbcH5c involvement in HRD1-mediated ubiquitination (Nadav et al., 2003). Incubation of the reaction mixture with all of the components promoted APP ubiquitination (Fig. 4a, lane 6; supplemental Fig. 5, available at www.jneurosci.org as supplemental material), whereas if any of the components was removed, APP failed to ubiquitinate (Fig. 4a, lanes 1–5).
Figure 4.

Ubiquitination and degradation of APP by HRD1. a, In vitro ubiquitination assay. The reaction products, containing APP protein produced by a transcription/translation system, were mixed in the reaction buffer with E1 (rabbit), E2 (GST-UbcH5c), E3 (GST-RP-HRD1), and GST-ubiquitin. The reaction mixture was immunoprecipitated with anti-FLAG antibody and analyzed by Western blotting using anti-GST antibody. b, Degradation of APP by HRD1. Normal HEK293 cells and those stably expressing wt-HRD1, two stable cell lines (3 and 4), or M-HRD1 were transiently transfected with APP-FLAG and incubated for the indicated periods in the presence or absence of 10 μm MG132 for 12 h. The total cell lysates were analyzed by Western blotting with anti-FLAG (first panel), anti-myc (second panel), anti-α-tubulin (third panel) antibodies. c, d, Aβ levels were measured by a sandwich ELISA in b. HEK293 cells and those expressing wt- and M-HRD1 were transiently transfected with APP-FLAG. Data (pg/ml Aβ peptide) are normalized to the amount of APP mRNA quantified by real-time PCR. Results are expressed as a fold increase compared with normal cells (mean ± SEM; n = 3). Statistical analysis was performed with ANOVA followed by Bonferroni correction (M-HRD1 vs wt-HRD1; **p < 0.01).
We next investigated whether HRD1 promotes APP degradation through its E3 activity. APP-FLAG was transfected into normal HEK293 cells and into those stably expressing wt-HRD1 or M-HRD1. In the presence of wt-HRD1, the APP levels of both clones (#3 and #4), were reduced at 12 and 36 h posttransfection (Fig. 4b, first panel, lanes 5, 6, 8, 9), compared with control cells (Fig. 4b, first panel, lanes 2, 3). In contrast, expression of M-HRD1, which lacks cytosolic domains, increased in APP compared with the absence of effect observed in normal cells (Fig. 4b, first panel, lanes 11, 12). Moreover, to determine whether this APP decrease was due to HRD1-mediated protein degradation by the proteasome system, these cell lines were treated with the reversible proteasome inhibitor MG132. The wt-HRD1-induced decrease in APP was inhibited by MG132, indicating that the HRD1-induced decrease in APP resulted from proteasome-dependent protein degradation (Fig. 4b, first panel, lanes 4, 7, 10, 13). Furthermore, we demonstrated that, in the presence of wt-HRD1, the APP level was dependent on protein-level changes and not on mRNA expression (data not shown).
APP is processed into Aβ peptides (Aβ40 and Aβ42) by β- and γ-secretase. We hypothesized that the HRD1-induced decrease in APP holo-protein results in suppression of Aβ generation and secretion. To quantify the Aβ secreted into the culture medium from the APP degradation experiment (Fig. 4b), we performed ELISA using specific anti-Aβ antibodies against the Aβ40 and Aβ42 peptides. The concentration of Aβ was normalized by APP mRNA expression levels to exclude a difference in APP expression among the clones. Consistent with the results of the APP degradation experiments, ELISA revealed that wt-HRD1 (clones #3 and #4) significantly reduced Aβ40 and Aβ42 levels compared with normal HEK293 cells and to those expressing M-HRD1 (Fig. 4c,d).
Reduction of HRD1 expression induces APP accumulation and Aβ generation
Using siRNA against HRD1 in SH-SY5Y cells stably expressing APP, we examined the effects of decreased HRD1 expression on APP and Aβ. The amount of APP was increased by the suppression of HRD1 expression (Fig. 5a, first and second panels, lane 3) compared with untransfected (Control) and non-targeted siRNA controls (NC). Interestingly, levels of the ER chaperones, GRP78 and GRP94, were elevated in HRD1-suppressed cells, indicating ER stress induction (Fig. 5a, third panel, lane 3). Next, to confirm the involvement of HRD1 in APP protein degradation, we performed a cycloheximide (CHX) assay. As shown in Figure 5b, the APP protein decay rates were reduced in the absence of HRD1 (NC 32%, HRD1 50%; at 1 h after CHX addition), indicating that HRD1 accelerates APP protein degradation.
Figure 5.

APP accumulation and Aβ generation by suppression of HRD1 expression. a, Induction of APP accumulation by HRD1 siRNA. SH-SY5Y cells stably expressing APP-FLAG were transiently transfected with NC or HRD1 siRNA. The total cell lysates were analyzed by Western blotting with the indicated antibodies. GRP78 and GRP94 were detected with anti-KDEL antibody. b, CHX assay. SH-SY5Y cells stably expressing APP-FLAG were transfected with NC or HRD1 siRNA. At 48 h after transfection, the cells were treated with 20 μg/ml CHX for the indicated periods. Total cell lysates were analyzed by Western blotting using anti-FLAG M2 antibody. Levels of APP at each time point were plotted relative to the amount present at time 0 (mean ± SEM; n = 5). Asterisk represents a significant difference (Student's t test, *p < 0.05). c, d, Aβ levels were measured by a sandwich ELISA using the culture medium of a. Results are expressed as a ratio of the total amount of Aβ peptides (pg) to the total amount of protein from whole-cell extracts (mg) (mean ± SEM; n = 4). Statistical analysis was performed with ANOVA followed by Bonferroni correction (control and NC vs HRD1; *p < 0.05, **p < 0.01).
Using the culture medium from the foregoing APP accumulation experiment (Fig. 5a), we further determined whether the APP accumulation induced by HRD1 suppression leads to increased Aβ generation. As APP accumulation increased, the secretion of Aβ40 and Aβ42 was significantly augmented by the suppression of HRD1 expression (Fig. 5c,d). Together, these findings suggest that the disruption of HRD1-mediated ERAD resulted in APP accumulation followed by Aβ generation.
Suppression of HRD1 expression inhibits APP aggresome formation resulting in apoptosis
Using APP-overexpressed cells, we conducted an immunocytochemical analysis to observe the localization of accumulated APP under HRD1-suppressed conditions. When APP was overexpressed in SH-SY5Y cells, an inclusion body was formed in the perinuclear region (Fig. 6a, top). Interestingly, the inclusion body disappeared in the HRD1-reduced cells treated with siRNA (Fig. 6a, bottom). Under these conditions, caspase-3 was activated indicating apoptosis (Fig. 6a, bottom center), whereas overexpression of APP did not affect control cells transfected with NC siRNA (Fig. 6a, top center). Furthermore, to statistically estimate the apoptosis induced by HRD1 suppression, cells activating caspase-3 were counted in normal SH-SY5Y cells and those expressing APP. APP-overexpressed cells treated with HRD1-siRNA showed significantly increased apoptosis compared with the HRD1-suppressed normal cells and APP-overexpressed cells treated with NC siRNA (Fig. 6b), suggesting that HRD1 is involved in the formation of APP inclusion bodies and prevents APP-induced apoptosis.
Figure 6.

Inhibition of APP aggresome formation by suppression of HRD1 expression. a, SH-SY5Y cells stably expressing APP-FLAG were transiently transfected with NC (top) or HRD1 (bottom) siRNA. The cells were subjected to immunofluorescence staining with anti-FLAG (green) and anti-cleaved caspase-3 (red) antibodies. Scale bars, 100 μm. b, Statistical analysis of Figure 6a. The percentage of apoptotic cells was counted in three different areas. The results are expressed as mean ± SEM (n = 3). Statistical analysis was performed with ANOVA followed by Bonferroni correction (*p < 0.05, **p < 0.01). c, Localization of APP and γ-tubulin protein. d, Localization of HRD1 and γ-tubulin. SH-SY5Y cells were treated with 5 μm MG132 for 12 h and then subjected to immunofluorescence staining using APP (C-terminal), HRD1 (Otsuka), and γ-tubulin antibodies. Scale bars, 20 μm.
We also found a smaller object in the perinuclear region of normal SH-SY5Y cells, which was similar to the inclusion body seen in the APP overexpressed cells (Fig. 2b). Interestingly, endogenous HRD1 and APP were highly colocalized in this aggregate (Fig. 2b). Ubiquitinated proteins that are not degraded by proteasomes are transported to the centrosome, wherein they form perinuclear aggresomes (Kopito, 2000; Kawaguchi et al., 2003). To determine whether this body was an aggresome, we immunocytochemically demonstrated that APP was colocalized with γ-tubulin, a component of the centrosome, at this region (Fig. 6c, top). Interestingly, HRD1 was also colocalized with γ-tubulin at the centrosome (Fig. 6d, top). In addition, MG132 inhibition of the proteasome led to concentration of APP and HRD1 at the centrosome (Fig. 6c,d, bottom), suggesting that APP and HRD1 escaped from proteasome degradation are collected on the aggresome. Thus, the inclusion body was an aggresome that included APP and HRD1.
UPR activation induces APP degradation and decreased Aβ generation
We found that overexpression of transcription factors (ATF6 and XBP1) involved in the UPR induces HRD1 expression (Kaneko et al., 2007), and that Pael-R degradation is promoted by ATF6 and XBP1 overexpression, presumably due to ATF6- and XBP1-induced expression of a number of ERAD genes, including HRD1 (Omura et al., 2006). HEK293 cells transiently transfected with APP, and an empty vector (mock), ATF6 (1-373, cytoplasmic domain working as a transcription factor), or XBP1 (spliced form) were incubated in the presence or absence of MG132. The increased expression of GRP78 and GRP94 indicated UPR induction by ATF6 and XBP1 (Fig. 7a, middle, lanes 5, 6, 8, 9). The HRD1 protein was also upregulated by ATF6 and XBP1 (Fig. 7a, bottom, lanes 3, 4). Under such conditions, APP was markedly decreased in cells expressing ATF6 (Fig. 7a, top, lanes 5, 6). Although XBP1 was less effective than ATF6, XBP1 overexpression also reduced APP (Fig. 7a, top, lanes 8, 9). Moreover, MG132 inhibited the decrease in APP by ATF6 and XBP1 overexpression, indicating that the decrease in APP by ATF6 and XBP1 was proteasome-dependent protein degradation (Fig. 7a, top, lanes 7, 10).
Figure 7.
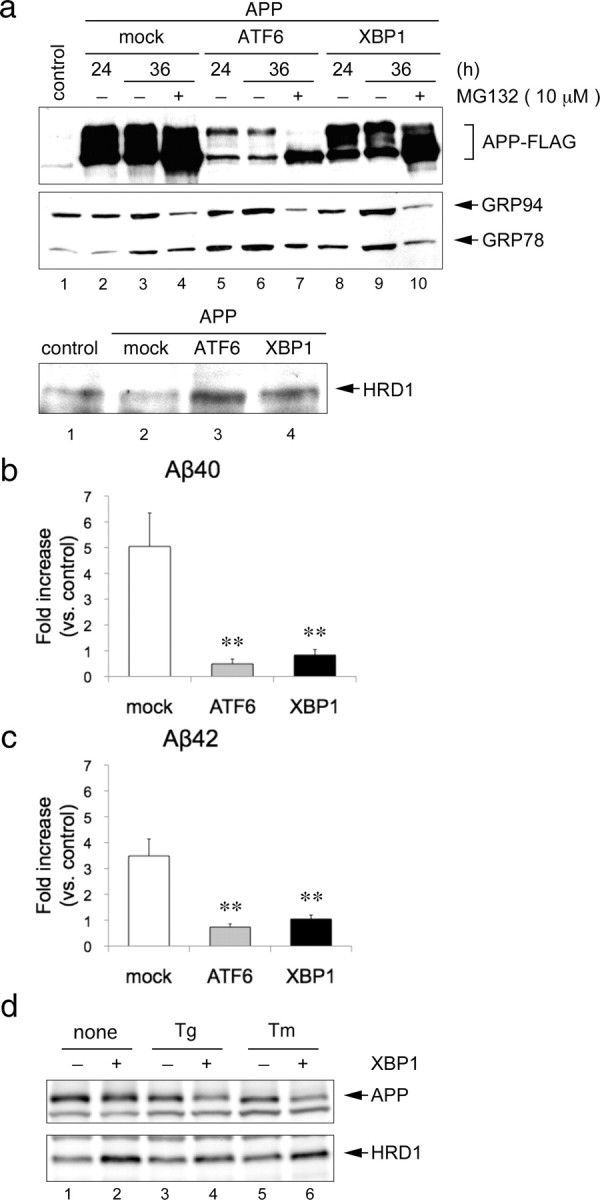
Degradation of APP by UPR activation. a, Degradation of overexpressed APP protein by ATF6 and XBP1. HEK293 cells were transiently transfected with APP-FLAG and an empty vector (mock), HA-ATF6, or HA-XBP1 for the indicated periods. The total cell lysates were analyzed by Western blotting using FLAG, KDEL and HRD1 (Otsuka) antibodies. b, c, Aβ levels were measured by sandwich ELISA using the culture medium of a at 36 h. Data (pg/ml Aβ peptide) are normalized to the amount of APP mRNA quantified by real-time PCR. Results are expressed as a fold increase compared with normal cells (mean ± SEM; n = 4). Statistical analysis was performed with ANOVA followed by Bonferroni correction (control vs mock, ATF6, and XBP1; **p < 0.01). d, Degradation of endogenous APP protein by XBP1 under ER stress conditions. XBP1 were induced using a tetracycline-inducible expression system (Tet-on) in HEK293 cells. The cells were treated with 1 μm Tg and 5 μg/ml Tm for 12 h. Total cell lysates were analyzed by Western blotting using APP (C-terminal) and HRD1 (Sigma) antibodies.
To ascertain whether secreted Aβs were concomitantly decreased, ELISA was performed with the culture medium used in the above experiment. Overexpression of ATF6 and XBP1 significantly reduced Aβ-peptide (Aβ40 and Aβ42) production compared with mock-transfected control cells (Fig. 7b,c).
Furthermore, we examined the effects of XBP1 overexpression on endogenous APP. As a result, XBP1 overexpression-induced UPR activation did not affect APP under steady-state conditions (Fig. 7d, top, lanes 1, 2). Interestingly, APP protein levels were reduced by XBP1 overexpression in the presence of ER stress reagents, such as thapsigargin (Tg) and Tm (Fig. 7d, top, lanes 4, 6), while ER stress reagents alone did not alter APP protein levels (Fig. 7d, top, lanes 3, 5). This indicates that unfolded APP protein, but not folded APP, is degraded by UPR activation.
Discussion
In this study, we found that protein levels of HRD1 were reduced in the cerebral cortex of AD patients. We demonstrated that HRD1 is coexpressed with APP in brain neurons and colocalizes and interacts with APP in neuronal cells. Furthermore, the present study indicated that HRD1 promotes ubiquitination and degradation of APP and leads to decreased Aβ production. We also found that suppression of HRD1 expression causes APP accumulation and Aβ generation accompanied by ER stress and apoptosis that was concomitant with the disappearance of the APP- and HRD1-containing aggresomes. Therefore, these results suggest that HRD1 promoted APP degradation and aggresome formation through ERAD, resulting in the prevention of Aβ generation.
Although APP appears to be degraded by the ubiquitin-proteasome system (Bunnell et al., 1998; Hare, 2006), with the exception of α-, β-, and γ-secretases, few molecules have been shown to be involved in APP metabolism. MOCA (also known as Dock3), which interacts with presenilin, induces proteasome-dependent degradation of APP (Chen et al., 2002). However, it has hitherto not been reported whether MOCA is involved in ERAD. Most recently, the stress-responsive chaperone-protease HtrA2 (also known as Omi) has been reported to participate in APP metabolism through ERAD-mediated degradation; however, there has been no E3 ligase associated with this degradation (Park et al., 2006; Huttunen et al., 2007). Because HRD1 and Derlin-1 constitute a complex and coordinately serve in ERAD (Schulze et al., 2005; Carvalho et al., 2006; Denic et al., 2006), the finding that APP retrotranslocation to the cytosol was mediated by Derlin-1 strongly supports our model in which HRD1 ubiquitinates APP. In addition, the result that HtrA2 inhibition increased Aβ secretion are consistent with our finding that inhibition of HRD1-mediated ERAD resulted in APP accumulation and increased Aβ production (Huttunen et al., 2007). They proposed that increased Aβ production by HtrA2 inhibition causes enhanced ER exit (protein transport from the ER to the Golgi) of APP, which is mediated by COPII cargo proteins (Huttunen et al., 2007). Based on these findings, disruption of HRD1-mediated ERAD also appears to steer APP toward the pathway for ER exit, resulting in elevated processing of APP to Aβ.
A large number of studies have focused on Aβ production by β- and γ-secretases, but it is unknown why APP processing is induced in sporadic AD. As a possible mechanism, ERAD dysfunction may cause increased Aβ production that is triggered by a functional decline of E3 ubiquitin ligase for APP. Surprisingly, the cerebral cortex of AD patients showed a loss of HRD1 protein. On the other hand, to exclude the possibility that HRD1 depletion is merely due to neuronal loss in the later event of AD, we examined SEL1 protein levels in the AD brains (Fig. 1a, second panel). Since SEL1 interacts with HRD1 to stabilize it, SEL1 was almost completely colocalized with HRD1 in brain neurons (supplemental Fig. 3a,b, available at www.jneurosci.org as supplemental material). SEL1 protein levels were not significantly different between the normal and AD brains, suggesting that HRD1 depletion does not result from neuronal loss. However, it remains unclear whether the reduction of HRD1 protein precedes increased Aβ production. To clarify whether loss of HRD1 causes an increase in Aβ generation that leads to AD onset, the effects of an HRD1 defect on accumulation of APP and Aβ should be examined during aging in HRD1 knock-out mice.
It is also important to understand how HRD1 is decreased in AD patients. Because HRD1 is partially localized to the aggresome, it appears to be eliminated from the ER for ubiquitination and either degraded by the proteasome or transported to the aggresome. In fact, overexpression of APP and Pael-R induced a decrease in HRD1 (Figs. 4b, second panel, lanes 6, 9, 12; 7a, third panel, lanes 2). Hence, we speculate that an increase in substrate load could lead to a decline of HRD1 protein, ERAD dysfunction, Aβ generation, and ER stress.
We found that inhibition of HRD1 expression caused ER stress and caspase-3 activation in SH-SY5Y cells stably expressing APP. Aβ has been shown to induce ER stress, which is proposed to be the cause of Aβ-mediated toxicity in AD (Nakagawa et al., 2000; Kadowaki et al., 2005); however, APP overexpression in SH-SY5Y cells failed to induce ER stress and apoptosis despite increased Aβ production resulting in the APP-containing aggresome (Fig. 6a,b). In contrast, under HRD1-suppressed conditions, cell death and caspase-3 activation were induced and accompanied by the disappearance of the aggresome (Fig. 6a,b). The disappearance of the aggresome was most likely due to APP accumulation in the ER resulting from a breakdown in the retrograde transport and ubiquitination of APP due the lack of HRD1. However, it remains to be clarified whether ER accumulation of APP or Aβ generation in the absence of HRD1 was the direct cause of cell death.
The relationship between AD and ER stress has been reported. Presenilin mutation attenuated the UPR and caused ER stress in familial AD (Katayama et al., 1999) and S-nitrosylation of PDI reduced its chaperone activity and induced ER stress as observed in sporadic AD (Uehara et al., 2006). On the other hand, because mRNA levels of the ER-response genes, grp78 and chop, were elevated in HRD1-deficient brains, we found significantly induced ER stress in the AD brain associated with a decrease in HRD1. Therefore, the decrease in HRD1 may directly cause ER stress and neuronal cell death in AD patients. Based on these findings, it will be important to understand ER stress caused by the dysfunction of the ER quality control system as well as the toxicity of Aβ.
We additionally demonstrated that UPR upregulation mediated by ATF6 and XBP1 induces APP degradation and leads to a decrease in Aβ generation. Furthermore, we found that ERAD activated by the UPR preferentially promotes the degradation of unfolded APP, suggesting that the ERAD system plays important roles in the quality control of APP. Based on these findings, we speculate that abnormal maturation of APP in the ER could cause increased Aβ generation in a post-ER compartment because mutations in APP are linked to increased Aβ production in familial AD. Therefore, acceleration of the UPR or solo augmentation of HRD1 may prevent APP accumulation and an increase in Aβ before an irreversible loss of HRD1. In conclusion, our findings not only propose a new model for AD pathogenesis but may also provide an AD therapeutic strategy through ERAD-mediated APP degradation.
Footnotes
This study was supported by Grants-in-Aid for Scientific Research (KAKENHI) 19790070, 19300135, and 20659013 from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank T. Suzuki and Otsuka GEN Research Institute for providing materials. We also thank M. Itano-Nakazawa, K. Takata, and T. Omura for helpful discussions. We are grateful to U. Yokose, M. Matsuo, K. Murahashi, E. Tominaga, M. Onoguchi, N. Sugioka, and S. Touno for technical assistance.
References
- Amano T, Yamasaki S, Yagishita N, Tsuchimochi K, Shin H, Kawahara K, Aratani S, Fujita H, Zhang L, Ikeda R, Fujii R, Miura N, Komiya S, Nishioka K, Maruyama I, Fukamizu A, Nakajima T. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003;17:2436–2449. doi: 10.1101/gad.1096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apodaca J, Kim I, Rao H. Cellular tolerance of prion protein PrP in yeast involves proteolysis and the unfolded protein response. Biochem Biophys Res Commun. 2006;347:319–326. doi: 10.1016/j.bbrc.2006.06.078. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell WL, Pham HV, Glabe CG. gamma-secretase cleavage is distinct from endoplasmic reticulum degradation of the transmembrane domain of the amyloid precursor protein. J Biol Chem. 1998;273:31947–31955. doi: 10.1074/jbc.273.48.31947. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kimura H, Schubert D. A novel mechanism for the regulation of amyloid precursor protein metabolism. J Cell Biol. 2002;158:79–89. doi: 10.1083/jcb.200110151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare JF. Intracellular pathways of folded and misfolded amyloid precursor protein degradation. Arch Biochem Biophys. 2006;451:79–90. doi: 10.1016/j.abb.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Guénette SY, Peach C, Greco C, Xia W, Kim DY, Barren C, Tanzi RE, Kovacs DM. HtrA2 regulates beta-amyloid precursor protein (APP) metabolism through endoplasmic reticulum-associated degradation. J Biol Chem. 2007;282:28285–28295. doi: 10.1074/jbc.M702951200. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Ismail N, Ng DT. Have you HRD? Understanding ERAD is DOAble! Cell. 2006;126:237–239. doi: 10.1016/j.cell.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Nomura Y. ER signaling in unfolded protein response. Life Sci. 2003;74:199–205. doi: 10.1016/j.lfs.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Ishiguro M, Niinuma Y, Uesugi M, Nomura Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett. 2002;532:147–152. doi: 10.1016/s0014-5793(02)03660-8. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Yasui S, Niinuma Y, Arai K, Omura T, Okuma Y, Nomura Y. A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett. 2007;581:5355–5360. doi: 10.1016/j.febslet.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–485. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Nadav E, Shmueli A, Barr H, Gonen H, Ciechanover A, Reiss Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem Biophys Res Commun. 2003;303:91–97. doi: 10.1016/s0006-291x(03)00279-1. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Omura T, Kaneko M, Okuma Y, Orba Y, Nagashima K, Takahashi R, Fujitani N, Matsumura S, Hata A, Kubota K, Murahashi K, Uehara T, Nomura Y. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J Neurochem. 2006;99:1456–1469. doi: 10.1111/j.1471-4159.2006.04155.x. [DOI] [PubMed] [Google Scholar]
- Omura T, Kaneko M, Onoguchi M, Koizumi S, Itami M, Ueyama M, Okuma Y, Nomura Y. Novel functions of ubiquitin ligase HRD1 with transmembrane and proline-rich domains. J Pharmacol Sci. 2008a;106:512–519. doi: 10.1254/jphs.08005fp. [DOI] [PubMed] [Google Scholar]
- Omura T, Kaneko M, Tabei N, Okuma Y, Nomura Y. Immunohistochemical localization of a ubiquitin ligase HRD1 in murine brain. J Neurosci Res. 2008b;86:1577–1587. doi: 10.1002/jnr.21616. [DOI] [PubMed] [Google Scholar]
- Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, Min do S, Kang S, Rhim H. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem. 2006;281:34277–34287. doi: 10.1074/jbc.M603443200. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3:246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Yagishita N, Sasaki T, Nakazawa M, Kato Y, Yamadera T, Bae E, Toriyama S, Ikeda R, Zhang L, Fujitani K, Yoo E, Tsuchimochi K, Ohta T, Araya N, Fujita H, Aratani S, Eguchi K, Komiya S, Maruyama I, et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007;26:113–122. doi: 10.1038/sj.emboj.7601490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhong X, Ballar P, Luo S, Shen Y, Rubinsztein DC, Monteiro MJ, Fang S. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp Cell Res. 2007;313:538–550. doi: 10.1016/j.yexcr.2006.10.031. [DOI] [PubMed] [Google Scholar]